
Reinventing Therapeutic Proteins: Mining a Treasure of New Therapies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
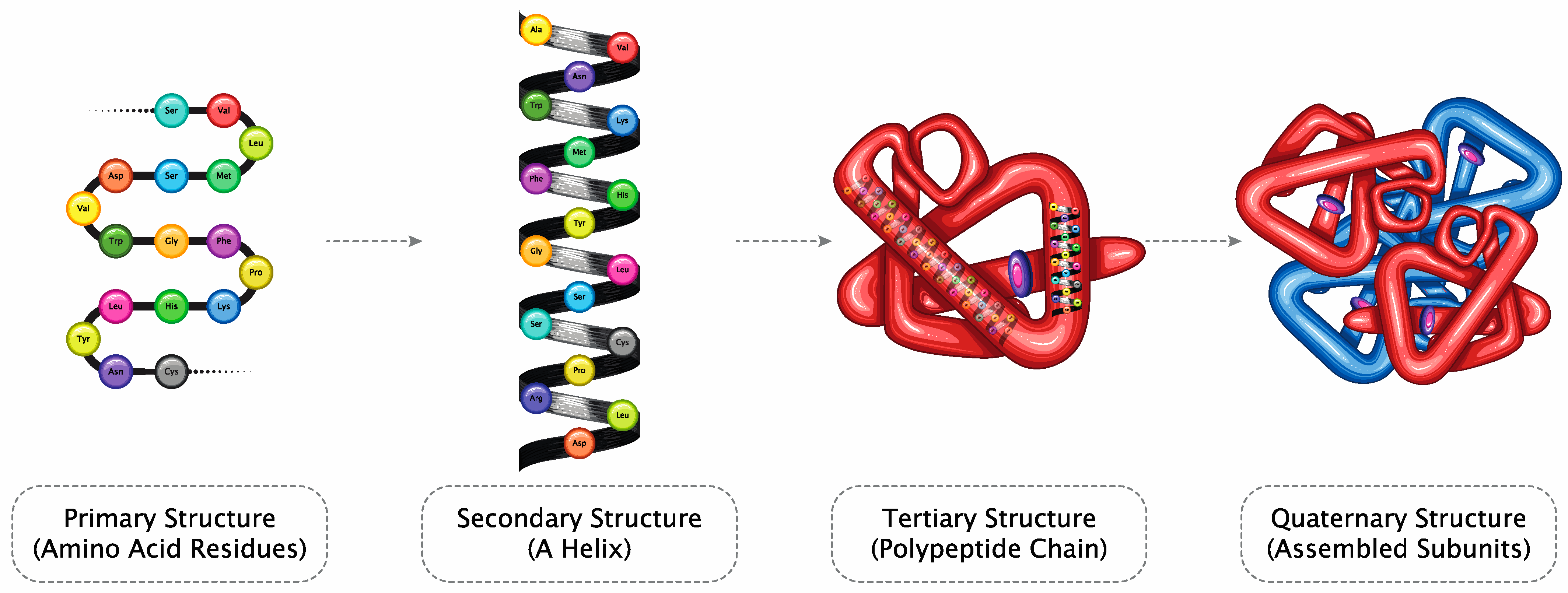
2. Understanding Therapeutic Proteins
3. Reinvention Scope
4. Intellectual Property
5. Artificial Intelligence (AI) and Machine Learning (ML)
5.1. Structure Prediction
5.2. Target Identification
- AtomNet is a convolutional neural network-based tool that applies the concepts of feature locality and hierarchical composition extracted through protein sequence, structure, and function to model bioactivity and chemical interactions of potential drug targets [74]. AtomNet’s parent AtomWise has recently enabled the rapid discovery of drugs against 27 disease targets. DeepDTA is also a deep-learning-based model that uses only sequence information of targets and drugs to predict drug–target interaction binding affinities and potential small molecules as drug candidates from given biological data [75].
- A commercially available natural compounds database and search engine that operates using machine learning, MolPort, when used with quantitative-structure-activity relationship (qsar), analyze the chemical structure and predicts the biological activity of potential targets in the early stages of drug discovery [76].
- Pathway analysis also enables the identification of potential targets. Some crucial biological pathways are available on the Kegg Pathway database [77], which provides insight into a disease mechanism. TargetNet [78] uses this pathways data and protein interaction profiles to predict potential drug targets against a specific disease.
