
Therapeutic Strategies for MASH: An Update on Drug Candidates Under Investigation in Late-Phase Clinical Trials


Abstract
1. Introduction
2. Promising MASH Therapeutic Agents in Late-Phase Clinical Trials and Their Targets
2.1. GLP-1R Agonists
2.2. FGF21 Analogues
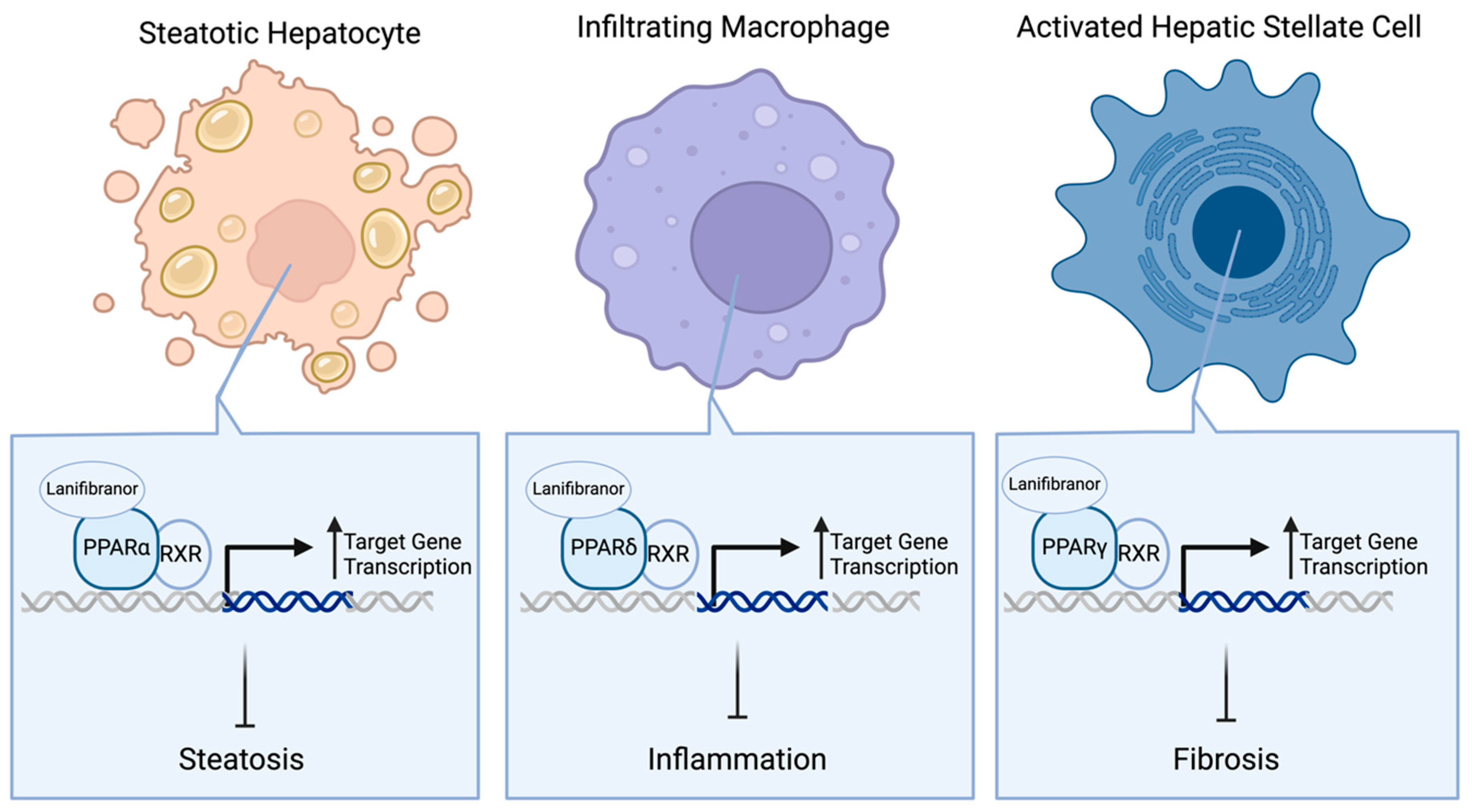
2.3. PPAR Agonists
2.4. SGLT2 Inhibitors
2.5. PDE Inhibitors
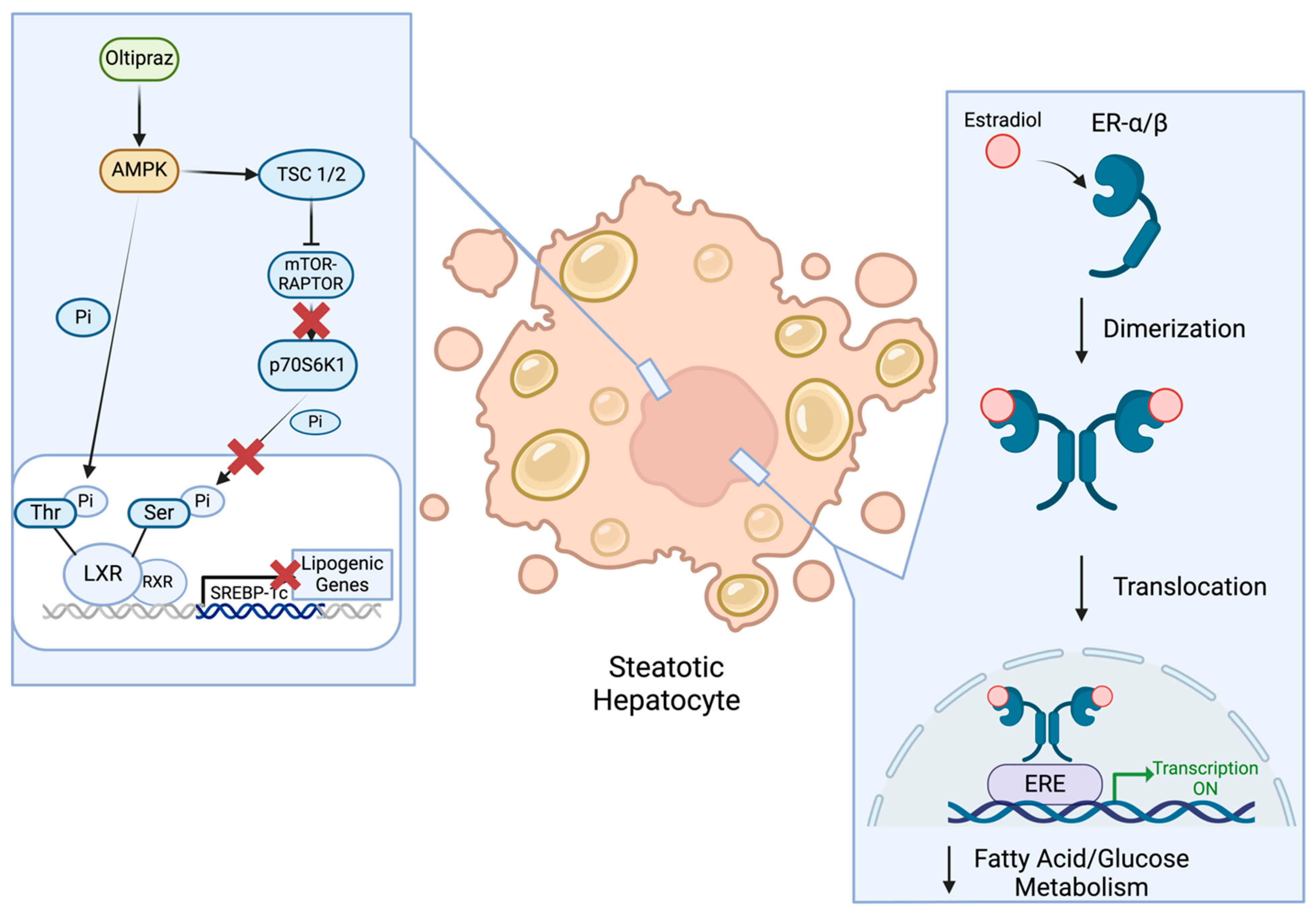
2.6. AMPK Activators
2.7. Voltage-Gated Chloride Channel Activators
2.8. GSH Precursors A
2.9. Estrogens
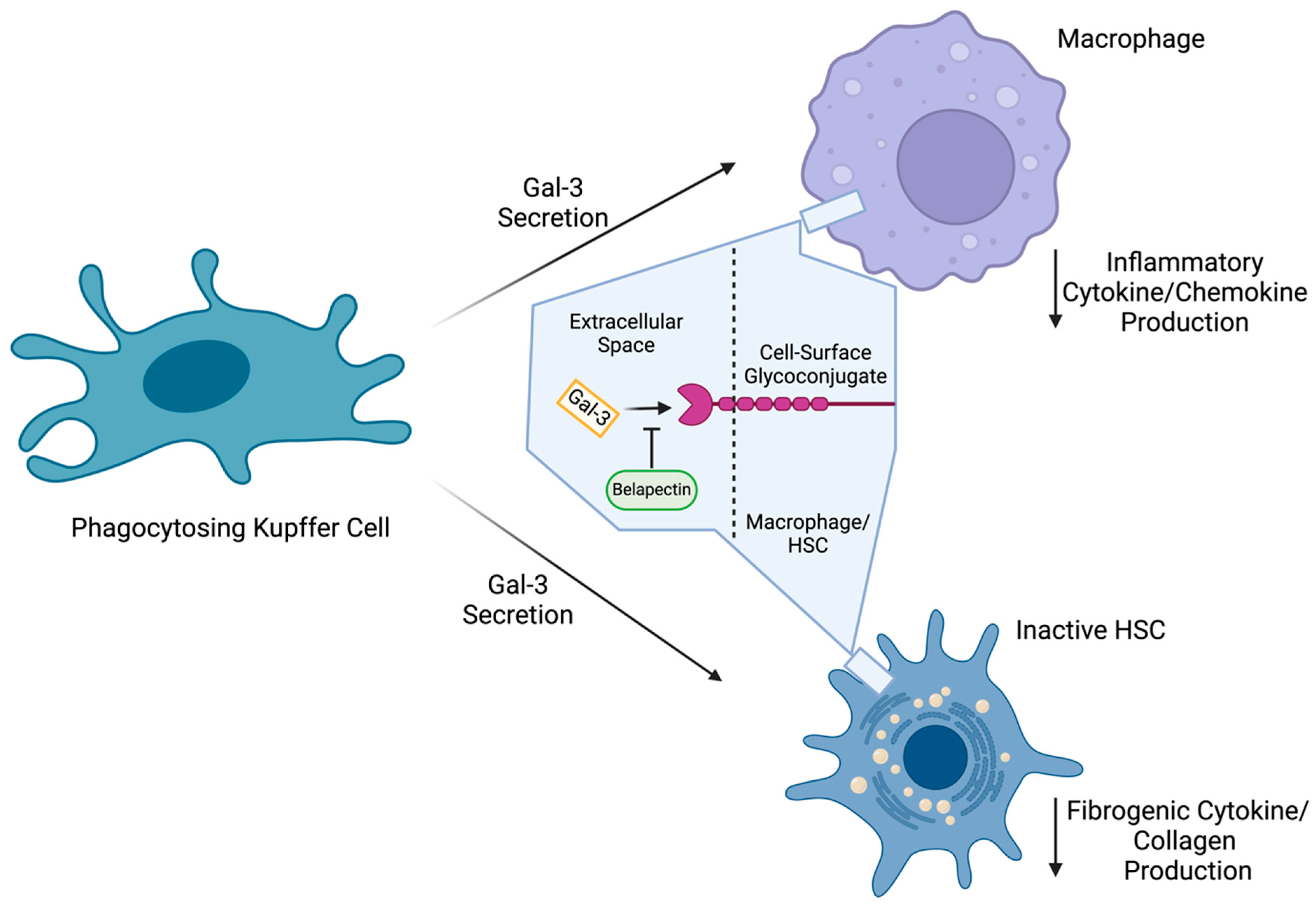
2.10. Galectin-3 Inhibitors
3. Suspended/Terminated Candidate Therapeutics for MASH
3.1. SCD1 Inhibitors
3.2. Antioxidants
3.3. FXR Agonists
3.4. CCR 2/5 Antagonists
3.5. ASK1 Inhibitors
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinella, M.E.; Sookoian, S. From NAFLD to MASLD: Updated Naming and Diagnosis Criteria for Fatty Liver Disease. J. Lipid Res. 2024, 65, 100485. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281.e4. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, A.; Jiang, Y.; Maidaiti, X.; Wu, Y.; Jin, Y. Global Burden of Metabolic Dysfunction-Associated Steatotic Liver Disease Attributable to High Fasting Plasma Glucose in 204 Countries and Territories from 1990 to 2021. Sci. Rep. 2024, 14, 22232. [Google Scholar] [CrossRef] [PubMed]
- Kabbany, M.N.; Selvakumar, P.K.C.; Watt, K.; Lopez, R.; Akras, Z.; Zein, N.; Carey, W.; Alkhouri, N. Prevalence of Nonalcoholic Steatohepatitis-Associated Cirrhosis in the United States: An Analysis of National Health and Nutrition Examination Survey Data. Am. J. Gastroenterol. 2017, 112, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Okanoue, T.; Nakajima, A. Phase 3 Drug Pipelines in the Treatment of Non-Alcoholic Steatohepatitis. Hepatol. Res. 2019, 49, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099.e1. [Google Scholar] [CrossRef] [PubMed]
- Leff, P.; Rich, N.E. First FDA-Approved NASH Treatment Produces NASH Resolution and Decreases Fibrosis: Results From the Landmark Phase 3 MAESTRO-NASH Trial; ACG Publications: Ipswich, UK, 2024. [Google Scholar]
- Thiagarajan, P.; Aithal, G.P. Drug Development for Nonalcoholic Fatty Liver Disease: Landscape and Challenges. J. Clin. Exp. Hepatol. 2019, 9, 515–521. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Yen, P.M. Actions of Thyroid Hormones and Thyromimetics on the Liver. Nat. Rev. Gastroenterol. Hepatol. 2025, 22, 9–22. [Google Scholar] [CrossRef]
- Au, K.; Zheng, M.-H.; Lee, W.-J.; Ghanem, O.M.; Mahawar, K.; Shabbir, A.; Le Roux, C.W.; Targher, G.; Byrne, C.D.; Yilmaz, Y.; et al. Resmetirom and Metabolic Dysfunction-Associated Steatohepatitis: Perspectives on Multidisciplinary Management from Global Healthcare Professionals. Curr. Obes. Rep. 2024, 13, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Charlton, M.R.; Harrison, S.A.; Bansal, M.B.; Alkhouri, N.; Loomba, R.; Sanyal, A.J.; Rinella, M.E. Expert Panel Recommendations: Practical Clinical Applications for Initiating and Monitoring Resmetirom in Patients With MASH/NASH and Moderate to Noncirrhotic Advanced Fibrosis. Clin. Gastroenterol. Hepatol. 2024, 22, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Tacke, F.; Sanyal, A.J.; Anstee, Q.M. Report on the AASLD/EASL Joint Workshop on Clinical Trial Endpoints in NAFLD. J. Hepatol. 2019, 71, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Dong, Y.; Li, X.; Zhong, W.; Lei, X.; Tang, J.; Mao, Y. Current Progress and Challenges in the Development of Pharmacotherapy for Metabolic Dysfunction-Associated Steatohepatitis. Diabetes Metab. Res. Rev. 2024, 40, e3846. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, M.; Birks, J.; Fryer, E.; Delaney, D.; Sarania, N.; Banerjee, R.; Neubauer, S.; Barnes, E.; Fleming, K.A.; Wang, L.M. Interobserver Variability in Histologic Evaluation of Liver Fibrosis Using Categorical and Quantitative Scores. Am. J. Clin. Pathol. 2017, 147, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Kuwashiro, T.; Takahashi, H.; Hyogo, H.; Ogawa, Y.; Imajo, K.; Yoneda, M.; Nakahara, T.; Oeda, S.; Tanaka, K.; Amano, Y.; et al. Discordant Pathological Diagnosis of Non-alcoholic Fatty Liver Disease: A Prospective Multicenter Study. JGH Open 2020, 4, 497–502. [Google Scholar] [CrossRef]
- Gawrieh, S.; Knoedler, D.M.; Saeian, K.; Wallace, J.R.; Komorowski, R.A. Effects of Interventions on Intra- and Interobserver Agreement on Interpretation of Nonalcoholic Fatty Liver Disease Histology. Ann. Diagn. Pathol. 2011, 15, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.; Sotoudehmanesh, R.; Nouraie, M.; Peikan-Heirati, M.; Sepanlou, S.G.; Malekzadeh, R.; Sotoudeh, M. Sampling Error in Histopathology Findings of Nonalcoholic Fatty Liver Disease: A Post Mortem Liver Histology Study. Arch. Iran. Med. 2012, 15, 418–421. [Google Scholar]
- Zhou, J.-H.; Cai, J.-J.; She, Z.-G.; Li, H.-L. Evaluation of NAFLD. World J. Gastroenterol. 2019, 25, 1289–1431. [Google Scholar]
