
Dysfunctional K+ Homeostasis as a Driver for Brain Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Potassium Homeostasis and Brain Inflammation
2.1. Microglia
2.1.1. Microglial Voltage-Gated Potassium Channels in Inflammation
2.1.2. Microglial 2KP Channels in Inflammation
2.1.3. Microglial Calcium-Activated Potassium Channels in Inflammation
2.1.4. Microglial Inward Rectifying Potassium Channels in Inflammation
2.1.5. Microglial ATP-Gated Cation Channels in Inflammation
2.1.6. Microglial Transporters in Inflammation
2.2. Neurons
2.3. Astrocytes
2.3.1. Astrocytic Voltage-Gated Potassium Channels in Inflammation
2.3.2. Astrocytic Inward Rectifying Potassium Channels in Inflammation
2.3.3. Astrocytic Calcium-Activated Potassium Channels in Inflammation
2.3.4. Other Astrocyte K+ Homeostatic Molecular Targets
2.4. Oligodendrocytes
3. Conclusions and Prospects
Funding
Conflicts of Interest
References
- Kettritz, R.; Loffing, J. Potassium homeostasis—Physiology and pharmacology in a clinical context. Pharmacol. Ther. 2023, 249, 108489. [Google Scholar] [CrossRef] [PubMed]
- Mcdonough, A.A.; Fenton, R.A. Potassium homeostasis: Sensors, mediators, and targets. Pflügers Arch.-Eur. J. Physiol. 2022, 474, 853–867. [Google Scholar] [CrossRef] [PubMed]
- Schmaul, S.; Hanuscheck, N.; Bittner, S. Astrocytic potassium and calcium channels as integrators of the inflammatory and ischemic CNS microenvironment. Biol. Chem. 2021, 402, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Sahranavard, T.; Carbone, F.; Montecucco, F.; Xu, S.; Al-Rasadi, K.; Jamialahmadi, T.; Sahebkar, A. The role of potassium in atherosclerosis. Eur. J. Clin. Investig. 2021, 51, e13454. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Fujikawa, D.G.; Kim, J.S.; Daniels, A.H.; Alcaraz, A.F.; Sohn, T.B. In vivo elevation of extracellular potassium in the rat amygdala increases extracellular glutamate and aspartate and damages neurons. Neuroscience 1996, 74, 695–706. [Google Scholar] [CrossRef]
- Villa, C.; Suphesiz, H.; Combi, R.; Akyuz, E. Potassium channels in the neuronal homeostasis and neurodegenerative pathways underlying Alzheimer’s disease: An update. Mech. Ageing Dev. 2020, 185, 111197. [Google Scholar] [CrossRef]
- Ding, F.; Sun, Q.; Long, C.; Rasmussen, R.N.; Peng, S.; Xu, Q.; Kang, N.; Song, W.; Weikop, P.; Goldman, S.A.; et al. Dysregulation of extracellular potassium distinguishes healthy ageing from neurodegeneration. Brain 2024, 147, 1726–1739. [Google Scholar] [CrossRef]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, M.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Canas, C.A.; Castano-Valencia, S.; Castro-Herrera, F. Pharmacological blockade of KV1.3 channel as a promising treatment in autoimmune diseases. J. Transl. Autoimmun. 2022, 5, 100146. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Feng, Y.; Quinn, R.J.; Pountney, D.L.; Richardson, D.R.; Mellick, G.D.; Ma, L. Potassium Channels in Parkinson’s Disease: Potential Roles in Its Pathogenesis and Innovative Molecular Targets for Treatment. Pharmacol. Rev. 2023, 75, 758–788. [Google Scholar] [CrossRef]
- Luo, Y.; Huang, L.; Liao, P.; Jiang, R. Contribution of Neuronal and Glial Two-Pore-Domain Potassium Channels in Health and Neurological Disorders. Neural Plasticity 2021, 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Kruys, V.; Vamecq, J.; Maze, M. The Role of Microglia in Perioperative Neuroinflammation and Neurocognitive Disorders. Front. Aging Neurosci. 2021, 13, 671499. [Google Scholar] [CrossRef] [PubMed]
- York, E.M.; Bernier, L.-P.; Macvicar, B.A. Microglial modulation of neuronal activity in the healthy brain. Dev. Neurobiol. 2018, 78, 593–603. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Grossinger, E.M.; Horiuchi, M.; Davis, K.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017, 65, 106–121. [Google Scholar] [CrossRef]