- DeepDock is the most recent AI-driven virtual screening platform with a vast library of small molecules. For example, DeepDock virtual screen results were used to identify 15% active molecules that led to the discovery of novel compounds against the Mpro protease of SARS-CoV2 [79].
5.3. Molecular Docking
- Higher binding affinity scores from an in-silico docking analysis of monoclonal antibodies (mAbs) against Alpha and Delta strains of SARS-CoV spike protein suggested that tixagevimab, regdanvimab, and cilgavimab can neutralize most Alpha strains efficiently and bamlanivimab, tixagevimab, and sotrovimab can be effective in suppressing the Delta strain [87]. Venetoclax [88], for treating chronic lymphocytic leukemia, was designed to target the overexpressed BCL-2 protein in cancer cells by binding to its hydrophobic groove. Its development involved optimizing the binding interactions between the drug and BCL-2 through in silico docking studies, highlighting the importance of docking in drug design.
- GOLD uses a genetic algorithm, and Autodock Vina uses a grid-based energy approach with a genetic algorithm.
- ICM [89] uses multiple stochastic runs.
- GLIDE SP [90] uses several sampling and scoring methods.
- DeepBSP, an ML-based sampling and evaluation tool, is very useful in generating and ranking profiles close to their respective native structures as a machine learning model-based pose sampling and evaluation [91].
- Identification of the correct view is crucial for higher binding affinity and lower steric hindrance, which can be efficiently achieved through precise AI-based tools. Structure prediction tools such as AlphaFold2 and trRosetta can be integrated with other ML-based approaches to identify and optimize potential poses. One such instance is identifying transition states between the active and inactive conformations of G-protein coupled receptors using multiple ML approaches [92].
- The effectiveness of interaction between the dynamic views and their binding partners can be weighted through scoring systems. Scoring functions are categorized into force-field-based, knowledge-based, and empirical scoring functions.
- Force-field-based scoring functions utilize molecular mechanics to evaluate complex energetic affinities based on their interactions, i.e., weak Van der Waals, electrostatic forces, bond stretching, bending, and torsional angles [93].
- Knowledge-based scoring functions include statistical analysis of distance-dependent atom-pair potentials of protein–ligand or protein–protein complexes generated directly from experimental structures [94,95]. Empirical scoring functions, e.g., LUDI [96], ID-Score [97], and GlideScore [90], are based on empirical data. They correlate binding free energies to weak Van der Waals energy, electrostatic energy, desolvation, entropy, enthalpy, H-bonding, rotational and translational degrees of freedom, polar and lipophilic effects, and hydrophobicity in the form of simple equations to reproduce experimental affinity data.
- These scores are used in combinations for better optimization, i.e., DockThor programs DockTScore [94,98] and blends empirical and force-field-based scoring methods, SMoG2016 [99] fuses empirical and knowledge-based scoring methods, and GalaxyDock BP2 Score [100] uses all three: force-field-based, knowledge-based, and empirical scoring methods [94].
- The recent integration of physics-based terms and ML in DockTScore has further enhanced binding energy prediction and conformation ranking [101].
- GNINA docking software, based on an ensemble of convolutional neural networks as a scoring function for scoring the sample view, has outperformed AutoDock Vina [102], once again proving that the paradigm shift from conventional methods to AI-based methods has significantly increased the impartial interpretations of scientific evidence leading to the discovery of targets.
5.4. Limitations
6. Structure Modifications
7. Drug Conjugates
8. Radioimmunoconjugates (RIC)

9. Regulatory Perspective
10. Regulatory Submission
10.1. Nonclinical Testing
10.2. Pharmacokinetics–Pharmacodynamics
10.3. Function Testing
10.4. Immunogenic Response
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raju, T.N. The nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905-98). Lancet 2000, 355, 1022. [Google Scholar] [CrossRef]
- Sean. The Process and Costs of Drug Development. FTLOScience (5 February 2023). 2022. Available online: https://ftloscience.com/process-costs-drug-development/ (accessed on 30 March 2023).
