
Oxidative Stress and Its Role in Cd-Induced Epigenetic Modifications: Use of Antioxidants as a Possible Preventive Strategy

 ,
,  ,
, 

Abstract
:1. Introduction
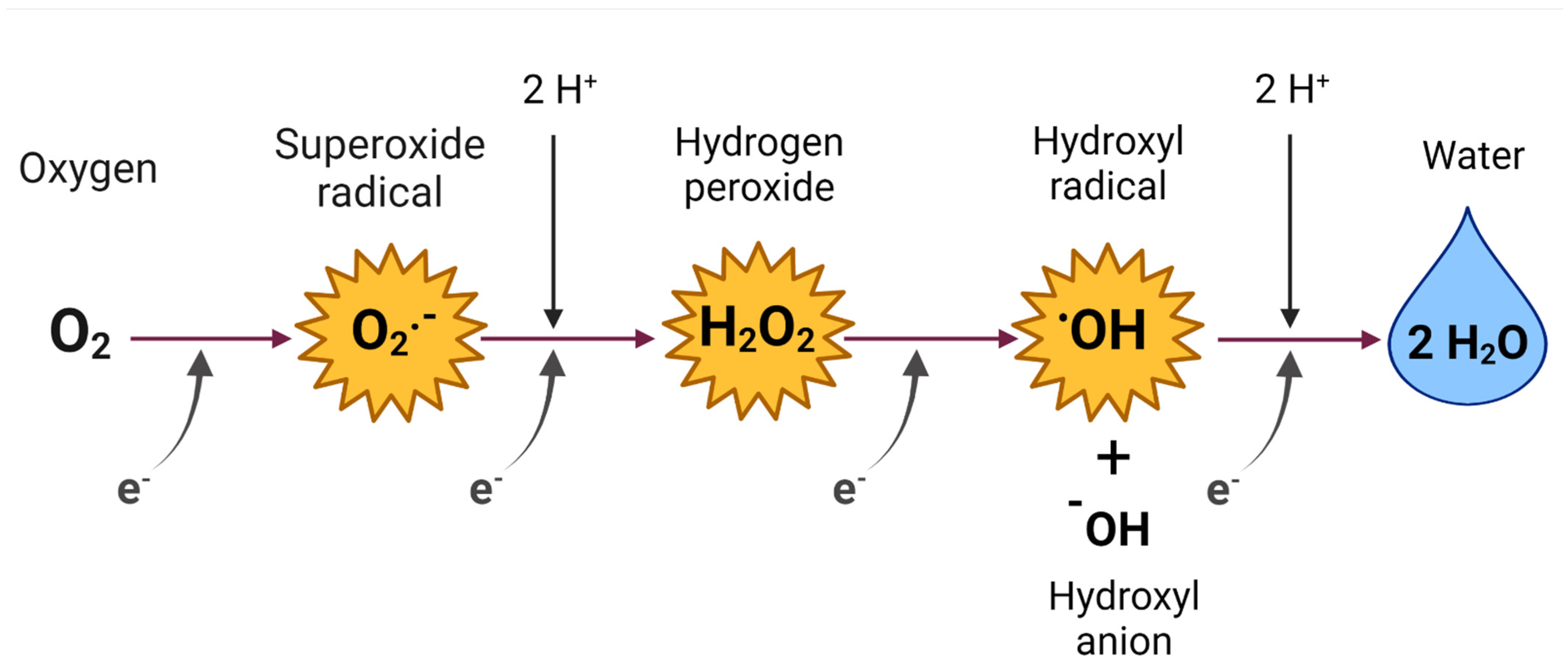
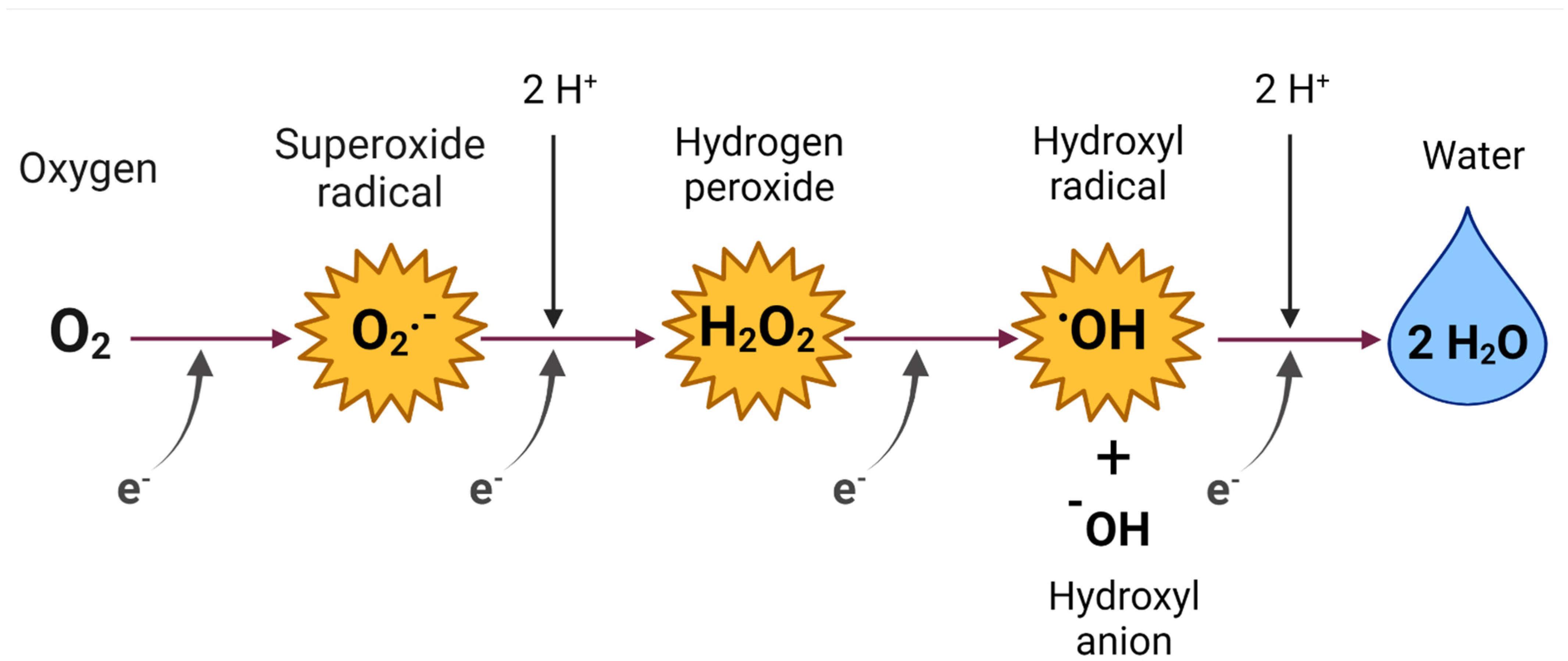
2. Reactive Oxygen Species (ROS)
ROS of Medical and Biological Importance
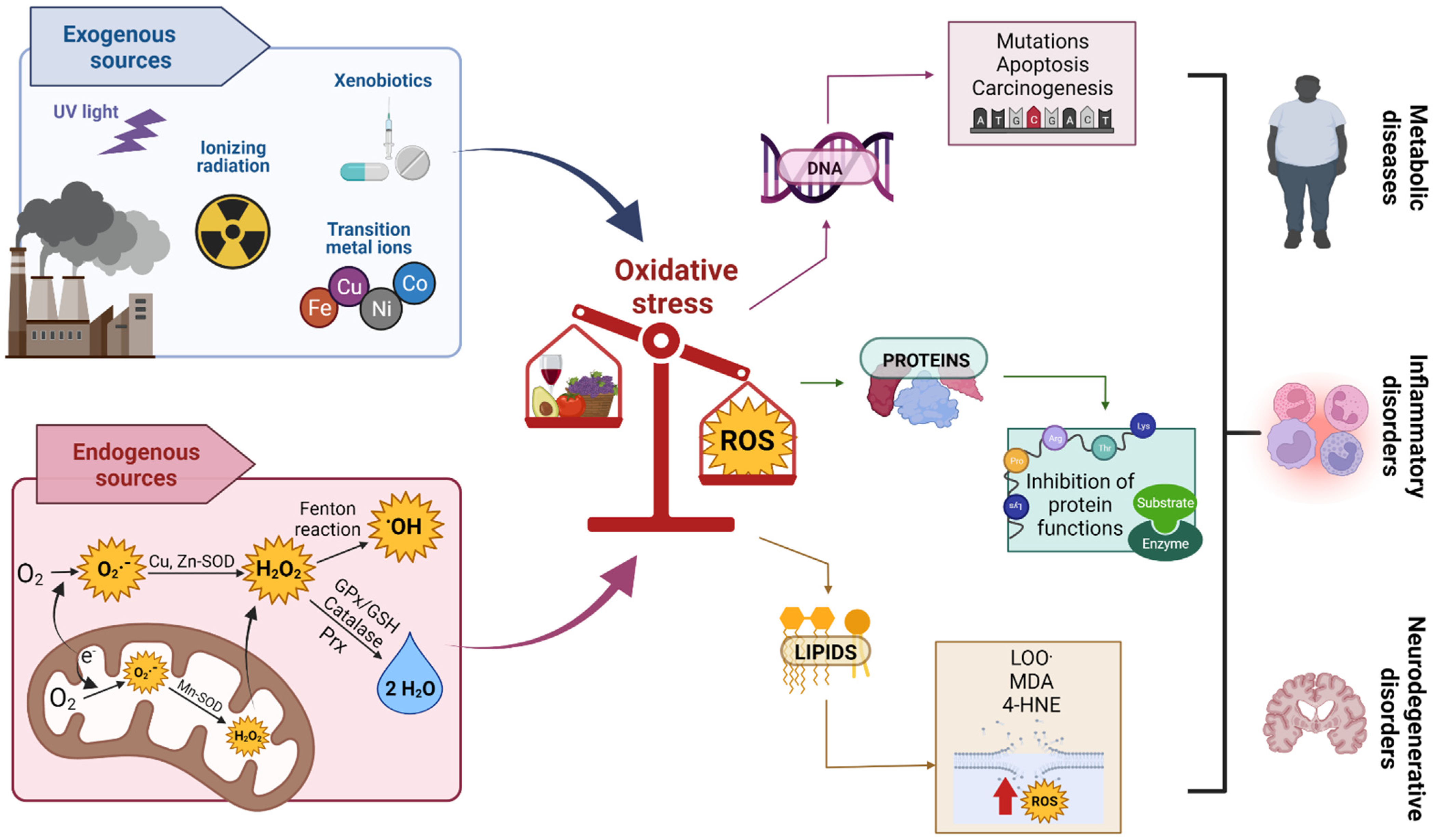
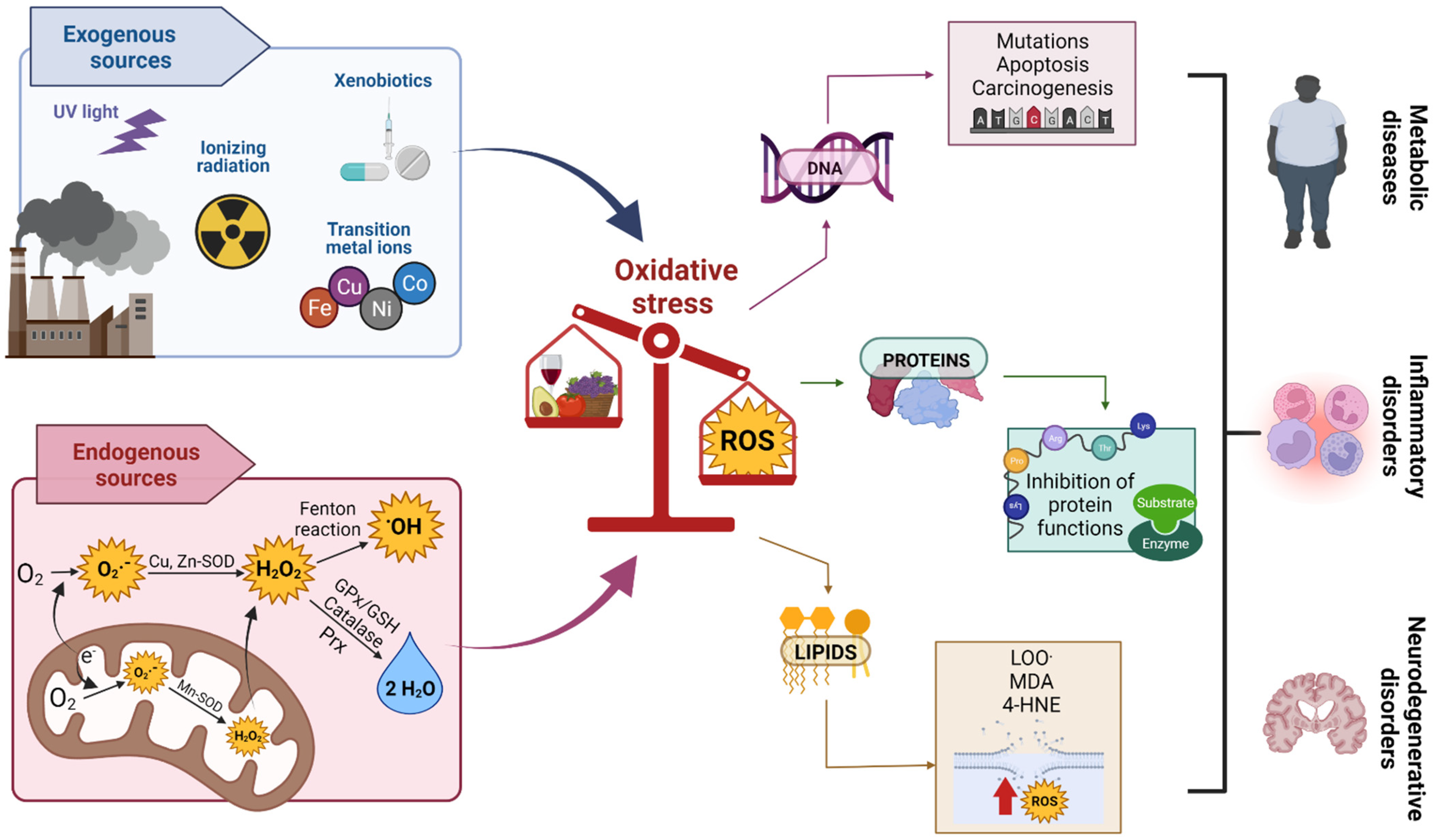
3. OS
Oxidant Damage Caused by OS
4. Antioxidants
4.1. Classification and Description of Antioxidants
4.1.1. Endogenous Antioxidants
Enzymatic Antioxidants
Nonenzymatic Antioxidants
4.1.2. Exogenous Antioxidants
5. Role of OS in Cd Toxicity
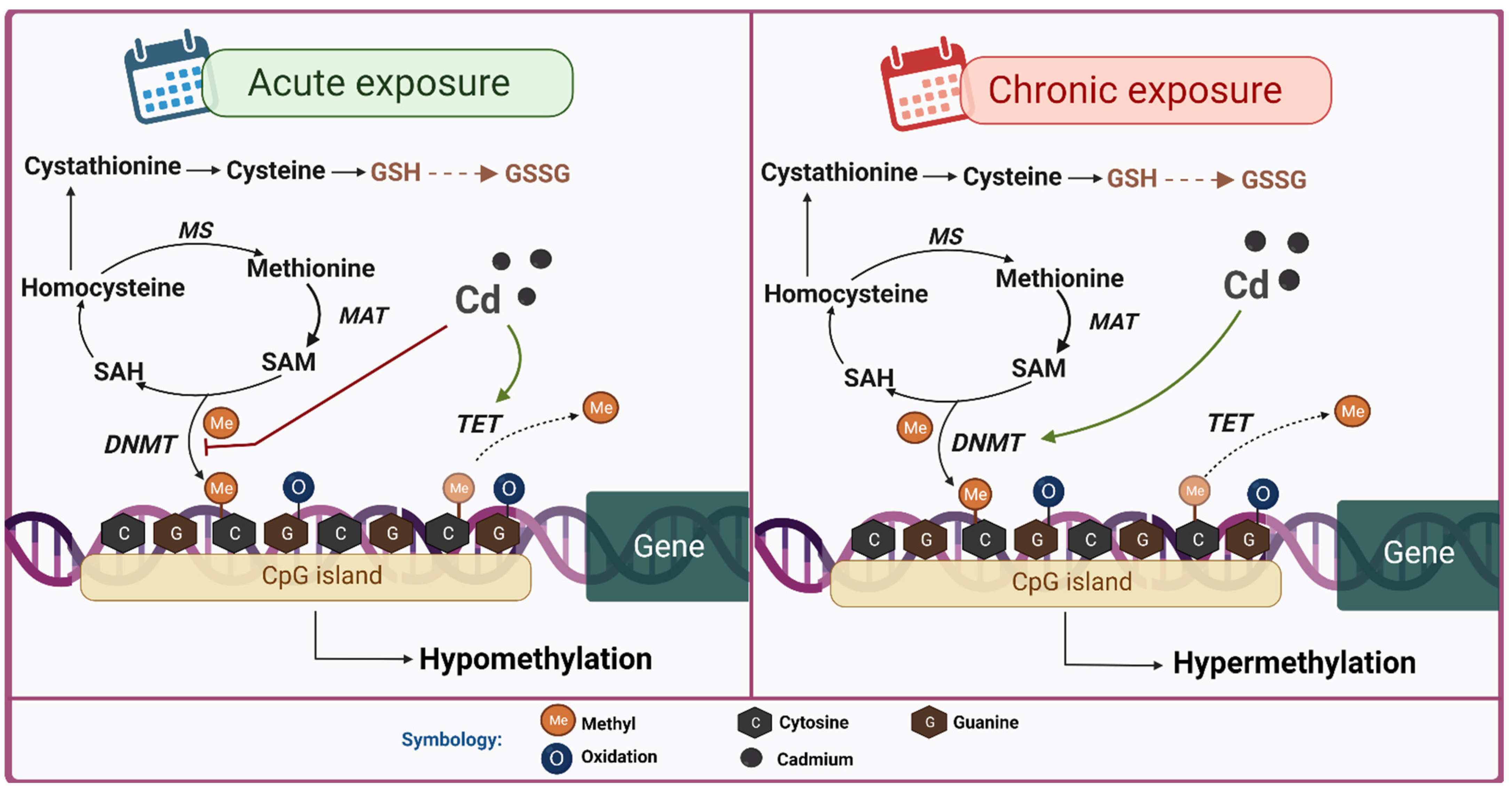
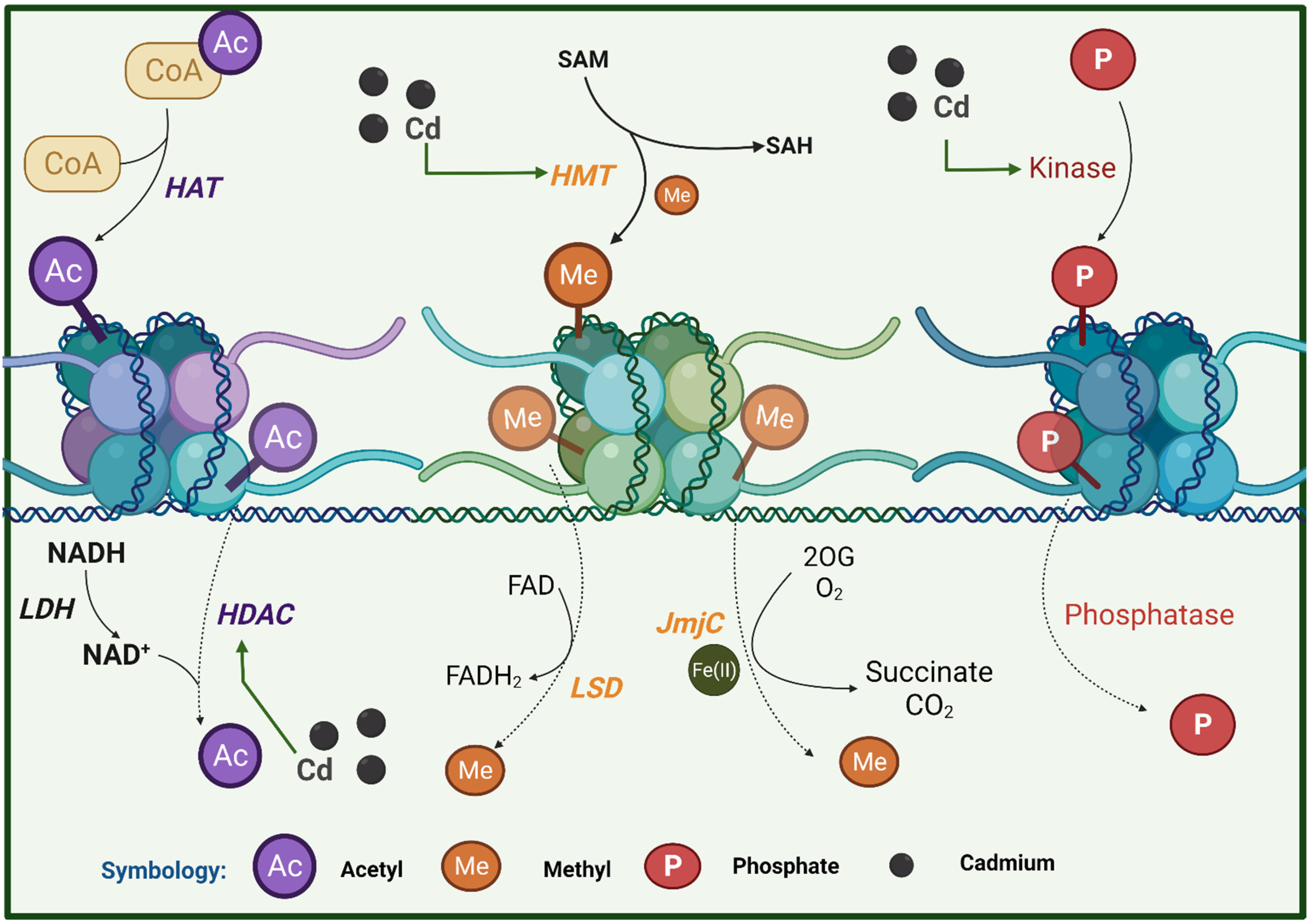
6. Epigenetic Effects of Cd Exposure
6.1. DNA Methylation
6.2. Histone Modification
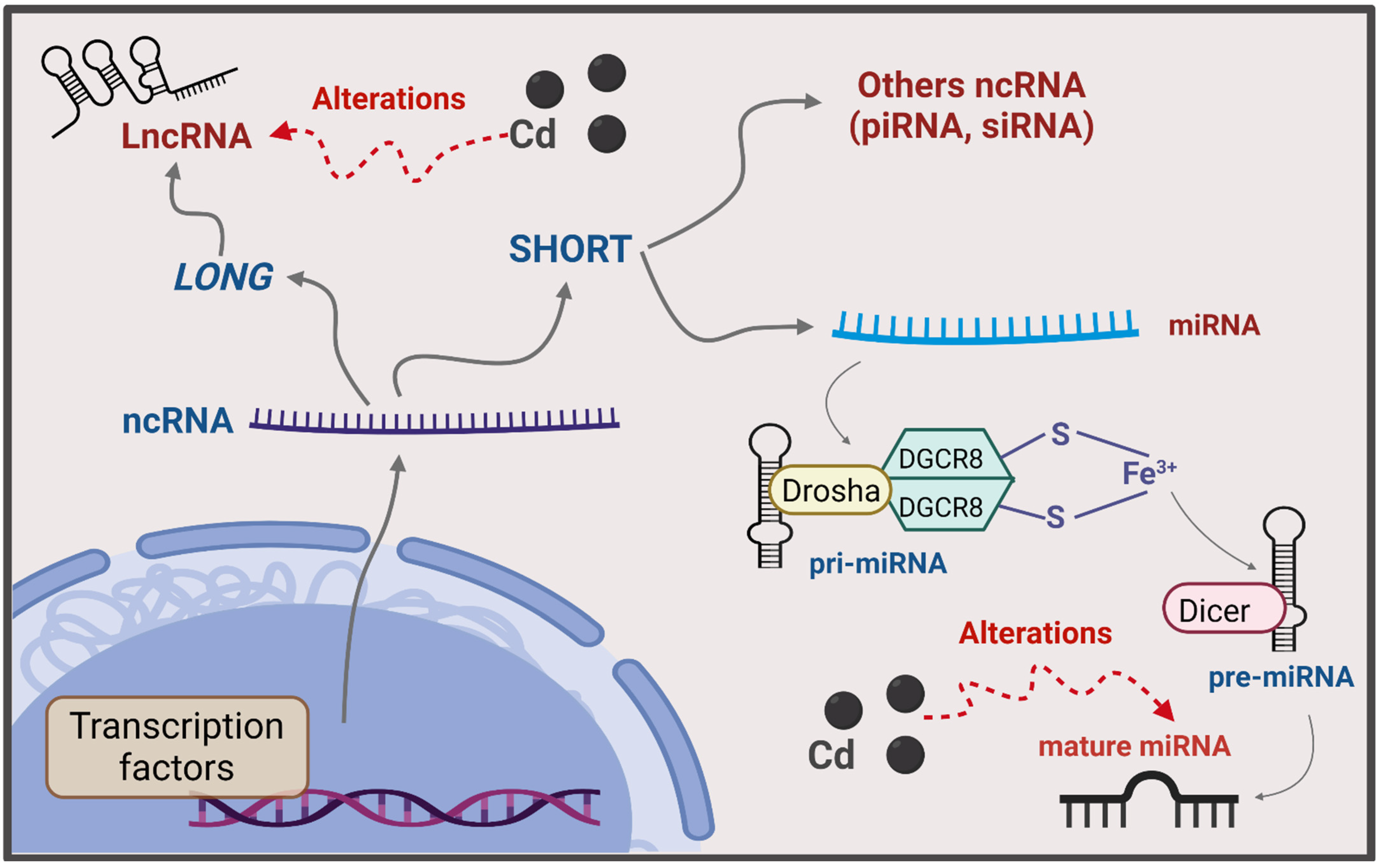
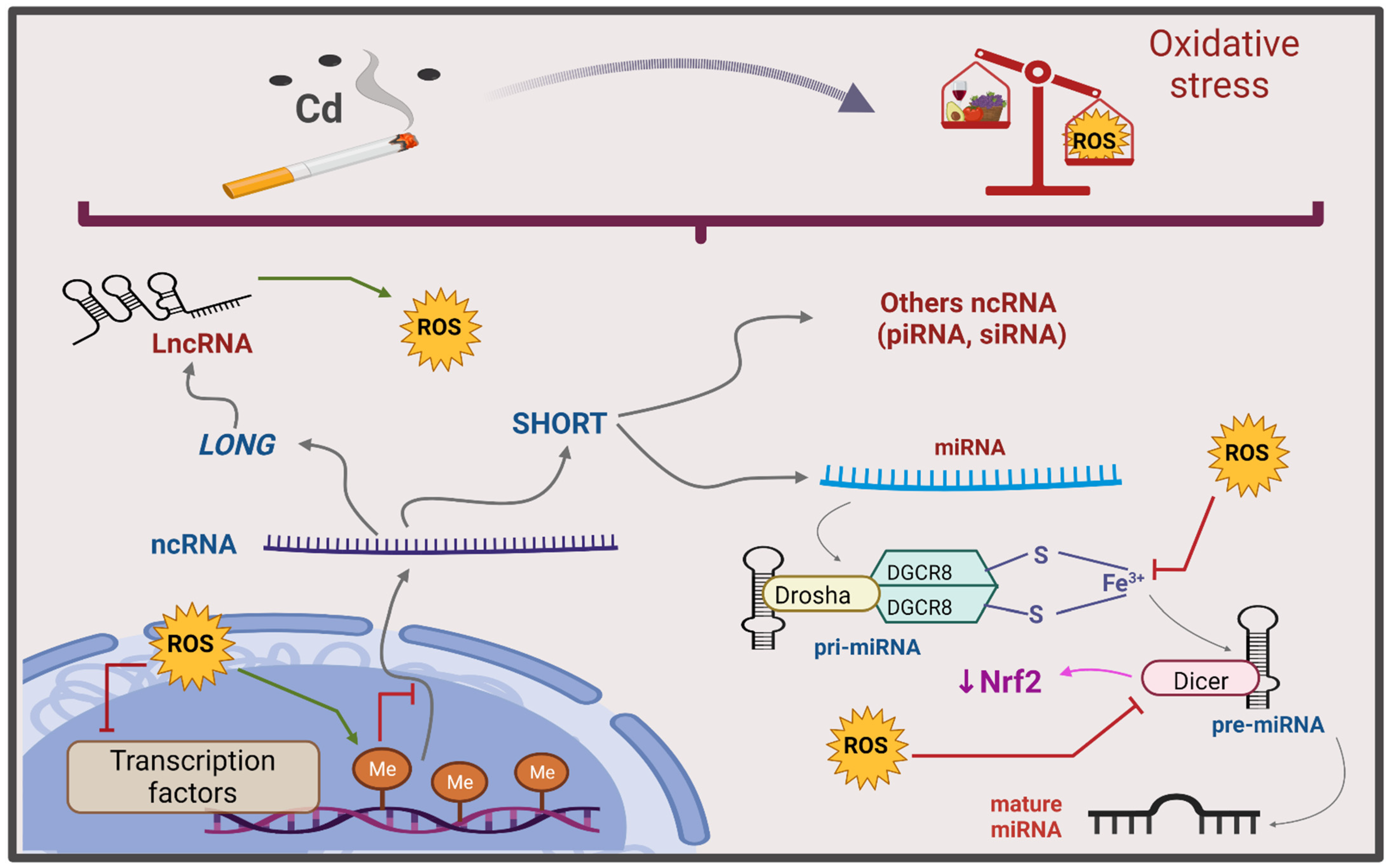
6.3. ncRNA
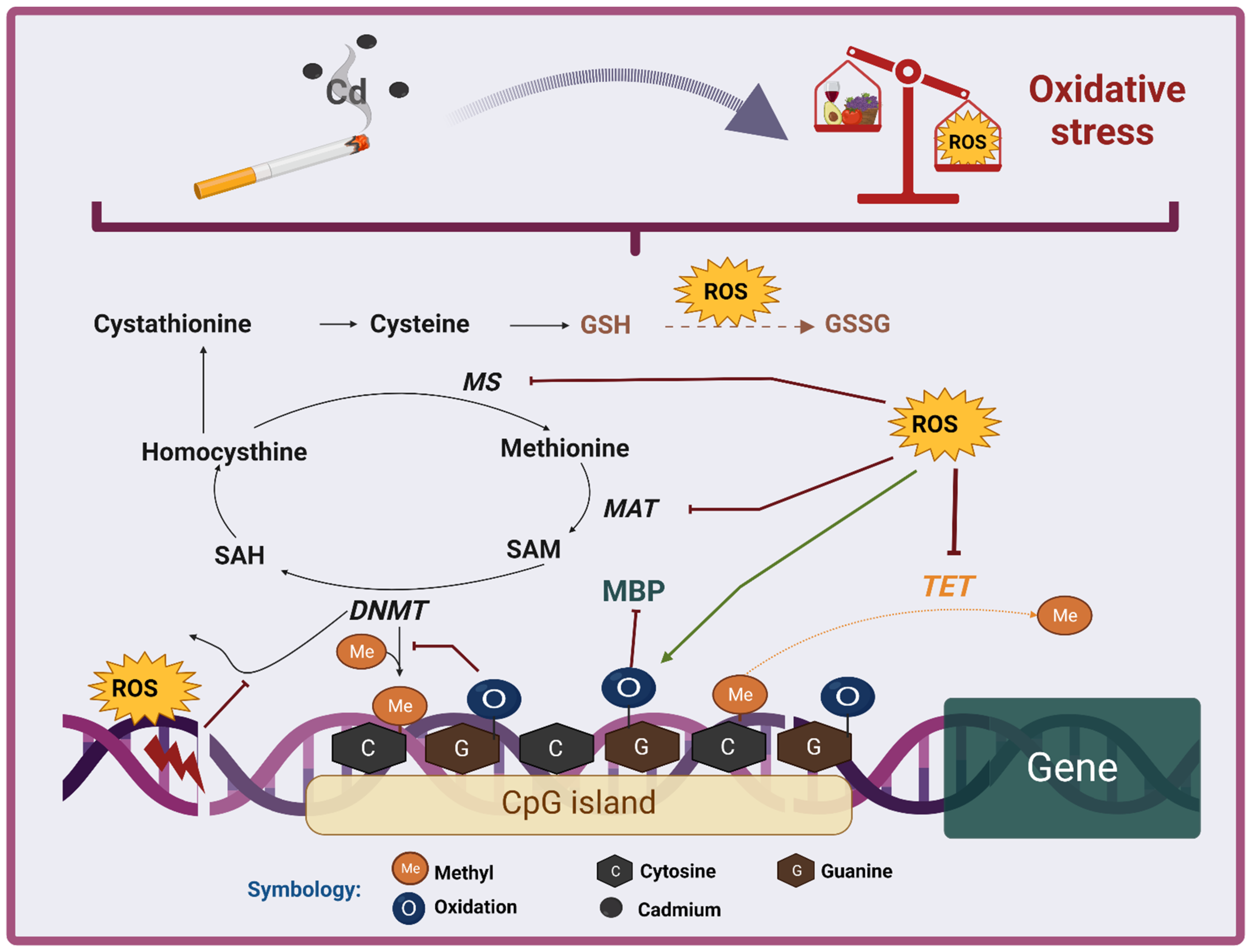
7. Relationship of Epigenetic Modifications with Cd-Induced OS
7.1. DNA Methylation
7.2. Histone Modification
7.3. ncRNA
8. Use of Antioxidants to Mitigate Cd-Induced Epigenetic Alterations