- Younossi, Z.M.; Loomba, R.; Anstee, Q.M.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; et al. Diagnostic Modalities for Nonalcoholic Fatty Liver Disease, Nonalcoholic Steatohepatitis, and Associated Fibrosis. Hepatology 2018, 68, 349–360. [Google Scholar] [CrossRef]
- Wattacheril, J.J.; Abdelmalek, M.F.; Lim, J.K.; Sanyal, A.J. AGA Clinical Practice Update on the Role of Noninvasive Biomarkers in the Evaluation and Management of Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2023, 165, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Patel, K.; Duarte-Rojo, A.; Asrani, S.K.; Alsawas, M.; Dranoff, J.A.; Fiel, M.I.; Murad, M.H.; Leung, D.H.; Levine, D.; et al. AASLD Practice Guideline on Blood-Based Noninvasive Liver Disease Assessment of Hepatic Fibrosis and Steatosis. Hepatology 2025, 81, 321–357. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Duarte-Rojo, A.; Patel, K.; Asrani, S.K.; Alsawas, M.; Dranoff, J.A.; Fiel, M.I.; Murad, M.H.; Leung, D.H.; Levine, D.; et al. AASLD Practice Guideline on Imaging-Based Noninvasive Liver Disease Assessment of Hepatic Fibrosis and Steatosis. Hepatology 2024, 81, 672–724. [Google Scholar] [CrossRef]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Zachou, M.; Flevari, P.; Nasiri-Ansari, N.; Varytimiadis, C.; Kalaitzakis, E.; Kassi, E.; Androutsakos, T. The Role of Anti-Diabetic Drugs in NAFLD. Have We Found the Holy Grail? A Narrative Review. Eur. J. Clin. Pharmacol. 2023, 80, 127–150. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rolph, T.; Knott, M.; Dubourg, J. FGF21 Agonists: An Emerging Therapeutic for Metabolic Dysfunction-Associated Steatohepatitis and Beyond. J. Hepatol. 2024, 81, 562–576. [Google Scholar] [CrossRef]
- Staels, B.; Butruille, L.; Francque, S. Treating NASH by Targeting Peroxisome Proliferator-Activated Receptors. J. Hepatol. 2023, 79, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Hwahng, S.H.; Ki, S.H.; Bae, E.J.; Kim, H.E.; Kim, S.G. Role of Adenosine Monophosphate-Activated Protein Kinase-P70 Ribosomal S6 Kinase-1 Pathway in Repression of Liver X Receptor-Alpha-Dependent Lipogenic Gene Induction and Hepatic Steatosis by a Novel Class of Dithiolethiones. Hepatology 2009, 49, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Kobayashi, T.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Iwaki, M.; Ozaki, A.; Kasai, Y.; Nogami, A.; Honda, Y.; et al. The Role of Leaky Gut in Nonalcoholic Fatty Liver Disease: A Novel Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 8161. [Google Scholar] [CrossRef] [PubMed]
- Tetsi, L.; Charles, A.-L.; Paradis, S.; Lejay, A.; Talha, S.; Geny, B.; Lugnier, C. Effects of Cyclic Nucleotide Phosphodiesterases (PDEs) on Mitochondrial Skeletal Muscle Functions. Cell. Mol. Life Sci. 2017, 74, 1883–1893. [Google Scholar] [CrossRef]
- Tuell, D.; Ford, G.; Los, E.; Stone, W. The Role of Glutathione and Its Precursors in Type 2 Diabetes. Antioxidants 2024, 13, 184. [Google Scholar] [CrossRef]
- Kasarinaite, A.; Sinton, M.; Saunders, P.T.K.; Hay, D.C. The Influence of Sex Hormones in Liver Function and Disease. Cells 2023, 12, 1604. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Zomer, E. Therapy of Experimental NASH and Fibrosis with Galectin Inhibitors. PLoS ONE 2013, 8, e83481. [Google Scholar] [CrossRef]
- Hinrichs, G.R.; Hovind, P.; Asmar, A. The GLP-1-Mediated Gut-Kidney Cross Talk in Humans: Mechanistic Insight. Am. J. Physiol.-Cell Physiol. 2023, 326, C567–C572. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The Physiology of Glucagon-like Peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Shilleh, A.H.; Viloria, K.; Broichhagen, J.; Campbell, J.E.; Hodson, D.J. GLP1R and GIPR Expression and Signaling in Pancreatic Alpha Cells, Beta Cells and Delta Cells. Peptides 2024, 175, 171179. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Expression Cloning of the Pancreatic Beta Cell Receptor for the Gluco-Incretin Hormone Glucagon-like Peptide 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8641–8645. [Google Scholar] [CrossRef]
- Ämmälä, C.; Ashcroft, F.M.; Rorsman, P. Calcium-Independent Potentiation of Insulin Release by Cyclic AMP in Single β-Cells. Nature 1993, 363, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Yabut, J.M.; Drucker, D.J. Glucagon-like Peptide-1 Receptor-basedTherapeutics for Metabolic Liver Disease. Endocr. Rev. 2023, 44, 14–32. [Google Scholar] [CrossRef]