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.M.; Meijboom, F.L.B. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 223. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.M. Chance and the prepared mind. Science 1912, 35, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. Selective optimization of side activities: The SOSA approach. Drug Discov. Today 2006, 11, 160–164. [Google Scholar] [CrossRef]
- Prosdocimi, M.; Zuccato, C.; Cosenza, L.C.; Borgatti, M.; Lampronti, I.; Finotti, A.; Gambari, R. A Rational Approach to Drug Repositioning in β-thalassemia: Induction of Fetal Hemoglobin by Established Drugs. Wellcome Open Res. 2022, 7, 150. [Google Scholar] [CrossRef]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef]
- Blair, H.A. Dimethyl fumarate: A review in moderate to severe plaque psoriasis. Drugs 2018, 78, 123–130. [Google Scholar] [CrossRef]
- Santoro, M.G.; Carafoli, E. Remdesivir: From Ebola to COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. [Google Scholar] [CrossRef]
- Gilvary, C.; Elkhader, J.; Madhukar, N.; Henchcliffe, C.; Goncalves, M.D.; Elemento, O. A machine learning and network framework to discover new indications for small molecules. PLoS Comput. Biol. 2020, 16, e1008098. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, M.; Xu, Y.; Wu, Z.; Wang, J.; Zhang, C.; Liu, G.; Li, W.; Li, J.; Tang, Y. Drug repositioning by prediction of drug’s anatomical therapeutic chemical code via network-based inference approaches. Briefings Bioinform. 2021, 22, 2058–2072. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Shintani, M.; Imanari, F.; Osada, N.; Endo, T. A New Approach to Drug Repurposing with Two-Stage Prediction, Machine Learning, and Unsupervised Clustering of Gene Expression. OMICS A J. Integr. Biol. 2022, 26, 339–347. [Google Scholar] [CrossRef]
- Available online: https://www.drugs.com/new-indications.html (accessed on 30 March 2023).
- Jackson, D.A.; Symons, R.H.; Berg, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Berg, P.; Baltimore, D.; Boyer, H.W.; Cohen, S.N.; Davis, R.W.; Hogness, D.S.; Nathans, D.; Roblin, R.; Watson, J.D.; Weissman, S.; et al. Letter: Potential biohazards of recombinant DNA molecules. Science 1974, 185, 303. [Google Scholar] [CrossRef]
- Landgraf, W.; Sandow, J. Recombinant Human Insulins—Clinical Efficacy and Safety in Diabetes Therapy. Eur. Endocrinol. 2016, 12, 12–17. [Google Scholar] [CrossRef]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [CrossRef]
- Available online: https://www.biospace.com/article/biologics-market-size-to-hit-usd-719-94-billion-by-2030-/ (accessed on 30 March 2023).
- FDA. Available online: https://www.fda.gov/media/107622/download (accessed on 30 March 2023).
- Available online: https://www.ncbi.nlm.nih.gov/books/NBK562260/#:~:text=A%20peptide%20is%20a%20short,the%20building%20block%20of%20proteins (accessed on 30 March 2023).
- Niazi, S.K. Molecular Biosimilarity—An AI-Driven Paradigm Shift. Int. J. Mol. Sci. 2022, 23, 10690. [Google Scholar] [CrossRef]
- Zwanzig, R.; Szabo, A.; Bagchi, B. Levinthal’s paradox. Proc. Natl. Acad. Sci. USA 1992, 89, 20–22. [Google Scholar] [CrossRef]
- Schmidt, T.; Bergner, A.; Schwede, T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today 2014, 19, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.cms.gov/monoclonal#:~:text=Monoclonal%20Antibodies%20to%20Treat%20Mild%2Dto%2DModerate%20COVID%2D19&text=On%20December%2023%2C%202022%2C%20the,with%20severe%20COVID%2D19%20illness (accessed on 30 March 2023).
- Available online: https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf (accessed on 30 March 2023).
- Available online: https://www.ajmc.com/view/considerations-for-use-of-bevacizumab-vikg-in-wet-amd (accessed on 30 March 2023).
- Hotzel, I.; Theil, F.-P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Bumbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.W.; Patapoff, T.W.; Kabakoff, B.; Pai, S.; Hilario, E.; Zhang, B.; Li, C.; Borisov, O.; Kelley, R.F.; Chorny, I.; et al. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 2014, 111, 18601–18606. [Google Scholar] [CrossRef]
- Lagassé, H.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug reinventing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug reinventing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Available online: https://www.greyb.com/blog/biologics-patents-expiring-2022-2023-2024-2025-2026-2027/ (accessed on 30 March 2023).