- Maezawa, I.; Nguyen, H.M.; Di Lucente, J.; Jenkins, D.P.; Singh, V.; Hilt, S.; Kim, K.; Rangaraju, S.; Levey, A.I.; Wulff, H.; et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: Preclinical proof of concept. Brain 2018, 141, 596–612. [Google Scholar] [CrossRef]
- Rangaraju, S.; Raza, S.A.; Pennati, A.; Deng, Q.; Dammer, E.B.; Duong, D.; Pennington, M.W.; Tansey, M.G.; Lah, J.J.; Betarbet, R.; et al. A systems pharmacology-based approach to identify novel Kv1.3 channel-dependent mechanisms in microglial activation. J. Neuroinflamm. 2017, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Kaur, C.; Sivakumar, V.; Lu, J.; Ling, E.A. Kv1.1 expression in microglia regulates production and release of proinflammatory cytokines, endothelins and nitric oxide. Neuroscience 2009, 158, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Wu, C.Y.; Kaur, C.; Sivakumar, V.; Sun, J.; Li, S.; Ling, E.A. Expression of Kv1.2 in microglia and its putative roles in modulating production of proinflammatory cytokines and reactive oxygen species. J. Neurochem. 2008, 106, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Charolidi, N.; Schilling, T.; Eder, C. Microglial Kv1.3 Channels and P2Y12 Receptors Differentially Regulate Cytokine and Chemokine Release from Brain Slices of Young Adult and Aged Mice. PLoS ONE 2015, 10, e0128463. [Google Scholar] [CrossRef]
- Fordyce, C.B. Microglia Kv1.3 Channels Contribute to Their Ability to Kill Neurons. J. Neurosci. 2005, 25, 7139–7149. [Google Scholar] [CrossRef]
- Khanna, R.; Roy, L.; Zhu, X.; Schlichter, L.C. K+ channels and the microglial respiratory burst. Am. J. Physiol.-Cell Physiol. 2001, 280, C796–C806. [Google Scholar] [CrossRef]
- Kotecha, S.A.; Schlichter, L.C. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J. Neurosci. 1999, 19, 10680–10693. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Blomster, L.V.; Christophersen, P.; Wulff, H. Potassium channel expression and function in microglia: Plasticity and possible species variations. Channels 2017, 11, 305–315. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Effects of kinase inhibitors on TGF-β induced upregulation of Kv1.3 K+ channels in brain macrophages. Pflügers Arch. Eur. J. Physiol. 2003, 447, 312–315. [Google Scholar] [CrossRef]
- Kust, B.M.; Biber, K.; van Calker, D.; Gebicke-Haerter, P.J. Regulation of K+ channel mRNA expression by stimulation of adenosine A2a-receptors in cultured rat microglia. Glia 1999, 25, 120–130. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Di Lucente, J.; Nguyen, H.M.; Wulff, H.; Jin, L.W.; Maezawa, I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 2018, 66, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; di Lucente, J.; Chen, Y.J.; Cui, Y.; Ibrahim, R.H.; Pennington, M.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 2020, 68, 2377–2394. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.A.; Nguyen, H.M.; Lin, Y.; Bagchi, P.; Natu, A.; Espinosa-Garcia, C.; Werner, E.; Kumari, R.; Brandelli, A.D.; Kumar, P.; et al. Proximity Labeling Proteomics Reveals Kv1.3 Potassium Channel Immune Interactors in Microglia. Mol. Cell. Proteom. 2024, 23, 100809. [Google Scholar] [CrossRef]
- Ramesha, S.; Rayaprolu, S.; Bowen, C.A.; Giver, C.R.; Bitarafan, S.; Nguyen, H.M.; Gao, T.; Chen, M.J.; Nwabueze, N.; Dammer, E.B.; et al. Unique molecular characteristics and microglial origin of Kv1.3 channel-positive brain myeloid cells in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2013545118. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Rangaraju, S.; Gearing, M.; Jin, L.W.; Levey, A. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 797–808. [Google Scholar] [CrossRef]
- Chung, S.; Lee, J.; Joe, E.H.; Uhm, D.Y. Beta-amyloid peptide induces the expression of voltage dependent outward rectifying K+ channels in rat microglia. Neurosci. Lett. 2001, 300, 67–70. [Google Scholar] [CrossRef]