9. Final Comments and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rafati Rahimzadeh, M.; Rafati Rahimzadeh, M.; Kazemi, S.; Moghadamnia, A.-A. Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135–145. [Google Scholar] [CrossRef]
- Gamero Esparza, C. El rastro del Cadmio. Vivat Acad. 2003, 33, 10–28. [Google Scholar] [CrossRef]
- Chunhabundit, R. Cadmium Exposure and Potential Health Risk from Foods in Contaminated Area, Thailand. Toxicol. Res. 2016, 32, 65–72. [Google Scholar] [CrossRef]
- Kim, K.; Melough, M.; Vance, T.; Noh, H.; Koo, S.; Chun, O. Dietary Cadmium Intake and Sources in the US. Nutrients 2018, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Zheng, N.; Tang, L.; Ji, X.; Li, Y.; Hua, X. Pollution characteristics, sources, and health risk assessment of human exposure to Cu, Zn, Cd and Pb pollution in urban street dust across China between 2009 and 2018. Environ. Int. 2019, 128, 430–437. [Google Scholar] [CrossRef]
- Zhang, T.; Ruan, J.; Zhang, B.; Lu, S.; Gao, C.; Huang, L.; Bai, X.; Xie, L.; Gui, M.; Qiu, R. Heavy metals in human urine, foods and drinking water from an e-waste dismantling area: Identification of exposure sources and metal-induced health risk. Ecotoxicol. Environ. Saf. 2019, 169, 707–713. [Google Scholar] [CrossRef]