- Jin, T.; Weng, J. Hepatic Functions of GLP-1 and Its Based Drugs: Current Disputes and Perspectives. Am. J. Physiol. Endocrinol. Metab. 2016, 311, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Bernsmeier, C.; Meyer-Gerspach, A.C.; Blaser, L.S.; Jeker, L.; Steinert, R.E.; Heim, M.H.; Beglinger, C. Glucose-Induced Glucagon-like Peptide 1 Secretion Is Deficient in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2014, 9, e87488. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Annuzzi, G.; Ragucci, M.; Di Donato, O.; Della Pepa, G.; Della Corte, G.; Griffo, E.; Anniballi, G.; Giacco, A.; Mancini, M.; et al. Insulin Resistance, Postprandial GLP-1 and Adaptive Immunity Are the Main Predictors of NAFLD in a Homogeneous Population at High Cardiovascular Risk. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 623–629. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like Peptide-1 Receptor Agonism Improves Metabolic, Biochemical, and Histopathological Indices of Nonalcoholic Steatohepatitis in Mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Sanyal, A.J.; Engebretsen, K.A.; Kliers, I.; Østergaard, L.; Vanni, D.; Bugianesi, E.; Rinella, M.E.; Roden, M.; Ratziu, V. Semaglutide 2.4 Mg in Participants With Metabolic Dysfunction-Associated Steatohepatitis: Baseline Characteristics and Design of the Phase 3 ESSENCE Trial. Aliment. Pharmacol. Ther. 2024, 60, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Bedossa, P.; Fraessdorf, M.; Neff, G.W.; Lawitz, E.; Bugianesi, E.; Anstee, Q.M.; Hussain, S.A.; Newsome, P.N.; Ratziu, V.; et al. A Phase 2 Randomized Trial of Survodutide in MASH and Fibrosis. N. Engl. J. Med. 2024, 391, 311–319. [Google Scholar] [CrossRef]
- Tan, H.; Yue, T.; Chen, Z.; Wu, W.; Xu, S.; Weng, J. Targeting FGF21 in Cardiovascular and Metabolic Diseases: From Mechanism to Medicine. Int. J. Biol. Sci. 2023, 19, 66–88. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Dolegowska, K.; Marchelek-Mysliwiec, M.; Nowosiad-Magda, M.; Slawinski, M.; Dolegowska, B. FGF19 Subfamily Members: FGF19 and FGF21. J. Physiol. Biochem. 2019, 75, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Li, X. The FGF Metabolic Axis. Front. Med. 2019, 13, 511–530. [Google Scholar] [CrossRef]
- Kamm, D.R.; Mccommis, K.S.; Forsythe, I.; Chondronikola, M.; Kamm, D.R.; Mccommis, K.S. Hepatic Stellate Cells in Physiology and Pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of Its Aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef]
- Fisher, F.M.; Chui, P.C.; Nasser, I.A.; Popov, Y.; Cunniff, J.C.; Lundasen, T.; Kharitonenkov, A.; Schuppan, D.; Flier, J.S.; Maratos-Flier, E. Fibroblast Growth Factor 21 Limits Lipotoxicity by Promoting Hepatic Fatty Acid Activation in Mice on Methionine and Choline-Deficient Diets. Gastroenterology 2014, 147, 1073–1083.e6. [Google Scholar] [CrossRef] [PubMed]
- Dunshee, D.R.; Bainbridge, T.W.; Kljavin, N.M.; Zavala-Solorio, J.; Schroeder, A.C.; Chan, R.; Corpuz, R.; Wong, M.; Zhou, W.; Deshmukh, G.; et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J. Biol. Chem. 2016, 291, 5986–5996. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, J.; Mohanty, J.; Sousa, L.P.; Tome, F.; Pardon, E.; Steyaert, J.; Lemmon, M.A.; Lax, I.; Schlessinger, J. Structures of β-Klotho Reveal a ‘Zip Code’-like Mechanism for Endocrine FGF Signalling. Nature 2018, 553, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in Non-Alcoholic Steatohepatitis: A Randomized, Double-Blind, Placebo-Controlled, Phase 2a Trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Stanislaus, S.; Hecht, R.; Yie, J.; Hager, T.; Hall, M.; Spahr, C.; Wang, W.; Weiszmann, J.; Li, Y.; Deng, L.; et al. A Novel Fc-FGF21 With Improved Resistance to Proteolysis, Increased Affinity Toward β-Klotho, and Enhanced Efficacy in Mice and Cynomolgus Monkeys. Endocrinology 2017, 158, 1314–1327. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Kowdley, K.V.; Bhatt, D.L.; Alkhouri, N.; Frias, J.P.; Bedossa, P.; Harrison, S.A.; Lazas, D.; Barish, R.; et al. Randomized, Controlled Trial of the FGF21 Analogue Pegozafermin in NASH. N. Engl. J. Med. 2023, 389, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications—A Review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- López-Velázquez, J.A.; Carrillo-Córdova, L.D.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. Nuclear Receptors in Nonalcoholic Fatty Liver Disease. J. Lipids 2012, 2012, 139875. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential Effects of Selective- and Pan-PPAR Agonists on Experimental Steatohepatitis and Hepatic Macrophages☆. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Tacke, F. Hepatic Macrophages in Homeostasis and Liver Diseases: From Pathogenesis to Novel Therapeutic Strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Vonderlin, J.; Chavakis, T.; Sieweke, M.; Tacke, F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1311–1324. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Gamez, D.; Deb, S. SGLT2 Inhibitors as Potential Anticancer Agents. Biomedicines 2023, 11, 1867. [Google Scholar] [CrossRef]
- Oku, A.; Ueta, K.; Arakawa, K.; Ishihara, T.; Nawano, M.; Kuronuma, Y.; Matsumoto, M.; Saito, A.; Tsujihara, K.; Anai, M.; et al. T-1095, an Inhibitor of Renal Na+-Glucose Cotransporters, May Provide a Novel Approach to Treating Diabetes. Diabetes 1999, 48, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Kim, E.R.; Lee, M.; Choi, D.H.; Kim, S.H.; Shin, E.; Kim, J.H.; Cho, J.W.; Han, D.H.; Cha, B.S.; et al. Increased Expression of Sodium-Glucose Cotransporter 2 and O-GlcNAcylation in Hepatocytes Drives Non-Alcoholic Steatohepatitis. Metabolism 2023, 145, 155612. [Google Scholar] [CrossRef]