- Sarkar, S.; Nguyen, H.M.; Malovic, E.; Luo, J.; Langley, M.; Palanisamy, B.N.; Singh, N.; Manne, S.; Neal, M.; Gabrielle, M.; et al. Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J. Clin. Investig. 2020, 130, 4195–4212. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, P.; Zhang, Y.; Wu, Y.; Song, Y.; Wang, X.; Chen, T.; Peng, B.; Liu, W.; Yin, J.; et al. Blockade of Kv1.3 Potassium Channel Inhibits Microglia-Mediated Neuroinflammation in Epilepsy. Int. J. Mol. Sci. 2022, 23, 14693. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, K.; Li, Z.; Zhao, Y.; Wang, Y.; Hu, B.; Xu, P.; Shi, X.; Zhou, B.; Pennington, M.; et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro-Oncology 2014, 16, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Jou, I.; Pyo, H.; Chung, S.; Jung, S.Y.; Gwag, B.J.; Joe, E.H. Expression of Kv1.5 K+ channels in activated microglia in vivo. Glia 1998, 24, 408–414. [Google Scholar] [CrossRef]
- Beeton, C.; Pennington, M.W.; Wulff, H.; Singh, S.; Nugent, D.; Crossley, G.; Khaytin, I.; Calabresi, P.A.; Chen, C.-Y.; Gutman, G.A.; et al. Targeting Effector Memory T Cells with a Selective Peptide Inhibitor of Kv1.3 Channels for Therapy of Autoimmune Diseases. Mol. Pharmacol. 2005, 67, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Kundu-Raychaudhuri, S.; Chen, Y.-J.; Wulff, H.; Raychaudhuri, S.P. Kv1.3 in psoriatic disease: PAP-1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis—Xenograft model. J. Autoimmun. 2014, 55, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Tajti, G.; Panyi, G. The Kv1.3 K+ channel in the immune system and its “precision pharmacology” using peptide toxins. Biol. Futur. 2021, 72, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tarcha, E.J.; Olsen, C.M.; Probst, P.; Peckham, D.; Muñoz-Elías, E.J.; Kruger, J.G.; Iadonato, S.P. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS ONE 2017, 12, e0180762. [Google Scholar] [CrossRef]
- Tarcha, E.J.; Chi, V.; Muñoz-Elías, E.J.; Bailey, D.; Londono, L.M.; Upadhyay, S.K.; Norton, K.; Banks, A.; Tjong, I.; Nguyen, H.; et al. Durable Pharmacological Responses from the Peptide ShK-186, a Specific Kv1.3 Channel Inhibitor That Suppresses T Cell Mediators of Autoimmune Disease. J. Pharmacol. Exp. Ther. 2012, 342, 642–653. [Google Scholar] [CrossRef]
- Madry, C.; Kyrargyri, V.; Arancibia-Carcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron 2018, 97, 299–312.e296. [Google Scholar] [CrossRef]
- Drinkall, S.; Lawrence, C.B.; Ossola, B.; Russell, S.; Bender, C.; Brice, N.B.; Dawson, L.A.; Harte, M.; Brough, D. The two pore potassium channel THIK-1 regulates NLRP3 inflammasome activation. Glia 2022, 70, 1301–1316. [Google Scholar] [CrossRef]
- Ossola, B.; Rifat, A.; Rowland, A.; Hunter, H.; Drinkall, S.; Bender, C.; Hamlischer, M.; Teall, M.; Burley, R.; Barker, D.F.; et al. Characterisation of C101248: A novel selective THIK-1 channel inhibitor for the modulation of microglial NLRP3-inflammasome. Neuropharmacology 2023, 224, 109330. [Google Scholar] [CrossRef]
- Rifat, A.; Ossola, B.; Bürli, R.W.; Dawson, L.A.; Brice, N.L.; Rowland, A.; Lizio, M.; Xu, X.; Page, K.; Fidzinski, P.; et al. Differential contribution of THIK-1 K+ channels and P2X7 receptors to ATP-mediated neuroinflammation by human microglia. J. Neuroinflamm. 2024, 21, 58. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Lively, S.; Schlichter, L.C. IL-4 type 1 receptor signaling up-regulates KCNN4 expression, and increases the KCa3.1 current and its contribution to migration of alternative-activated microglia. Front. Cell. Neurosci. 2014, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Schlichter, L.C. Selective Activation of KCa3.1 and CRAC Channels by P2Y2 Receptors Promotes Ca2+ Signaling, Store Refilling and Migration of Rat Microglial Cells. PLoS ONE 2013, 8, e62345. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Koeberle, P.D.; Wang, Y.; Schlichter, L.C. The Ca2+-Activated K+ Channel KCNN4/KCa3.1 Contributes to Microglia Activation and Nitric Oxide-Dependent Neurodegeneration. J. Neurosci. 2007, 27, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Bouhy, D.; Ghasemlou, N.; Lively, S.; Redensek, A.; Rathore, K.I.; Schlichter, L.C.; David, S. Inhibition of the Ca2+-Dependent K+Channel, KCNN4/KCa3.1, Improves Tissue Protection and Locomotor Recovery after Spinal Cord Injury. J. Neurosci. 2011, 31, 16298–16308. [Google Scholar] [CrossRef]