- Prokopowicz, A.; Sobczak, A.; Szuła-Chraplewska, M.; Ochota, P.; Kośmider, L. Exposure to Cadmium and Lead in Cigarette Smokers Who Switched to Electronic Cigarettes. Nicotine Tob. Res. 2019, 21, 1198–1205. [Google Scholar] [CrossRef]
- Dinh, Q.P.; Novirsa, R.; Jeong, H.; Nugraha, W.C.; Addai-Arhin, S.; Viet, P.H.; Tominaga, N.; Ishibashi, Y.; Arizono, K. Mercury, cadmium, and lead in cigarettes from international markets: Concentrations, distributions and absorption ability of filters. J. Toxicol. Sci. 2021, 46, 401–411. [Google Scholar] [CrossRef]
- Schoeters, G.; Den Hond, E.; Zuurbier, M.; Naginiene, R.; Van Den Hazel, P.; Stilianakis, N.; Ronchetti, R.; Koppe, J. Cadmium and children: Exposure and health effects. Acta Paediatr. 2006, 95, 50–54. [Google Scholar] [CrossRef]
- Fatima, G.; Raza, A.M.; Hadi, N.; Nigam, N.; Mahdi, A.A. Cadmium in Human Diseases: It’s More than Just a Mere Metal. Indian J. Clin. Biochem. 2019, 34, 371–378. [Google Scholar] [CrossRef]