- Li, L.; Li, Q.; Huang, W.; Han, Y.; Tan, H.; An, M.; Xiang, Q.; Zhou, R.; Yang, L.; Cheng, Y. Dapagliflozin Alleviates Hepatic Steatosis by Restoring Autophagy via the AMPK-mTOR Pathway. Front. Pharmacol. 2021, 12, 589273. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M.K. Autophagy: A Critical Regulator of Cellular Metabolism and Homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Perkovic, V.; Wheeler, D.C.; Hantel, S.; George, J.T.; Von Eynatten, M.; Koitka-Weber, A.; Wanner, C.; on behalf of the EMPA-REG OUTCOME Investigators. Empagliflozin and Cardiovascular and Kidney Outcomes Across KDIGO Risk Categories: Post Hoc Analysis of a Randomized, Double-Blind, Placebo-Controlled, Multinational Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1433–1444. [Google Scholar] [CrossRef]
- van der Aart-van der Beek, A.B.; de Boer, R.A.; Heerspink, H.J.L. Kidney and Heart Failure Outcomes Associated with SGLT2 Inhibitor Use. Nat. Rev. Nephrol. 2022, 18, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 Inhibitors in Patients with Heart Failure with Reduced Ejection Fraction: A Meta-Analysis of the EMPEROR-Reduced and DAPA-HF Trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Anker, S.D.; Usman, M.S.; Butler, J. SGLT2 Inhibitors: From Antihyperglycemic Agents to All-Around Heart Failure Therapy. Circulation 2022, 146, 299–302. [Google Scholar] [CrossRef]
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An Update on Sodium-Glucose Co-Transporter-2 Inhibitors for the Treatment of Diabetes Mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Takasu, T. Therapeutic Effects of SGLT2 Inhibitor Ipragliflozin and Metformin on NASH in Type 2 Diabetic Mice. Endocr. Res. 2020, 45, 147–161. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sun, P.; Wang, Y.; Chen, Y.; Niu, Y.; Ding, Y.; Xu, N.; Zhang, Y.; Xie, W. Dapagliflozin Attenuates Steatosis in Livers of High-Fat Diet-Induced Mice and Oleic Acid-Treated L02 Cells via Regulating AMPK/mTOR Pathway. Eur. J. Pharmacol. 2021, 907, 174304. [Google Scholar] [CrossRef]
- Daniele, G.; Xiong, J.; Solis-Herrera, C.; Merovci, A.; Eldor, R.; Tripathy, D.; DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Dapagliflozin Enhances Fat Oxidation and Ketone Production in Patients With Type 2 Diabetes. Diabetes Care 2016, 39, 2036–2041. [Google Scholar] [CrossRef]
- Bonner, C.; Kerr-Conte, J.; Gmyr, V.; Queniat, G.; Moerman, E.; Thévenet, J.; Beaucamps, C.; Delalleau, N.; Popescu, I.; Malaisse, W.J.; et al. Inhibition of the Glucose Transporter SGLT2 with Dapagliflozin in Pancreatic Alpha Cells Triggers Glucagon Secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium–Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Ozutsumi, T.; Namisaki, T.; Shimozato, N.; Kaji, K.; Tsuji, Y.; Kaya, D.; Fujinaga, Y.; Furukawa, M.; Nakanishi, K.; Sato, S.; et al. Combined Treatment with Sodium-Glucose Cotransporter-2 Inhibitor (Canagliflozin) and Dipeptidyl Peptidase-4 Inhibitor (Teneligliptin) Alleviates NASH Progression in A Non-Diabetic Rat Model of Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 2164. [Google Scholar] [CrossRef]
- Petito-da-Silva, T.I.; Souza-Mello, V.; Barbosa-da-Silva, S. Empaglifozin Mitigates NAFLD in High-Fat-Fed Mice by Alleviating Insulin Resistance, Lipogenesis and ER Stress. Mol. Cell. Endocrinol. 2019, 498, 110539. [Google Scholar] [CrossRef]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 Inhibitor, Attenuates the Development of Hepatocellular Carcinoma in a Mouse Model of Human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef] [PubMed]
- Salah, H.M.; Fudim, M. Sodium-Glucose Cotransporter 2 Inhibitors and Nonalcoholic Fatty Liver Disease. Heart Fail. Clin. 2022, 18, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-H.; Jung, C.-H.; Mok, J.-O.; Kim, C.-H.; Kang, S.-K.; Kim, B.-Y. Effect of Dapagliflozin on Alanine Aminotransferase Improvement in Type 2 Diabetes Mellitus with Non-Alcoholic Fatty Liver Disease. Endocrinol. Metab. 2018, 33, 387. [Google Scholar] [CrossRef] [PubMed]
- Marjot, T.; Green, C.J.; Charlton, C.A.; Cornfield, T.; Hazlehurst, J.; Moolla, A.; White, S.; Francis, J.; Neubauer, S.; Cobbold, J.F.; et al. Sodium-glucose Cotransporter 2 Inhibition Does Not Reduce Hepatic Steatosis in Overweight, Insulin-resistant Patients without Type 2 Diabetes. JGH Open 2020, 4, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. The Complexity and Multiplicity of the Specific cAMP Phosphodiesterase Family: PDE4, Open New Adapted Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 10616. [Google Scholar] [CrossRef] [PubMed]
- Möllmann, J.; Kahles, F.; Lebherz, C.; Kappel, B.; Baeck, C.; Tacke, F.; Werner, C.; Federici, M.; Marx, N.; Lehrke, M. The PDE4 Inhibitor Roflumilast Reduces Weight Gain by Increasing Energy Expenditure and Leads to Improved Glucose Metabolism. Diabetes Obes. Metab. 2017, 19, 496–508. [Google Scholar] [CrossRef]