- Lam, D.; Schlichter, L.C. Expression and contributions of the Kir2.1 inward-rectifier K+ channel to proliferation, migration and chemotaxis of microglia in unstimulated and anti-inflammatory states. Front. Cell. Neurosci. 2015, 9, 185. [Google Scholar] [CrossRef]
- Gattlen, C.; Deftu, A.F.; Tonello, R.; Ling, Y.; Berta, T.; Ristoiu, V.; Suter, M.R. The inhibition of Kir2.1 potassium channels depolarizes spinal microglial cells, reduces their proliferation, and attenuates neuropathic pain. Glia 2020, 68, 2119–2135. [Google Scholar] [CrossRef]
- Du, R.-H.; Sun, H.-B.; Hu, Z.-L.; Lu, M.; Ding, J.-H.; Hu, G. Kir6.1/K-ATP channel modulates microglia phenotypes: Implication in Parkinson’s disease. Cell Death Dis. 2018, 9, 404. [Google Scholar] [CrossRef]
- Tang, Z.; Shao, X.; Wu, J.; Chen, H.; Zhang, A.; Xu, F.; Ping, H.; Li, S.; Liu, C.; Li, Y.; et al. Naloxone Protects against Lipopolysaccharide-Induced Neuroinflammation and Microglial Activation via Inhibiting ATP-Sensitive Potassium Channel. Comput. Math. Methods Med. 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Li, H.; Chen, D.; Sun, W.; Chen, J.; Luo, C.; Xu, H.; Ma, J.H.; Tang, S. KATP Opener Attenuates Diabetic-Induced Muller Gliosis and Inflammation by Modulating Kir6.1 in Microglia. Investig. Ophthalmol. Vis. Sci. 2021, 62, 3. [Google Scholar] [CrossRef]
- Shu, Q.; Liu, J.; Liu, X.; Zhao, S.; Li, H.; Tan, Y.; Xu, J. GABABR/GSK-3β/NF-κB signaling pathway regulates the proliferation of colorectal cancer cells. Cancer Med. 2016, 5, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Chen, K.; Zhou, M.; Liu, Z.; Chen, H. Kir2.1 and K2P1 channels reconstitute two levels of resting membrane potential in cardiomyocytes. J. Physiol. 2017, 595, 5129–5142. [Google Scholar] [CrossRef] [PubMed]
- Reilly, L.; Eckhardt, L.L. Cardiac potassium inward rectifier Kir2: Review of structure, regulation, pharmacology, and arrhythmogenesis. Heart Rhythm. 2021, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Van Der Heyden, M.A.G. The network of cardiac KIR2.1: Its function, cellular regulation, electrical signaling, diseases and new drug avenues. Naunyn-Schmiedebergs Arch. Pharmacol. 2024, 397, 6369–6389. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Wang, S.-H.; Xu, L.-N.; Su, X.-L.; Zeng, Y.-F.; Wang, P.; Zhang, L.-R.; Han, S.-N. Glycogen synthase kinase 3 beta inhibitor SB216763 improves Kir2.1 expression after myocardia infraction in rats. J. Interv. Card. Electrophysiol. 2022, 63, 239–248. [Google Scholar] [CrossRef]
- Koide, M.; Harraz, O.F.; Dabertrand, F.; Longden, T.A.; Ferris, H.R.; Wellman, G.C.; Hill-Eubanks, D.C.; Greenstein, A.S.; Nelson, M.T. Differential restoration of functional hyperemia by antihypertensive drug classes in hypertension-related cerebral small vessel disease. J. Clin. Investig. 2021, 131(18), e149029. [Google Scholar] [CrossRef]
- Barbera-Cremades, M.; Gomez, A.I.; Baroja-Mazo, A.; Martinez-Alarcon, L.; Martinez, C.M.; de Torre-Minguela, C.; Pelegrin, P. P2X7 Receptor Induces Tumor Necrosis Factor-alpha Converting Enzyme Activation and Release to Boost TNF-alpha Production. Front. Immunol. 2017, 8, 862. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Z.M.; Wu, X.; Zhang, L.; Cao, Y.; Zhou, P. Distinct Molecular Mechanisms Underlying Potassium Efflux for NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609441. [Google Scholar] [CrossRef]
- Burm, S.M.; Zuiderwijk-Sick, E.A.; Weert, P.M.; Bajramovic, J.J. ATP-induced IL-1beta secretion is selectively impaired in microglia as compared to hematopoietic macrophages. Glia 2016, 64, 2231–2246. [Google Scholar] [CrossRef]