- Goode, R.; Chao, B. Biological patent thickets and delayed access to biosimilars, an American problem. J. Law Biosci. 2022, 9, lsac022. [Google Scholar] [CrossRef]
- Beneke, F.; Mackenrodt, M.-O. Artificial intelligence and collusion. IIC Int. Rev. Intellect. Prop. Compet. Law 2019, 50, 109–134. [Google Scholar] [CrossRef]
- Bielecki, A.; Bielecki, A. Foundations of artificial neural networks. In Models of Neurons and Perceptrons: Selected Problems and Challenges; Janusz, K., Ed.; Springer International Publishing; Polish Academy of Sciences: Warsaw, Poland, 2019; pp. 15–28. [Google Scholar]
- Da Silva, I.N. Artificial Neural Networks; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Cui, H.; Huang, T.; Wang, D.; Lian, B.; Li, W.; Qin, G.; Chen, L.; Xie, L. Prediction of synergistic anti-cancer drug combinations based on the drug target network and drug-induced gene expression profiles. Artif. Intell. Med. 2017, 83, 35–43. [Google Scholar] [CrossRef]
- Choudhury, C.; Murugan, N.A.; Priyakumar, U.D. Structure-based drug repurposing: Traditional and advanced AI/ML-aided methods. Drug Discov. Today 2022, 27, 1847–1861. [Google Scholar] [CrossRef] [PubMed]
- Moll, S.; Desmoulière, A.; Moeller, M.J.; Pache, J.-C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118474. [Google Scholar] [CrossRef]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef]
- Deloitte—Intelligent Drug Discovery. (n.d.). Deloitte. Available online: https://www2.deloitte.com/content/dam/Deloitte/my/Documents/risk/my-risk-sdg3-intelligent-drug-discovery.pdf (accessed on 8 March 2023).
- Dokholyan, N.V. Experimentally-Driven Protein Structure Modeling. J. Proteom. 2020, 220, 103777. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Bank, R.P.D. (n.d.). PDB Statistics: Protein-Only Structures Released per Year. Available online: https://www.rcsb.org/stats/growth/growth-protein (accessed on 30 March 2023).
- Mirdita, M.; Driesch, L.V.D.; Galiez, C.; Martin, M.-J.; Söding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef]
- BFD. (n.d.). Available online: https://bfd.mmseqs.com/ (accessed on 30 March 2023).
- Mitchell, A.L.; Scheremetjew, M.; Denise, H.; Potter, S.; Tarkowska, A.; Qureshi, M.; A Salazar, G.; Pesseat, S.; A Boland, M.; Hunter, F.; et al. EBI Metagenomics in 2017: Enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res. 2018, 46, D726–D735. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Database, A.P.S. (n.d.). AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 23 March 2023).
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Šali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Jia, Y.; Zhang, Y. Protein structure prediction. Int. J. Mod. Phys. B 2018, 32, 1840009. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.-K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Ohue, M. Solubility-Aware Protein Binding Peptide Design Using AlphaFold. Biomedicines 2022, 10, 1626. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold—Enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- Available online: https://pandaomics.com/access (accessed on 23 March 2023).