- Zhang, R.; Maratos-Flier, E.; Flier, J.S. Reduced Adiposity and High-Fat Diet-Induced Adipose Inflammation in Mice Deficient for Phosphodiesterase 4B. Endocrinology 2009, 150, 3076–3082. [Google Scholar] [CrossRef]
- Ott, E.; Lechner, H.; Fazekas, F. Hemorheological Effects of Pentoxifylline on Disturbed Flow Behavior of Blood in Patients with Cerebrovascular Insufficiency. Eur. Neurol. 1983, 22, 105–107. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Tagua, V.G.; Ferri, C.; Martín-Núñez, E.; Hernández-Carballo, C.; Ureña-Torres, P.; Ruiz-Ortega, M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons. J. Clin. Med. 2019, 8, 287. [Google Scholar] [CrossRef]
- Chen, Y.M.; Chiang, W.C.; Lin, S.L.; Tsai, T.J. Therapeutic Efficacy of Pentoxifylline on Proteinuria and Renal Progression: An Update. J. Biomed. Sci. 2017, 24, 84. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-Β1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Wu, K.-D.; Tsai, T.-J.; Hsieh, B.-S. Pentoxifylline Inhibits PDGF-Induced Proliferation of and TGF-β-Stimulated Collagen Synthesis by Vascular Smooth Muscle Cells. J. Mol. Cell. Cardiol. 1999, 31, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Chiang, W.C.; Yang, Y.; Lai, C.F.; Wu, K.D.; Lin, S.L. Pentoxifylline Attenuates Proteinuria in Anti-Thy1 Glomerulonephritis via Downregulation of Nuclear Factor-κB and Smad2/3 Signaling. Mol. Med. 2015, 21, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline Improves Nonalcoholic Steatohepatitis: A Randomized Placebo-Controlled Trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef]
- Van Wagner, L.B.; Koppe, S.W.P.; Brunt, E.M.; Gottstein, J.; Gardikiotes, K.; Green, R.M.; Rinella, M.E. Pentoxifylline for the Treatment of Non-Alcoholic Steatohepatitis: A Randomized Controlled Trial. Ann. Hepatol. 2011, 10, 277–286. [Google Scholar] [CrossRef]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. AMP-Activated Protein Kinase: An Energy Sensor and Survival Mechanism in the Reinstatement of Metabolic Homeostasis. Exp. Cell Res. 2023, 428, 113614. [Google Scholar] [CrossRef]
- Zhao, P.; Saltiel, A.R. From Overnutrition to Liver Injury: AMP-Activated Protein Kinase in Nonalcoholic Fatty Liver Diseases. J. Biol. Chem. 2020, 295, 12279–12289. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, Z.; Xie, M.; Xue, J. Involvement of Adenosine Monophosphate-Activated Protein Kinase in the Influence of Timed High-Fat Evening Diet on the Hepatic Clock and Lipogenic Gene Expression in Mice. Nutr. Res. 2015, 35, 792–799. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Hardie, D.G. Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef]
- Liang, Z.; Li, T.; Jiang, S.; Xu, J.; Di, W.; Yang, Z.; Hu, W.; Yang, Y. AMPK: A Novel Target for Treating Hepatic Fibrosis. Oncotarget 2017, 8, 62780–62792. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Bertolani, C.; Guerra, C.T.; Aleffi, S.; Galastri, S.; Trappoliere, M.; Vizzutti, F.; Gelmini, S.; Laffi, G.; Pinzani, M.; et al. Adenosine Monophosphate–Activated Protein Kinase Modulates the Activated Phenotype of Hepatic Stellate Cells. Hepatology 2008, 47, 668–676. [Google Scholar] [CrossRef]
- Xu, M.; Neelands, T.; Powers, A.S.; Liu, Y.; Miller, S.D.; Pintilie, G.D.; Bois, J.D.; Dror, R.O.; Chiu, W.; Maduke, M. CryoEM Structures of the Human CLC-2 Voltage-Gated Chloride Channel Reveal a Ball-and-Chain Gating Mechanism. eLife 2024, 12, RP90648. [Google Scholar] [CrossRef] [PubMed]
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the Selectivity Filter in ClC Chloride Channels. Science 2003, 300, 108–112. [Google Scholar] [CrossRef]
- Thiemann, A.; Gründer, S.; Pusch, M.; Jentsch, T.J. A Chloride Channel Widely Expressed in Epithelial and Non-Epithelial Cells. Nature 1992, 356, 57–60. [Google Scholar] [CrossRef]
- Stölting, G.; Teodorescu, G.; Begemann, B.; Schubert, J.; Nabbout, R.; Toliat, M.R.; Sander, T.; Nürnberg, P.; Lerche, H.; Fahlke, C. Regulation of ClC-2 Gating by Intracellular ATP. Pflüg. Arch. Eur. J. Physiol. 2013, 465, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Chen, Z.; Ma, J.; Shan, Y.; Wang, Y.; Xie, B.; Zheng, D.; Guo, F.; Song, X.; Gao, G.; et al. Biallelic CLCN2 Mutations Cause Retinal Degeneration by Impairing Retinal Pigment Epithelium Phagocytosis and Chloride Channel Function. Hum. Genet. 2023, 142, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; Yan, Y.; Ahlberg, G.; Vad, O.B.; Refsgaard, L.; Dos Santos, J.L.; Mutsaers, N.; Svendsen, J.H.; Olesen, M.S.; Bentzen, B.H.; et al. A Novel Loss-of-Function Variant in the Chloride Ion Channel Gene Clcn2 Associates with Atrial Fibrillation. Sci. Rep. 2020, 10, 1453. [Google Scholar] [CrossRef] [PubMed]