- Ronning, K.E.; Déchelle-Marquet, P.-A.; Che, Y.; Guillonneau, X.; Sennlaub, F.; Delarasse, C. The P2X7 Receptor, a Multifaceted Receptor in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11747. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, J.; Zhang, W.; Zhang, J.; Yang, J.; Li, K.; He, Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 2013, 45, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulos, J.M.; Almeida-Da-Silva, C.L.C.; Rüütel Boudinot, S.; Ojcius, D.M. Structural and Functional Features of the P2X4 Receptor: An Immunological Perspective. Front. Immunol. 2021, 12, 645834. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Chico, A.; Manterola, A.; Cipriani, R.; Katona, I.; Matute, C.; Mato, S. P2x7 receptors control demyelination and inflammation in the cuprizone model. Brain Behav. Immun. Health 2020, 4, 100062. [Google Scholar] [CrossRef]
- Yuan, H.; Lu, B.; Ji, Y.; Meng, B.; Wang, R.; Sun, D.; Liu, R.; Zhai, X.; Li, X.; Qin, J.; et al. Role of P2X4/NLRP3 Pathway-Mediated Neuroinflammation in Perioperative Neurocognitive Disorders. Mediat. Inflamm. 2022, 2022, 6355805. [Google Scholar] [CrossRef]
- He, W.; Wang, Q.; Sha, W.; Wang, L.; Li, D.; Chen, G. P2X4 Inhibition reduces microglia inflammation and apoptosis by NLRP3 and improves nervous system defects in rat brain trauma model. J. Clin. Neurosci. 2022, 99, 224–232. [Google Scholar] [CrossRef]
- Schilling, T.; Stock, C.; Schwab, A.; Eder, C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur. J. Neurosci. 2004, 19, 1469–1474. [Google Scholar] [CrossRef]
- Annunziato, L.; Boscia, F.; Pignataro, G. Ionic transporter activity in astrocytes, microglia, and oligodendrocytes during brain ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 969–982. [Google Scholar] [CrossRef]
- Tóth, K.; Lénárt, N.; Berki, P.; Fekete, R.; Szabadits, E.; Pósfai, B.; Cserép, C.; Alatshan, A.; Benkő, S.; Kiss, D.; et al. The NKCC1 ion transporter modulates microglial phenotype and inflammatory response to brain injury in a cell-autonomous manner. PLoS Biol. 2022, 20, e3001526. [Google Scholar] [CrossRef]
- Tessier, M.; Saez Garcia, M.; Goubert, E.; Blasco, E.; Consumi, A.; Dehapiot, B.; Tian, L.; Molinari, F.; Laurin, J.; Guillemot, F.; et al. Bumetanide induces post-traumatic microglia-interneuron contact to promote neurogenesis and recovery. Brain 2023, 146, 4247–4261. [Google Scholar] [CrossRef]
- Georgoula, C.; Ferrin, M.; Pietraszczyk-Kedziora, B.; Hervas, A.; Marret, S.; Oliveira, G.; Rosier, A.; Crutel, V.; Besse, E.; Severo, C.A.; et al. A Phase III Study of Bumetanide Oral Liquid Formulation for the Treatment of Children and Adolescents Aged Between 7 and 17 Years with Autism Spectrum Disorder (SIGN 1 Trial): Participant Baseline Characteristics. Child Psychiatry Hum. Dev. 2023, 54, 1360–1372. [Google Scholar] [CrossRef]
- Wang, T.; Shan, L.; Miao, C.; Xu, Z.; Jia, F. Treatment Effect of Bumetanide in Children With Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2021, 12, 751575. [Google Scholar] [CrossRef] [PubMed]
- Taubes, A.; Nova, P.; Zalocusky, K.A.; Kosti, I.; Bicak, M.; Zilberter, M.Y.; Hao, Y.; Yoon, S.Y.; Oskotsky, T.; Pineda, S.; et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nat. Aging 2021, 1, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Boyarko, B.; Podvin, S.; Greenberg, B.; Momper, J.D.; Huang, Y.; Gerwick, W.H.; Bang, A.G.; Quinti, L.; Griciuc, A.; Kim, D.Y.; et al. Evaluation of bumetanide as a potential therapeutic agent for Alzheimer’s disease. Front. Pharmacol. 2023, 14, 1190402. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Kam, T.-I.; Wang, H.; Neifert, S.; Chou, S.-C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 2022, 110, 2422–2437.e2429. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Wang, W.; Nguyen, L.T.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; Leblanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.C.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar] [CrossRef]