- Grioni, S.; Agnoli, C.; Krogh, V.; Pala, V.; Rinaldi, S.; Vinceti, M.; Contiero, P.; Vescovi, L.; Malavolti, M.; Sieri, S. Dietary cadmium and risk of breast cancer subtypes defined by hormone receptor status: A prospective cohort study. Int. J. Cancer 2019, 144, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. Cadmium and Cancer. In Cadmium: From Toxicity to Essentiality. Metal Ions in Life Sciences; Springer: Berlin/Heidelberg, Germany, 2013; Volume 11, pp. 491–507. [Google Scholar]
- IARC. Cadmium and Cadmium Compounds. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; Arsenic, Metals, Fibres, and Dusts: Lyon, France, 2012. [Google Scholar]
- Nordberg, M.; Nordberg, G.F. Metallothionein and Cadmium Toxicology—Historical Review and Commentary. Biomolecules 2022, 12, 360. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, A. Toxicología del cadmio. Conceptos actuales para evaluar exposición ambiental u ocupacional con indicadores biológicos. An. Fac. Med. 2013, 63, 51. [Google Scholar] [CrossRef] [Green Version]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. BioMetals 2010, 23, 927–940. [Google Scholar] [CrossRef]
- Yan, L.-J.; Allen, D.C. Cadmium-Induced Kidney Injury: Oxidative Damage as a Unifying Mechanism. Biomolecules 2021, 11, 1575. [Google Scholar] [CrossRef]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Takiguchi, M.; Achanzar, W.E.; Qu, W.; Li, G.; Waalkes, M.P. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp. Cell Res. 2003, 286, 355–365. [Google Scholar] [CrossRef]
- Somji, S.; Garrett, S.H.; Toni, C.; Zhou, X.; Zheng, Y.; Ajjimaporn, A.; Sens, M.; Sens, D.A. Differences in the epigenetic regulation of MT-3 gene expression between parental and Cd+2 or As+3 transformed human urothelial cells. Cancer Cell Int. 2011, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Mortoglou, M.; Buha Djordjevic, A.; Djordjevic, V.; Collins, H.; York, L.; Mani, K.; Valle, E.; Wallace, D.; Uysal-Onganer, P. Role of microRNAs in response to cadmium chloride in pancreatic ductal adenocarcinoma. Arch. Toxicol. 2022, 96, 467–485. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Ren, L.; Yan, J.; Bai, Z.; Zhang, L.; Zhang, H.; Xie, Y.; Li, X. Cadmium causes hepatopathy by changing the status of DNA methylation in the metabolic pathway. Toxicol. Lett. 2021, 340, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Benbrahim-Tallaa, L.; Waterland, R.A.; Dill, A.L.; Webber, M.M.; Waalkes, M.P. Tumor