- Fei, G.; Raehal, K.; Liu, S.; Qu, M.-H.; Sun, X.; Wang, G.-D.; Wang, X.-Y.; Xia, Y.; Schmid, C.L.; Bohn, L.M.; et al. Lubiprostone Reverses the Inhibitory Action of Morphine on Intestinal Secretion in Guinea Pig and Mouse. J. Pharmacol. Exp. Ther. 2010, 334, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; De Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-Driven Gut Vascular Barrier Disruption Is a Prerequisite for Non-Alcoholic Steatohepatitis Development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Imajo, K.; Kobayashi, T.; Ozaki, A.; Iwaki, M.; Honda, Y.; Kato, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; et al. Lubiprostone in Patients with Non-Alcoholic Fatty Liver Disease: A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Trial. Lancet Gastroenterol. Hepatol. 2020, 5, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Walenbergh, S.M.A.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Hladik, A.; Göderle, L.; et al. Malondialdehyde Epitopes Are Sterile Mediators of Hepatic Inflammation in Hypercholesterolemic Mice. Hepatology 2017, 65, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Cannella, L.; De Martin, S.; Shiri-Sverdlov, R.; Squadrito, G. Biomedicines The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef]
- Wierman, M.E. Sex Steroid Effects at Target Tissues: Mechanisms of Action. Adv. Physiol. Educ. 2007, 31, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sayaf, K.; Gabbia, D.; Russo, F.P.; De Martin, S. The Role of Sex in Acute and Chronic Liver Damage. Int. J. Mol. Sci. 2022, 23, 10654. [Google Scholar] [CrossRef] [PubMed]
- Bryzgalova, G.; Gao, H.; Ahren, B.; Zierath, J.R.; Galuska, D.; Steiler, T.L.; Dahlman-Wright, K.; Nilsson, S.; Gustafsson, J.-Å.; Efendic, S.; et al. Evidence That Oestrogen Receptor-α Plays an Important Role in the Regulation of Glucose Homeostasis in Mice: Insulin Sensitivity in the Liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef]
- Ahmed-Sowur, H.; Bailey, C.J. Role of Ovarian Hormones in the Long-Term Control of Glucose Homeostasis Glycogen Formation and Gluconeogenesis. Ann. Nutr. Metab. 1981, 25, 208–212. [Google Scholar] [CrossRef]
- Bulun, S.E.; Cheng, Y.-H.; Yin, P.; Imir, G.; Utsunomiya, H.; Attar, E.; Innes, J.; Julie Kim, J. Progesterone Resistance in Endometriosis: Link to Failure to Metabolize Estradiol. Mol. Cell. Endocrinol. 2006, 248, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Chlebowski, R.T.; Stefanick, M.L.; Aragaki, A.K.; Rossouw, J.E.; Prentice, R.L.; Anderson, G.; Howard, B.V.; Thomson, C.A.; LaCroix, A.Z.; et al. Menopausal Hormone Therapy and Health Outcomes During the Intervention and Extended Poststopping Phases of the Women’s Health Initiative Randomized Trials. JAMA 2013, 310, 1353. [Google Scholar] [CrossRef]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.W.; Cummings, R.D.; Drickamer, K.; Felzi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.I.; et al. Galectins: A Family of Animal β-Galactoside-Binding Lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Bouffette, S.; Botez, I.; De Ceuninck, F. Targeting Galectin-3 in Inflammatory and Fibrotic Diseases. Trends Pharmacol. Sci. 2023, 44, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Farhad, M.; Rolig, A.S.; Redmond, W.L. The Role of Galectin-3 in Modulating Tumor Growth and Immunosuppression within the Tumor Microenvironment. OncoImmunology 2018, 7, e1434467. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 Regulates Myofibroblast Activation and Hepatic Fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Ricci, C.; Fantauzzi, C.B.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 Ablation Protects Mice from Diet-Induced NASH: A Major Scavenging Role for Galectin-3 in Liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, A.C.; Tonev, D.; Jacoby, B.; Pinzani, M.; Slack, R.J. Galectin-3: Therapeutic Targeting in Liver Disease. Expert Opin. Ther. Targets 2023, 27, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, F.; Zhong, W.; Jiang, X.; Zhang, F.; Ji, X.; Xue, M.; Qiu, Y.; Yu, J.; Hu, X.; et al. Secretory Galectin-3 Promotes Hepatic Steatosis via Regulation of the PPARγ/CD36 Signaling Pathway. Cell. Signal. 2021, 84, 110043. [Google Scholar] [CrossRef] [PubMed]
- Jeftic, I.; Jovicic, N.; Pantic, J.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Galectin-3 Ablation Enhances Liver Steatosis, but Attenuates Inflammation and IL-33-Dependent Fibrosis in Obesogenic Mouse Model of Nonalcoholic Steatohepatitis. Mol. Med. 2015, 21, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marcos, L.V.; Martínez-Beamonte, R.; Macías-Herranz, M.; Arnal, C.; Barranquero, C.; Puente-Lanzarote, J.J.; Gascón, S.; Herrero-Continente, T.; Gonzalo-Romeo, G.; Alastrué-Vera, V.; et al. Hepatic Galectin-3 Is Associated with Lipid Droplet Area in Non-Alcoholic Steatohepatitis in a New Swine Model. Sci. Rep. 2022, 12, 1024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Chen, X.; Hsu, D.K.; Baghy, K.; Serizawa, N.; Scott, F.; Takada, Y.; Takada, Y.; Fukada, H.; Chen, J.; et al. Galectin-3 Modulates Phagocytosis-Induced Stellate Cell Activation and Liver Fibrosis in Vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediators Inflamm. 2017, 2017, 9247574. [Google Scholar] [CrossRef]