- Fann, D.Y.-W.; Lim, Y.-A.; Cheng, Y.-L.; Lok, K.-Z.; Chunduri, P.; Baik, S.-H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.-G.; et al. Evidence that NF-κB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons Following Ischemic Stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef]
- Noh, W.; Pak, S.; Choi, G.; Yang, S.; Yang, S. Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Front. Cell. Neurosci. 2019, 13, 265. [Google Scholar] [CrossRef]
- Goldstein, S.A.N.; Bayliss, D.A.; Kim, D.; Lesage, F.; Plant, L.D.; Rajan, S. International Union of Pharmacology. LV. Nomenclature and Molecular Relationships of Two-P Potassium Channels. Pharmacol. Rev. 2005, 57, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; Mckinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and Molecular Relationships of Voltage-Gated Potassium Channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Adelman, J.P.; Clapham, D.E.; Jan, L.Y.; Karschin, A.; Kurachi, Y.; Lazdunski, M.; Nichols, C.G.; Seino, S.; Vandenberg, C.A. International Union of Pharmacology. LIV. Nomenclature and Molecular Relationships of Inwardly Rectifying Potassium Channels. Pharmacol. Rev. 2005, 57, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef]
- Gao, F.; Liu, Z.; Ren, W.; Jiang, W. Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro. Neuropsychiatr. Dis. Treat. 2014, 10, 1489–1495. [Google Scholar] [CrossRef]
- Tzour, A.; Leibovich, H.; Barkai, O.; Biala, Y.; Lev, S.; Yaari, Y.; Binshtok, A.M. KV7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J. Physiol. 2017, 595, 713–738. [Google Scholar] [CrossRef]
- Hung, C.H.; Chin, Y.; Fong, Y.O.; Lee, C.H.; Han, D.S.; Lin, J.H.; Sun, W.H.; Chen, C.C. Acidosis-related pain and its receptors as targets for chronic pain. Pharmacol. Ther. 2023, 247, 108444. [Google Scholar] [CrossRef]
- Meuth, S.G.; Kanyshkov, T.; Melzer, N.; Bittner, S.; Kieseier, B.C.; Budde, T.; Wiendl, H. Altered neuronal expression of TASK1 and TASK3 potassium channels in rodent and human autoimmune CNS inflammation. Neurosci. Lett. 2008, 446, 133–138. [Google Scholar] [CrossRef]
- Liao, P.; Qiu, Y.; Mo, Y.; Fu, J.; Song, Z.; Huang, L.; Bai, S.; Wang, Y.; Zhu, J.J.; Tian, F.; et al. Selective activation of TWIK-related acid-sensitive K+ 3 subunit-containing channels is analgesic in rodent models. Sci. Transl. Med. 2019, 11, eaaw8434. [Google Scholar] [CrossRef]
- Harris, B.M.; Hughes, D.I.; Bolton, P.S.; Tadros, M.A.; Callister, R.J.; Graham, B.A. Contrasting Alterations to Synaptic and Intrinsic Properties in Upper-Cervical Superficial Dorsal Horn Neurons following Acute Neck Muscle Inflammation. Mol. Pain 2014, 10, 25. [Google Scholar] [CrossRef]
- Biet, M.; Dansereau, M.A.; Sarret, P.; Dumaine, R. The neuronal potassium current IA is a potential target for pain during chronic inflammation. Physiol. Rep. 2021, 9, e14975. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D.; Lamkanfi, M.; Kim, Y.-G.; Chen, G.; Park, J.-H.; Franchi, L.; Vandenabeele, P.; Núñez, G. Pannexin-1-Mediated Recognition of Bacterial Molecules Activates the Cryopyrin Inflammasome Independent of Toll-like Receptor Signaling. Immunity 2007, 26, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Purohit, R.; Bera, A.K. Pannexin 1 plays a pro-survival role by attenuating P2X7 receptor-mediated Ca2+ influx. Cell Calcium 2021, 99, 102458. [Google Scholar] [CrossRef]
- Silverman, W.R.; De Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The Pannexin 1 Channel Activates the Inflammasome in Neurons and Astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Pivoriunas, A. Astroglia support, regulate and reinforce brain barriers. Neurobiol. Dis. 2023, 179, 106054. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Mata-Martínez, E.; Díaz-Muñoz, M.; Vázquez-Cuevas, F.G. Glial Cells and Brain Diseases: Inflammasomes as Relevant Pathological Entities. Front. Cell. Neurosci. 2022, 16, 929529. [Google Scholar] [CrossRef]