- Matsuzaka, Y.; Yashiro, R. Applications of Deep Learning for Drug Discovery Systems with BigData. BioMedinformatics 2022, 2, 603–624. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Ferreira, L.; Borba, J.; Moreira-Filho, J.; Rimoldi, A.; Andrade, C.; Costa, F. QSAR-Based Virtual Screening of Natural Products Database for Identification of Potent Antimalarial Hits. Biomolecules 2021, 11, 459. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.-J.; Dong, J.; Che, Y.-J.; Zhu, M.-F.; Wen, M.; Wang, N.-N.; Wang, S.; Lu, A.-P.; Cao, D.-S. TargetNet: A web service for predicting potential drug–target interaction profiling via multi-target SAR models. J. Comput. Mol. Des. 2016, 30, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.-T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein–Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef] [PubMed]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Venkatachalam, C.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Das, N.C.; Chakraborty, P.; Bayry, J.; Mukherjee, S. In Silico Analyses on the Comparative Potential of Therapeutic Human Monoclonal Antibodies Against Newly Emerged SARS-CoV-2 Variants Bearing Mutant Spike Protein. Front. Immunol. 2022, 12, 782506. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.; Muthukumaran, J.; Freire, F.; Paquete-Ferreira, J.; Otrelo-Cardoso, A.R.; Svergun, D.; Panjkovich, A.; Santos-Silva, T. Shedding Light on the Interaction of Human Anti-Apoptotic Bcl-2 Protein with Ligands through Biophysical and in Silico Studies. Int. J. Mol. Sci. 2019, 20, 860. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; He, X.; Zhang, J.Z.H. DeepBSP—A Machine Learning Method for Accurate Prediction of Protein–Ligand Docking Structures. J. Chem. Inf. Model. 2021, 61, 2231–2240. [Google Scholar] [CrossRef]
- Yadav, P.; Mollaei, P.; Cao, Z.; Wang, Y.; Farimani, A.B. Prediction of GPCR activity using machine learning. Comput. Struct. Biotechnol. J. 2022, 20, 2564–2573. [Google Scholar] [CrossRef] [PubMed]
- Vemula, D.; Jayasurya, P.; Sushmitha, V.; Kumar, Y.N.; Bhandari, V. CADD, AI and ML in drug discovery: A comprehensive review. Eur. J. Pharm. Sci. 2023, 181, 106324. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Böhm, H.-J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J. Comput. Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.-S.; Wong, M.-H.; Ballester, P.J. Low-quality structural and interaction data improves binding affinity prediction via random forest. Molecules 2015, 20, 10947–10962. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, C.S.; Almeida, D.M.; Barbosa, H.J.C.; Dardenne, L.E. A dynamic niching genetic algorithm strategy for docking highly flexible ligands. Inf. Sci. 2014, 289, 206–224. [Google Scholar] [CrossRef]
- Debroise, T.; Shakhnovich, E.I.; Chéron, N. A Hybrid knowledge-based and empirical scoring function for protein–Ligand interaction: SMoG2016. J. Chem. Inf. Model. 2017, 57, 584–593. [Google Scholar] [CrossRef]
- Baek, M.; Shin, W.-H.; Chung, H.W.; Seok, C. GalaxyDock BP2 score: A hybrid scoring function for accurate protein–ligand docking. J. Comput. Mol. Des. 2017, 31, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.J. A study on the application and use of artificial intelligence to support drug development. Clin. Ther. 2019, 41, 1414–1426. [Google Scholar] [CrossRef]
- Choi, S.; Park, H.; Jung, S.; Kim, E.-K.; Cho, M.-L.; Min, J.-K.; Moon, S.-J.; Lee, S.-M.; Cho, J.-H.; Lee, D.-H.; et al. Therapeutic Effect of Exogenous Truncated IK Protein in Inflammatory Arthritis. Int. J. Mol. Sci. 2017, 18, 1976. [Google Scholar] [CrossRef]
- Rigi, G.; Kardar, G.; Hajizade, A.; Zamani, J.; Ahmadian, G. The effects of a truncated form of Staphylococcus aureus protein A (SpA) on the expression of cytokines of autoimmune patients and healthy individuals. Europe PMC, 2022; not peer-reviewed. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Véniant, M.M.; Komorowski, R.; Chen, P.; Stanislaus, S.; Winters, K.; Hager, T.; Zhou, L.; Wada, R.; Hecht, R.; Xu, J. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology 2012, 153, 4192–4203. [Google Scholar] [CrossRef]
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; van der Veen, L.A.; et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res. 2016, 22, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. Available online: https://pubmed.ncbi.nlm.nih.gov/26182432/ (accessed on 23 March 2023). [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 1–25. [Google Scholar] [CrossRef]
- Available online: https://www.bio-itworld.com/pressreleases/2022/11/28/fda-approved-adc-drugs-list-up-to-2022 (accessed on 23 March 2023).