- Sato, S.; St-Pierre, C.; Bhaumik, P.; Nieminen, J. Galectins in Innate Immunity: Dual Functions of Host Soluble β-Galactoside-Binding Lectins as Damage-Associated Molecular Patterns (DAMPs) and as Receptors for Pathogen-Associated Molecular Patterns (PAMPs). Immunol. Rev. 2009, 230, 172–187. [Google Scholar] [CrossRef]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- Nakamura, S.; Konishi, H.; Kishino, M.; Yatsuji, S.; Tokushige, K.; Hashimoto, E.; Shiratori, K. Prevalence of Esophagogastric Varices in Patients with Non-alcoholic Steatohepatitis. Hepatol. Res. 2008, 38, 572–579. [Google Scholar] [CrossRef]
- Liu, X.; Strable, M.S.; Ntambi, J.M. Stearoyl CoA Desaturase 1: Role in Cellular Inflammation and Stress. Adv. Nutr. 2011, 2, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Dobrzyn, A.; Man, W.C.; Chu, K.; Sampath, H.; Kim, H.J.; Ntambi, J.M. Stearoyl-CoA Desaturase 1 Gene Expression Is Necessary for Fructose-Mediated Induction of Lipogenic Gene Expression by Sterol Regulatory Element-Binding Protein-1c-Dependent and Independent Mechanisms. J. Biol. Chem. 2004, 279, 25164–25171. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Basta, B.; Mato, J.M.; Craig, A.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Tsvirkun, D.; Hayardeny, L.; Chandar, V.; Schwartz, R.E.; et al. Aramchol Downregulates Stearoyl CoA-Desaturase 1 in Hepatic Stellate Cells to Attenuate Cellular Fibrogenesis. JHEP Rep. 2021, 3, 100237. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Cohen, P.; Asilmaz, E.; Grahame, D.; Friedman, J.M.; Ntambi, J.M. Stearoyl-CoA Desaturase 1 Deficiency Increases Fatty Acid Oxidation by Activating AMP-Activated Protein Kinase in Liver. Proc. Natl. Acad. Sci. USA 2004, 101, 6409–6414. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Yilmaz, Y.; Lazas, D.; Friedman, S.L.; Lackner, C.; Behling, C.; Cummings, O.W.; Chen, L.; Petitjean, M.; Gilgun-Sherki, Y.; et al. Aramchol Improves Hepatic Fibrosis in Metabolic Dysfunction–Associated Steatohepatitis: Results of Multimodality Assessment Using Both Conventional and Digital Pathology. Hepatology 2024. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Jeong, S.W.; Jang, J.Y. Recent Updates on Pharmacologic Therapy in Non-Alcoholic Fatty Liver Disease. Clin. Mol. Hepatol. 2023, 30, 129–133. [Google Scholar] [CrossRef]
- Calabrese, V.; Calderone, A.; Ragusa, N.; Rizza, V. Effects of Metadoxine on Cellular Status of Glutathione and of Enzymatic Defence System Following Acute Ethanol Intoxication in Rats. Drugs Exp. Clin. Res. 1996, 22, 17–24. [Google Scholar] [PubMed]
- Shenoy, K.T.; Balakumaran, L.K.; Mathew, P.; Prasad, M.; Prabhakar, B.; Sood, A.; Singh, S.P.; Rao, N.P.; Zargar, S.A.; Bignamini, A.A. Metadoxine Versus Placebo for the Treatment of Non-Alcoholic Steatohepatitis: A Randomized Controlled Trial. J. Clin. Exp. Hepatol. 2014, 4, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Ruiz, M.C.; Bucio, L.; Correa, A.; Souza, V.; Hernández, E.; Gómez-Quiroz, L.E.; Kershenobich, D. Metadoxine Prevents Damage Produced by Ethanol and Acetaldehyde in Hepatocyte and Hepatic Stellate Cells in Culture. Pharmacol. Res. 2001, 44, 431–436. [Google Scholar] [CrossRef]
- Smith, S.M.; Pegram, A.H. Obeticholic Acid. J. Pharm. Technol. 2017, 33, 66–71. [Google Scholar] [CrossRef]
- Ali, A.H.; Carey, E.J.; Lindor, K.D. Recent Advances in the Development of Farnesoid X Receptor Agonists. Ann. Transl. Med. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X Receptor Activation Inhibits Inflammation and Preserves the Intestinal Barrier in Inflammatory Bowel Disease. Gut 2011, 60, 463. [Google Scholar] [CrossRef]
- Anderson, K.M.; Gayer, C.P. The Pathophysiology of Farnesoid x Receptor (Fxr) in the Gi Tract: Inflammation, Barrier Function and Innate Immunity. Cells 2021, 10, 3206. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Dufour, J.-F.; Anstee, Q.M.; Goodman, Z.; Younossi, Z.; Harrison, S.A.; Loomba, R.; Sanyal, A.J.; Bonacci, M.; Trylesinski, A.; et al. Non-Invasive Evaluation of Response to Obeticholic Acid in Patients with NASH: Results from the REGENERATE Study. J. Hepatol. 2022, 76, 536–548. [Google Scholar] [CrossRef]
- McKenzie, H. Intercept’s NASH Treatment Fails to Win Support of FDA Advisory Committee. Available online: https://www.biospace.com/article/intercept-shares-fall-as-fda-again-questions-risk-benefit-of-nash-treatment-/ (accessed on 1 July 2024).
- Puengel, T.; Krenkel, O.; Kohlhepp, M.; Lefebvre, E.; Luedde, T.; Trautwein, C.; Tacke, F. Differential Impact of the Dual CCR2/CCR5 Inhibitor Cenicriviroc on Migration of Monocyte and Lymphocyte Subsets in Acute Liver Injury. PLoS ONE 2017, 12, e0184694. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wai-Sun Wong, V.; Abdelmalek, M.F.; Rodriguez-Araujo, G.; Landgren, H.; Park, G.S.; Bedossa, P.; Alkhouri, N.; Tacke, F.; et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin. Gastroenterol. Hepatol. 2024, 22, 124–134.e1. [Google Scholar] [CrossRef] [PubMed]
- PubChem Compound Summary for CID 71245288, Selonsertib. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Selonsertib (accessed on 1 July 2024).