- Lénárt, N.; Brough, D.; Dénes, Á. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J. Cereb. Blood Flow Metab. 2016, 36, 1668–1685. [Google Scholar] [CrossRef]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef]
- Mészáros, Á.; Molnár, K.; Fazakas, C.; Nógrádi, B.; Lüvi, A.; Dudás, T.; Tiszlavicz, L.; Farkas, A.E.; Krizbai, I.A.; Wilhelm, I. Inflammasome activation in peritumoral astrocytes is a key player in breast cancer brain metastasis development. Acta Neuropathol. Commun. 2023, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Lee, B.W.L.; Sng, Y.J.; Poh, L.; Rajeev, V.; Selvaraji, S.; Drummond, G.R.; Sobey, C.G.; Arumugam, T.V.; Fann, D.Y. Inflammasome Activation Mediates Apoptotic and Pyroptotic Death in Astrocytes Under Ischemic Conditions. NeuroMol. Med. 2023, 25, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, Y.; He, Z.; Xu, Y.; Li, X.; Ding, J.; Lu, M.; Hu, G. Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav. Immun. 2020, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, N.; Zhang, L.; Meng, C.; Zhao, J.; Wu, J. NLRP6 expressed in astrocytes aggravates neurons injury after OGD/R through activating the inflammasome and inducing pyroptosis. Int. Immunopharmacol. 2020, 80, 106183. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.-Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Sim, J.; Ahn, J.W.; Park, J.; Kim, Y.J.; Jeong, J.-Y.; Lee, J.M.; Cho, K.; Ahn, H.J.; Sung, K.S.; Moon, J.-S.; et al. Non-canonical NLRC4 inflammasomes in astrocytes contribute to glioma malignancy. Inflamm. Res. 2023, 72, 813–827. [Google Scholar] [CrossRef]
- Barclay, W.E.; Aggarwal, N.; Deerhake, M.E.; Inoue, M.; Nonaka, T.; Nozaki, K.; Luzum, N.A.; Miao, E.A.; Shinohara, M.L. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight 2022, 7, e155563. [Google Scholar] [CrossRef]
- Mcneill, J.; Rudyk, C.; Hildebrand, M.E.; Salmaso, N. Ion Channels and Electrophysiological Properties of Astrocytes: Implications for Emergent Stimulation Technologies. Front. Cell. Neurosci. 2021, 15, 644126. [Google Scholar] [CrossRef]
- Bozic, I.; Tesovic, K.; Laketa, D.; Adzic, M.; Jakovljevic, M.; Bjelobaba, I.; Savic, D.; Nedeljkovic, N.; Pekovic, S.; Lavrnja, I. Voltage Gated Potassium Channel Kv1.3 Is Upregulated on Activated Astrocytes in Experimental Autoimmune Encephalomyelitis. Neurochem. Res. 2018, 43, 1020–1034. [Google Scholar] [CrossRef]
- Zurolo, E.; De Groot, M.; Iyer, A.; Anink, J.; Van Vliet, E.A.; Heimans, J.J.; Reijneveld, J.C.; Gorter, J.A.; Aronica, E. Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: A role for interleukin-1 β. J. Neuroinflamm. 2012, 9, 280. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Bian, Z.; Zhang, Z.; Wang, X.; Zhu, A.; Zhu, G. Astrocytic Kir4.1 regulates NMDAR/calpain signaling axis in lipopolysaccharide-induced depression-like behaviors in mice. Toxicol. Appl. Pharmacol. 2021, 429, 115711. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aslam, M.; Kalluri, S.R.; Schirmer, L.; Buck, D.; Tackenberg, B.; Rothhammer, V.; Chan, A.; Gold, R.; Berthele, A.; et al. Potassium Channel KIR4.1 as an Immune Target in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Mukai, T.; Nagao, Y.; Matsuba, Y.; Tsuji, Y.; Tanaka, S.; Tokudome, K.; Shimizu, S.; Ito, H.; Ikeda, A.; et al. Inhibition of Inwardly Rectifying Potassium (Kir) 4.1 Channels Facilitates Brain-Derived Neurotrophic Factor (BDNF) Expression in Astrocytes. Front. Mol. Neurosci. 2017, 10, 408. [Google Scholar] [CrossRef]
- Ohno, Y.; Hibino, H.; Lossin, C.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007, 1178, 44–51. [Google Scholar] [CrossRef]
- Su, S.; Ohno, Y.; Lossin, C.; Hibino, H.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J. Pharmacol. Exp. Ther. 2007, 320, 573–580. [Google Scholar] [CrossRef]
- Ohno, Y. Astrocytic Kir4.1 potassium channels as a novel therapeutic target for epilepsy and mood disorders. Neural Regen. Res. 2018, 13, 651–652. [Google Scholar] [CrossRef]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef]