Suppressor Gene Inactivation during Cadmium-Induced Malignant Transformation of Human Prostate Cells Correlates with Overexpression of de Novo DNA Methyltransferase. Environ. Health Perspect. 2007, 115, 1454–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Tariba Lovaković, B.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M.; et al. Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 2020, 9, 901. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015; ISBN 9780198717478. [Google Scholar]
- Ho, R.Y.N.; Liebman, J.F.; Valentine, J.S. Overview of the Energetics and Reactivity of Oxygen. In Active Oxygen in Chemistry; Springer: Dordrecht, The Netherlands, 1995; pp. 1–23. [Google Scholar]
- Di Meo, S.; Venditti, P. Evolution of the Knowledge of Free Radicals and Other Oxidants. Oxid. Med. Cell. Longev. 2020, 2020, 9829176. [Google Scholar] [CrossRef]
- Fridovich, I. Oxygen: How Do We Stand It? Med. Princ. Pract. 2013, 22, 131–137. [Google Scholar] [CrossRef]
- Bayr, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Tataranno, M.L. Oxygen toxicity: Chemistry and biology of reactive oxygen species. Semin. Fetal Neonatal Med. 2010, 15, 186–190. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Park, A.-M.; Kira, Y.; Imada, I.; Utsumi, K. Mitochondrial Generation of Reactive Oxygen Species and its Role in Aerobic Life. Curr. Med. Chem. 2003, 10, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1This review is based on the licentiate thesis “Thioredoxin reductase—Interactions with the redox active compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Havránková, R. Biological effects of ionizing radiation. Cas. Lek. Cesk. 2020, 159, 258–260. [Google Scholar]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative Stress, 1st ed.; Sies, H., Ed.; Academic Press: London, UK, 1985; ISBN 9781483289113. [Google Scholar]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D. Oxidative stress. In Encyclopedia of Stress; Fink, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 45–48. [Google Scholar]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.-R.; Rossner, P.; Haghdoost, S.; Jeng, H.A.; Hu, C.-W. Nucleic Acid Oxidation in Human Health and Disease. Oxid. Med. Cell. Longev. 2013, 2013, 368651. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.-M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 2022, 86, 101165. [Google Scholar] [CrossRef]