- Meijles, D.N.; Cull, J.J.; Markou, T.; Cooper, S.T.E.; Haines, Z.H.R.; Fuller, S.J.; O’Gara, P.; Sheppard, M.N.; Harding, S.E.; Sugden, P.H.; et al. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls P38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.-C.; Fang, Z.; Lee, J.E.; Park, J.H.; Ryu, J.-K.; Jung, K.H.; Hong, S.-S. Selonsertib Inhibits Liver Fibrosis via Downregulation of ASK1/ MAPK Pathway of Hepatic Stellate Cells. Biomol. Ther. 2020, 28, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Wong, V.W.-S.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for Patients with Bridging Fibrosis or Compensated Cirrhosis Due to NASH: Results from Randomized Phase III STELLAR Trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, L.; Gao, Y.; Ding, Y. The Prevalence of Nonalcoholic Fatty Liver Disease and Related Metabolic Comorbidities Was Associated with Age at Onset of Moderate to Severe Plaque Psoriasis: A Cross-Sectional Study. PLoS ONE 2017, 12, e0169952. [Google Scholar] [CrossRef] [PubMed]
- Prussick, R.B.; Miele, L. Nonalcoholic Fatty Liver Disease in Patients with Psoriasis: A Consequence of Systemic Inflammatory Burden? Br. J. Dermatol. 2018, 179, 16–29. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Gkaliagkousi, E.; Lazaridis, A.; Arelaki, S.; Pateinakis, P.; Ntinopoulou, M.; Mitsios, A.; Antoniadou, C.; Argyriou, C.; Georgiadis, G.S.; et al. Angiotensin II Triggers Release of Neutrophil Extracellular Traps, Linking Thromboinflammation with Essential Hypertension. JCI Insight 2021, 6, e148668. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.-J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or Not to NET:Current Opinions and State of the Science Regarding the Formation of Neutrophil Extracellular Traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Arelaki, S.; Koletsa, T.; Sinakos, E.; Papadopoulos, V.; Arvanitakis, K.; Skendros, P.; Akriviadis, E.; Ritis, K.; Germanidis, G.; Hytiroglou, P. Neutrophil Extracellular Traps Enriched with IL-1β and IL-17A Participate in the Hepatic Inflammatory Process of Patients with Non-Alcoholic Steatohepatitis. Virchows Arch. 2022, 481, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Abrar, M.; Islam, S.; Kamal, M.; Hasan, M.J.; Khan, A.S.; Ahmad, N. Effect of Telmisartan and Vitamin E on Liver Histopathology with Non-alcoholic Steatohepatitis: A Randomized, Open-label, Noninferiority Trial. JGH Open 2020, 4, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Wasta Esmail, V.A.; Al-Nimer, M.S.M.; Mohammed, M.O. Effects of Orlistat or Telmisartan on the Serum Free Fatty Acids in Non-Alcoholic Fatty Liver Disease Patients: An Open-Labeled Randomized Controlled Study. Turk. J. Gastroenterol. 2022, 33, 421. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlton, M. Rational Combination Therapy for NASH: Insights from Clinical Trials and Error. J. Hepatol. 2023, 78, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Veidal, S.S.; Fensholdt, L.K.D.; Rigbolt, K.T.G.; Papazyan, R.; Nielsen, J.C.; Feigh, M.; Vrang, N.; Young, M.; Jelsing, J.; et al. Combined Obeticholic Acid and Elafibranor Treatment Promotes Additive Liver Histological Improvements in a Diet-Induced Ob/Ob Mouse Model of Biopsy-Confirmed NASH. Sci. Rep. 2019, 9, 9046. [Google Scholar] [CrossRef] [PubMed]
- Galmed Pharmaceuticals Ltd. Galmed Announces Grant of New Patent for the Combination of Aramchol with Resmetirom (MGL-3196, REZDIFFRA) for the Treatment of NASH and Liver Fibrosis. Available online: https://www.prnewswire.com/news-releases/galmed-announces-grant-of-new-patent-for-the-combination-of-aramchol-with-resmetirom-mgl-3196-rezdiffra-for-the-treatment-of-nash-and-liver-fibrosis-302090340.html (accessed on 6 July 2024).
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and Efficacy of Combination Therapy with Semaglutide, Cilofexor and Firsocostat in Patients with Non-Alcoholic Steatohepatitis: A Randomised, Open-Label Phase II Trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Mark, H.E.; Allen, A.M.; Arab, J.P.; Carrieri, P.; Noureddin, M.; Alazawi, W.; Alkhouri, N.; Alqahtani, S.A.; Anstee, Q.M.; et al. A Global Action Agenda for Turning the Tide on Fatty Liver Disease. Hepatology 2024, 79, 502–523. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Candidate | Drug Class | Trial Phase | Trial Status | Clinical Trial Registration | Primary Endpoint |
|---|---|---|---|---|---|
| Semaglutide | GLP-1 Agonist [26] | 3 | Active | NCT04822181 | Part 1 (After 72 Weeks): Resolution of Steatohepatitis and No Worsening of Fibrosis Improvement in Liver Fibrosis and No Worsening of Steatohepatitis Part 2 (After 240 Weeks): Cirrhosis-Free Survival |
| Survodutide | GLP-1 Agonist [26] | 3 | Recruiting | NCT06309992 NCT06632444 | NCT06309992: After 48 Weeks: Relative Reduction in Liver Fat Content ≥30% Relative % Change in Body Weight NCT06632444: Part 1 (After 52 Weeks): MASH Resolution Without Worsening Fibrosis ≥1-Point Improvement in Fibrosis Stage Without Worsening MASH Part 2 (Up to 7 Years): Incidence of Liver-Related Adverse Event/All-Cause Mortality |
| Efruxifermin | FGF21 Analogue [27] | 3 | Recruiting | NCT06161571 NCT06215716 | NCT06161571: After 52 Weeks: Time/Incidence/Severity of Adverse Events Incidence of Clinically Significant Changes NCT06215716: After 52 Weeks: Resolution of MASH and ≥1 Stage Fibrosis Improvement After 240 Weeks: Event-Free Survival |
| Pegozafermin | FGF21 Analogue [27] | 3 | Recruiting | NCT06318169 | After 52 Weeks: Incidence of Fibrosis Improvement ≥ 1 Stage Without Worsening of MASH Incidence of MASH Resolution Without Worsening of Fibrosis |
| Lanifibranor | Pan-PPAR Agonist [28] | 3 | Recruiting | NCT04849728 | Part A (After 72 Weeks): Resolution of MASH/Fibrosis Improvement Part B (After 48 Weeks Post-Part A Completion): Incidence of Adverse Events |
| Dapagliflozin | SGLT2 Inhibitor [26] | 3 | Completed (Results Pending) | NCT03723252 | After 12 Months: Scored Liver Histological Improvement |
| Oltipraz | AMPK Activator [29] | 3 | Completed (Results Pending) | NCT04142749 | After 24 Weeks: Change in Assessed Liver Fat |
| Lubiprostone | ClC-2 Activator [30] | 3 | Completed (Results Pending) | NCT05768334 | After 48 Weeks: Change in Fat Quantification |
| Pentoxifylline (PTX) | PDE Inhibitor [31] | 3 | Completed (Results Pending) | NCT05284448 | After 6 Months: Improvement in Liver Aminotransferases (ALT and AST) Change in NAFLD Fibrosis Score (NFS) |
| N-Acetyl Cysteine (NAC) | Antioxidant [32] | 3 | Completed (Results Pending) | NCT05589584 | After 3 Months: Assessment of Leptin as Measure of Insulin Resistance |
| Estradiol | ER Agonist [33] | 3 | Recruiting | NCT04833140 | After 12 Months: Reduction of Liver Fibrosis Reduction in Liver Fat |
| Belapectin | Gal-3 Inhibitor [34] | 2b | Active | NCT04365868 | After 78 Weeks: Incidence of Newly Formed Esophageal Varices |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinerman, S.; Shu, Y. Therapeutic Strategies for MASH: An Update on Drug Candidates Under Investigation in Late-Phase Clinical Trials. Int. J. Transl. Med. 2025, 5, 7. https://doi.org/10.3390/ijtm5010007
Dinerman S, Shu Y. Therapeutic Strategies for MASH: An Update on Drug Candidates Under Investigation in Late-Phase Clinical Trials. International Journal of Translational Medicine. 2025; 5(1):7. https://doi.org/10.3390/ijtm5010007
Chicago/Turabian StyleDinerman, Samuel, and Yan Shu. 2025. "Therapeutic Strategies for MASH: An Update on Drug Candidates Under Investigation in Late-Phase Clinical Trials" International Journal of Translational Medicine 5, no. 1: 7. https://doi.org/10.3390/ijtm5010007
APA StyleDinerman, S., & Shu, Y. (2025). Therapeutic Strategies for MASH: An Update on Drug Candidates Under Investigation in Late-Phase Clinical Trials. International Journal of Translational Medicine, 5(1), 7. https://doi.org/10.3390/ijtm5010007