- McPherson, M.J.; Hobson, A.D. Pushing the Envelope: Advancement of ADCs Outside of Oncology In Antibody-Drug Conjugates; Tumey, L., Ed.; Humana: New York, NY, USA, 2020; Volume 2078, pp. 23–36. [Google Scholar] [CrossRef]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef]
- Beck, A.; Haeuw, J.-F.; Wurch, T.; Goetsch, L.; Bailly, C.; Corvaïa, N. The next generation of antibody-drug conjugates comes of age. Discov. Med. 2010, 10, 329–359. [Google Scholar]
- Ritter, A. Antibody-drug conjugates: Looking ahead to an emerging class of biotherapeutic. Pharm. Tech. 2012, 36, 42–47. [Google Scholar]
- Junttila, T.T.; Li, G.; Parson, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib-insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody–drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Hebbrecht, T.; Liu, J.; Zwaenepoel, O.; Boddin, G.; Van Leene, C.; Decoene, K.; Madder, A.; Braeckmans, K.; Gettemans, J. Nanobody click chemistry for convenient site-specific fluorescent labelling, single step immunocytochemistry and delivery into living cells by photoporation and live cell imaging. New Biotechnol. 2020, 59, 33–43. [Google Scholar] [CrossRef]
- Jaffray, D.A. Image-guided radiotherapy: From current concept to future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Nasr, D.; Kumar, P.A.; Zerdan, M.B.; Ghelani, G.; Dutta, D.; Graziano, S.; Lim, S.H. Radioimmunoconjugates in the age of modern immuno-oncology. Life Sci. 2022, 310, 121126. [Google Scholar] [CrossRef]
- Pouget, J.P.; Constanzo, J. Revisiting the radiobiology of targeted alpha therapy. Front. Med. 2018, 8, 692436. [Google Scholar] [CrossRef]
- Grillo-López, A.J. Zevalin: The first radioimmunotherapy approved for the treatment of lymphoma. Expert. Rev. Anticancer Ther. 2002, 2, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, J.; Kim, H.; Lee, Y.-J.; Kim, J.S.; Lim, S.M. Combination Radioimmunotherapy Strategies for Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5579. [Google Scholar] [CrossRef]
- Miranda, A.C.C.; Santos, S.N.D.; Fuscaldi, L.L.; Balieiro, L.M.; Bellini, M.H.; Guimarães, M.I.C.C.; de Araújo, E.B. Radioimmunotheranostic pair based on the anti-HER2 monoclonal antibody: Influence of chelating agents and radionuclides on biological properties. Pharmaceutics 2021, 13, 971. [Google Scholar] [CrossRef]
- Chiu, K.; Racz, R.; Burkhart, K.; Florian, J.; Ford, K.; Garcia, M.I.; Geiger, R.M.; Howard, K.E.; Hyland, P.L.; Ismaiel, O.A.; et al. New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front. Med. 2023, 9, 03. [Google Scholar] [CrossRef]
- U S Food and Drug Administration. Clinical Pharmacology Review for Application 214787Orig1S000 (Remdesivir). 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000ClinpharmR.pdf (accessed on 23 March 2023).
- Available online: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/division-applied-regulatory-science (accessed on 23 March 2023).
- Schotland, P.; Racz, R.; Jackson, D.B.; Soldatos, T.G.; Levin, R.; Strauss, D.G.; Burkhart, K. Target Adverse Event Profiles for Predictive Safety in the Postmarket Setting. Clin. Pharmacol. Ther. 2020, 109, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bhagwat, B.; Sanden, D.; Willingham, A.; Tan, A.; Knapton, A.D.; Weaver, J.L.; Howard, K.E. Evaluation of a TGN1412 analogue using in vitro assays and two immune humanized mouse models. Toxicol. Appl. Pharmacol. 2019, 372, 57–69. [Google Scholar] [CrossRef]
- Yan, H.; Semple, K.M.; Gonzaléz, C.M.; Howard, K.E. Bone marrow-liver-thymus (BLT) immune humanized mice as a model to predict cytokine release syndrome. Transl. Res. 2019, 210, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.L.; Zadrozny, L.M.; Gabrielson, K.; Semple, K.M.; I Shea, K.; E Howard, K. BLT-Immune humanized mice as a model for nivolumab-induced immune-mediated adverse events: Comparison of the NOG and NOG-EXL strains. Toxicol. Sci. 2019, 169, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Daluwatte, C.; Schotland, P.; Strauss, D.G.; Burkhart, K.K.; Racz, R. Predicting potential adverse events using safety data from marketed drugs. BMC Bioinform. 2020, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pistoiaalliance.org/ (accessed on 23 March 2023).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use [ICH]. Assessment and Control of DNA Reactive (mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. M7(R1). Current Step 4 Version. 2017. Available online: https://database.ich.org/sites/default/files/M7_R1_Guideline.pdf (accessed on 23 March 2023).