- Li, X.-T. The involvement of K+ channels in depression and pharmacological effects of antidepressants on these channels. Transl. Psychiatry 2024, 14, 411. [Google Scholar] [CrossRef]
- Hu, Z.L.; Sun, T.; Lu, M.; Ding, J.H.; Du, R.H.; Hu, G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain Behav. Immun. 2019, 81, 509–522. [Google Scholar] [CrossRef]
- Ng, K.-E.; Schwarzer, S.; Duchen, M.R.; Tinker, A. The Intracellular Localization and Function of the ATP-Sensitive K+ Channel Subunit Kir6.1. J. Membr. Biol. 2010, 234, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-F.; Chen, Z.-Z.; Zhao, Z.; Yang, D.-D.; Yan, H.; Ji, J.; Sun, X.-L. Potential role of microRNA-7 in the anti-neuroinflammation effects of nicorandil in astrocytes induced by oxygen-glucose deprivation. J. Neuroinflamm. 2016, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Wang, Y.; Xu, W.; Liu, Y.; Chen, H.; Yu, Z. KCa3.1 deficiency attenuates neuroinflammation by regulating an astrocyte phenotype switch involving the PI3K/AKT/GSK3beta pathway. Neurobiol. Dis. 2019, 132, 104588. [Google Scholar] [CrossRef] [PubMed]
- Cong, T.; Sun, Y.; Zhou, Y.; Wu, H.; Li, L.; Chu, Z.; Chen, X.; Li, J.; Zhao, D.; Wang, Y.; et al. Blocking Two-Pore Domain Potassium Channel TREK-1 Inhibits the Activation of A1-Like Reactive Astrocyte Through the NF-κB Signaling Pathway in a Rat Model of Major Depressive Disorder. Neurochem. Res. 2023, 48, 1737–1754. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-κB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.-L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: The role of TLR3 activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef]
- Boccazzi, M.S.R.; Raffaele, S.; Fumagalli, M. Not only myelination: The immuneinflammatory functions of oligodendrocytes. Neural Regen. Res. 2022, 17, 2661–2663. [Google Scholar]
- Zhang, X.; Wang, R.; Hu, D.; Sun, X.; Fujioka, H.; Lundberg, K.; Chan, E.R.; Wang, Q.; Xu, R.; Flanagan, M.E.; et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 2020, 6, eabb8680. [Google Scholar] [CrossRef]
- ELBini, I.; Neili, N.-E. Potassium channels at the crossroads of neuroinflammation and myelination in experimental models of multiple sclerosis. Biochem. Biophys. Res. Commun. 2023, 653, 140–146. [Google Scholar] [CrossRef]
- Xuan, Y.; Yan, G.; Wu, R.; Huang, Q.; Li, X.; Xu, H. The cuprizone-induced changes in (1)H-MRS metabolites and oxidative parameters in C57BL/6 mouse brain: Effects of quetiapine. Neurochem. Int. 2015, 90, 185–192. [Google Scholar] [CrossRef]
- Pasquini, L.A.; Calatayud, C.A.; Bertone Uña, A.L.; Millet, V.; Pasquini, J.M.; Soto, E.F. The Neurotoxic Effect of Cuprizone on Oligodendrocytes Depends on the Presence of Pro-inflammatory Cytokines Secreted by Microglia. Neurochem. Res. 2007, 32, 279–292. [Google Scholar] [CrossRef]
- Kapell, H.; Fazio, L.; Dyckow, J.; Schwarz, S.; Cruz-Herranz, A.; Mayer, C.; Campos, J.; D’Este, E.; Möbius, W.; Cordano, C.; et al. Neuron-oligodendrocyte potassium shuttling at nodes of Ranvier protects against inflammatory demyelination. J. Clin. Investig. 2023, 133, e164223. [Google Scholar] [CrossRef]
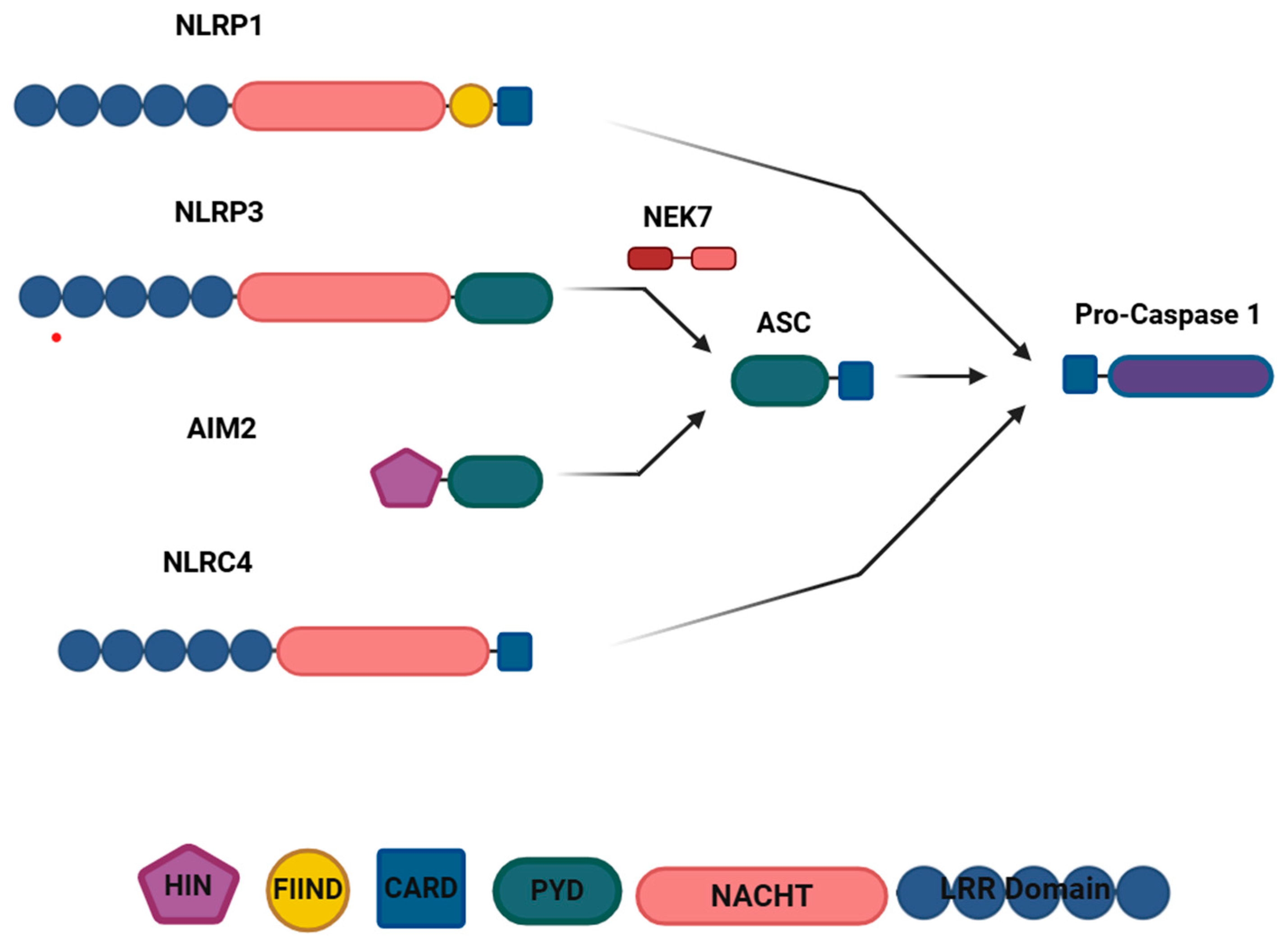
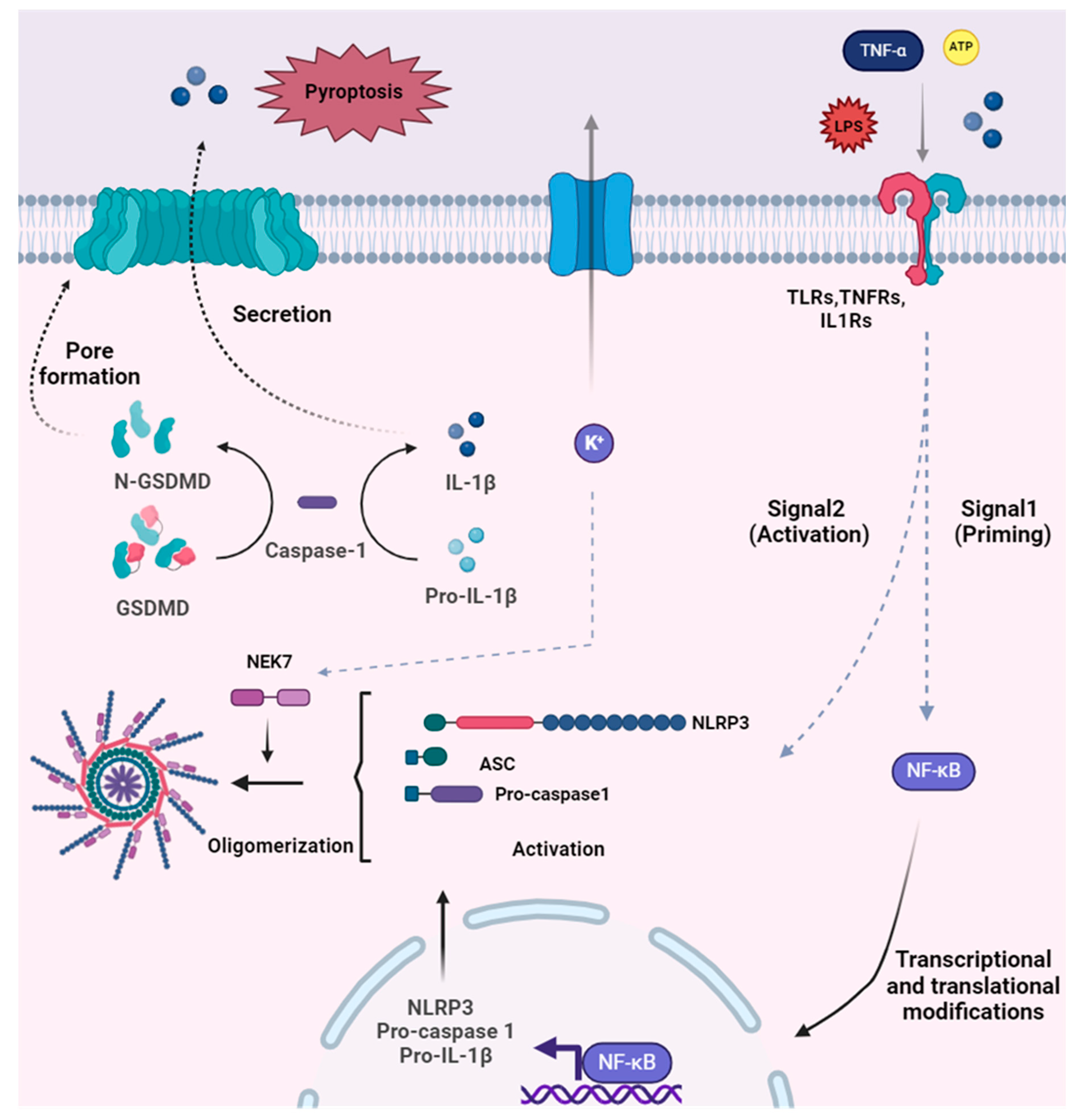


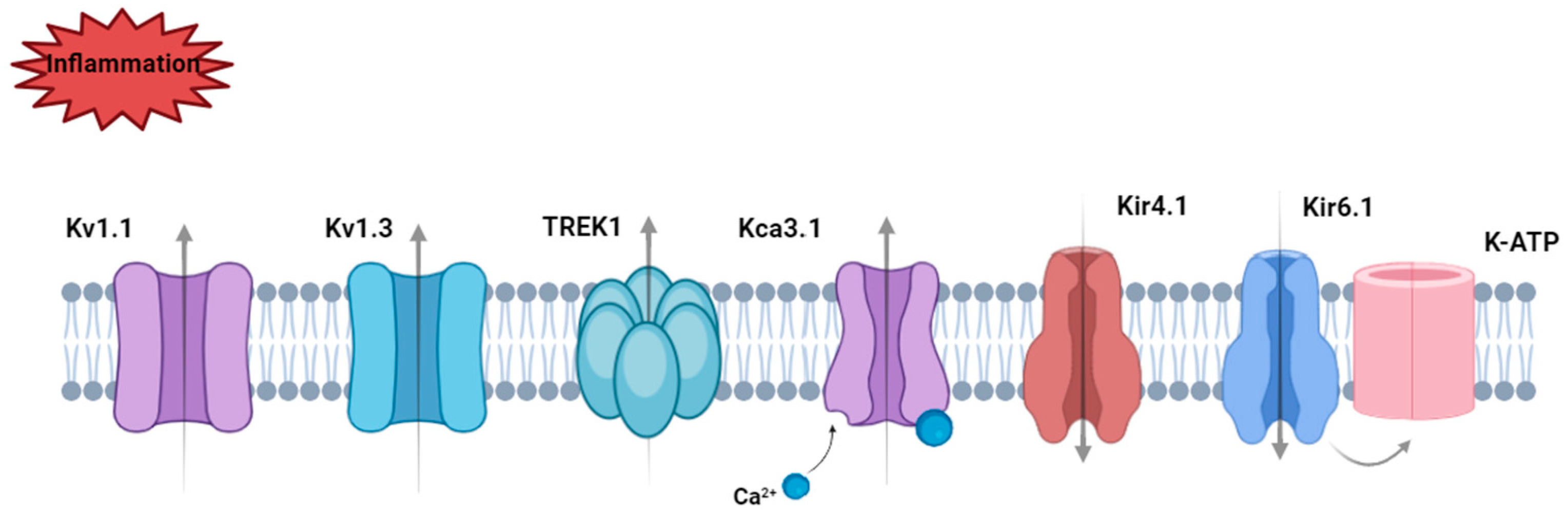
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozsoy, N.; Dallas, M.L. Dysfunctional K+ Homeostasis as a Driver for Brain Inflammation. Encyclopedia 2024, 4, 1681-1699. https://doi.org/10.3390/encyclopedia4040110
Ozsoy N, Dallas ML. Dysfunctional K+ Homeostasis as a Driver for Brain Inflammation. Encyclopedia. 2024; 4(4):1681-1699. https://doi.org/10.3390/encyclopedia4040110
Chicago/Turabian StyleOzsoy, Nagihan, and Mark L. Dallas. 2024. "Dysfunctional K+ Homeostasis as a Driver for Brain Inflammation" Encyclopedia 4, no. 4: 1681-1699. https://doi.org/10.3390/encyclopedia4040110
APA StyleOzsoy, N., & Dallas, M. L. (2024). Dysfunctional K+ Homeostasis as a Driver for Brain Inflammation. Encyclopedia, 4(4), 1681-1699. https://doi.org/10.3390/encyclopedia4040110