- Available online: https://www.youtube.com/watch?v=bNb2fEVKeEo&t=6sExternal_Link_Disclaimer (accessed on 23 March 2023).
- Available online: https://www.semitorr.com/specialties/particle-sentry-ai-quality-control-in-drug-product-manufacturing/ (accessed on 23 March 2023).
- Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 23 March 2023).
- Available online: https://www.ema.europa.eu/en/medicines/what-we-publish-when/european-public-assessment-reports-background-context (accessed on 23 March 2023).
- Available online: https://www.centerforbiosimilars.com/view/opinion-a-modified-351-a-licensing-pathway-for-biosimilars (accessed on 23 March 2023).
- Wang, Y.C.; Wang, Y.; Schrieber, S.J.; Earp, J.; Thway, T.M.; Huang, S.M.; Zineh, I.; Christl, L. Role of modeling and simulation in the development of novel and biosimilar therapeutic proteins. J. Pharm. Sci. 2019, 108, 73–77. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, S.M. Commentary on fit-for-purpose models for regulatory applications. J. Pharm. Sci. 2019, 108, 18–20. [Google Scholar] [CrossRef]
- Zhu, P.; Hsu, C.-H.; Liao, J.; Xu, S.; Zhang, L.; Zhou, H. Trial design and statistical considerations on the assessment of pharmacodynamic similarity. AAPS J. 2019, 21, 47. [Google Scholar] [CrossRef]
- Zhu, P.; Ji, P.; Wang, Y. Using Clinical PK/PD studies to support no clinically meaningful differences between a proposed biosimilar and the reference product. AAPS J. 2018, 20, 89. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Guidance: Bioanalytical Method Validation. 2018. Available online: https://www.fda.gov/media/70858/download (accessed on 1 April 2022).
- Lim, S.H.; Kim, K.; Choi, C.-I. Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis. J. Pers. Med. 2022, 12, 1265. [Google Scholar] [CrossRef]
- Niazi, S. Volume of Distribution as a Function of Time. J. Pharm. Sci. 1976, 65, 452–454. [Google Scholar] [CrossRef]
- Wesolowski, C.; Wesolowski, M.J.; Babyn, P.S.; Wanasundara, S.N. Time Varying Apparent Volume of Distribution and Drug Half-Lives Following Intravenous Bolus Injections. PLoS ONE 2016, 11, e0158798. [Google Scholar] [CrossRef] [PubMed]
- Gadkar, K.; Yadav, D.B.; Zuchero, J.Y.; Couch, J.A.; Kanodia, J.; Kenrick, M.K.; Atwal, J.K.; Dennis, M.S.; Prabhu, S.; Watts, R.J.; et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 2016, 101, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, K.D.; Thurber, G.M.; Schmidt, M.M.; Rhoden, J.J. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 2012, 503, 255–268. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef]
- Deng, R.; Loyet, K.M.; Lien, S.; Iyer, S.; Deforge, L.E.; Theil, F.-P.; Lowman, H.B.; Fielder, P.J.; Prabhu, S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-α antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab. Dispos. 2010, 38, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Robbie, G.J.; Criste, R.; Dall’Acqua, W.F.; Jensen, K.; Patel, N.K.; Losonsky, G.A.; Griffin, M.P. A Novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob. Agents Chemother. 2013, 57, 6147–6153. [Google Scholar] [CrossRef]
- Kamath, A.V. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 2016, 21-22, 75–83. [Google Scholar] [CrossRef]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef]
- Sweeney, G. Variability in the human drug response. Thromb. Res. 1983, 29, 3–15. [Google Scholar] [CrossRef]
- Marchant, B. Pharmacokinetic factors influencing variability in human drug response. Scand. J. Rheumatol. 1981, 10, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Babin, V.; Taran, F.; Audisio, D. Late-Stage Carbon-14 Labeling and Isotope Exchange: Emerging Opportunities and Future Challenges. JACS Au 2022, 2, 1234–1251. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.H.; Sheiner, L.B. Kinetics of pharmacologic response. Pharmacol. Ther. 1982, 16, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Keutzer, L.; You, H.; Farnoud, A.; Nyberg, J.; Wicha, S.G.; Maher-Edwards, G.; Vlasakakis, G.; Moghaddam, G.K.; Svensson, E.M.; Menden, M.P.; et al. Machine Learning and Pharmacometrics for Prediction of Pharmacokinetic Data: Differences, Similarities and Challenges Illustrated with Rifampicin. Pharmaceutics 2022, 14, 1530. [Google Scholar] [CrossRef]
- Cai, W.; Leil, T.; Gibiansky, L.; Krishna, M.; Zhang, H.; Gu, H.; Sun, H.; Throup, J.; Banerjee, S.; Girgis, I. Modeling and Simulation of the Pharmacokinetics and Target Engagement of an Antagonist Monoclonal Antibody to Interferon-γ–Induced Protein 10, BMS-986184, in Healthy Participants to Guide Therapeutic Dosing. Clin. Pharmacol. Drug Dev. 2020, 9, 689–698. [Google Scholar] [CrossRef]
- McClellan, J.E.; Conlon, H.D.; Bolt, M.W.; Kalfayan, V.; Palaparthy, R.; Rehman, M.I.; Kirchhoff, C.F. The ‘totality-of-the-evidence’ approach in the development of PF-06438179/GP1111, an infliximab biosimilar, and in support of its use in all indications of the reference product. Ther. Adv. Gastroenterol. 2019, 12, 1756284819852535. [Google Scholar] [CrossRef]
- Ryding, J.; Stahl, M.; Ullmann, M. Demonstrating biosimilar and originator antidrug antibody binding comparability in antidrug antibody assays: A practical approach. Bioanalysis 2017, 9, 1395–1406. [Google Scholar] [CrossRef]
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2018, 9, 74–85. [Google Scholar] [CrossRef]
- Todoroki, K.; Yamada, T.; Mizuno, H.; Toyo’Oka, T. Current Mass Spectrometric Tools for the Bioanalyses of Therapeutic Monoclonal Antibodies and Antibody-Drug Conjugates. Anal. Sci. 2018, 34, 397–406. [Google Scholar] [CrossRef]
- Láng, J.A.; Balogh, Z.C.; Nyitrai, M.F.; Juhász, C.; Gilicze, A.K.B.; Iliás, A.; Zólyomi, Z.; Bodor, C.; Rábai, E. In vitro functional characterization of biosimilar therapeutic antibodies. Drug Discov. Today Technol. 2020, 37, 41–50. [Google Scholar] [CrossRef]
- Cymera, F.; Becka, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2018, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Hufton, S.E.; Fox, B.; Dougall, T.; Rigsby, P.; Bristow, A. Participants of the study International standards for monoclonal antibodies to support pre- and post-marketing product consistency: Evaluation of a candidate international standard for the bioactivities of rituximab. Mabs 2018, 10, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.-P.; Kronthaler, U.; Fritsch, C.; Grau, R.; Müller, S.O.; Mayer, R.; Seidl, A.; Da Silva, A. Characterization and non-clinical assessment of the proposed etanercept biosimilar GP2015 with originator etanercept (Enbrel®). Expert Opin. Biol. Ther. 2016, 16, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-immunogenicity-considerations-biosimilar-and-interchangeable-insulin-products (accessed on 23 March 2023).
- Zhou, S.-F.; Zhong, W.-Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef]
- Available online: https://michaelschlander.com/publications-since-2020.html?file=files/downloads/publications/2018/Schlander-et-al-Cost-Drug-Development-2021-PharmacoEconomics.pdf&cid=5702#:~:text=Results%20Estimates%20of%20total%20average,%244.54%20billion%20(2019%20US%24) (accessed on 23 March 2023).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Mariam, Z. Reinventing Therapeutic Proteins: Mining a Treasure of New Therapies. Biologics 2023, 3, 72-94. https://doi.org/10.3390/biologics3020005
Niazi SK, Mariam Z. Reinventing Therapeutic Proteins: Mining a Treasure of New Therapies. Biologics. 2023; 3(2):72-94. https://doi.org/10.3390/biologics3020005
Chicago/Turabian StyleNiazi, Sarfaraz K., and Zamara Mariam. 2023. "Reinventing Therapeutic Proteins: Mining a Treasure of New Therapies" Biologics 3, no. 2: 72-94. https://doi.org/10.3390/biologics3020005