- Vangaveti, V.; Baune, B.T.; Kennedy, R.L. Review: Hydroxyoctadecadienoic acids: Novel regulators of macrophage differentiation and atherogenesis. Ther. Adv. Endocrinol. Metab. 2010, 1, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, C.E.; Ringel, A.; Feldstein, A.E.; Taha, A.Y.; MacIntosh, B.A.; Hibbeln, J.R.; Majchrzak-Hong, S.F.; Faurot, K.R.; Rapoport, S.I.; Cheon, Y.; et al. Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Shishehbor, M.H.; Zhang, R.; Medina, H.; Brennan, M.-L.; Brennan, D.M.; Ellis, S.G.; Topol, E.J.; Hazen, S.L. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 2006, 41, 1678–1683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Alhasani, R.H.; Zhou, X.; Reilly, J.; Zeng, Z.; Strang, N.; Shu, X. Oxysterols and retinal degeneration. Br. J. Pharmacol. 2021, 178, 3205–3219. [Google Scholar] [CrossRef]
- Samadi, A.; Sabuncuoglu, S.; Samadi, M.; Isikhan, S.Y.; Chirumbolo, S.; Peana, M.; Lay, I.; Yalcinkaya, A.; Bjørklund, G. A Comprehensive Review on Oxysterols and Related Diseases. Curr. Med. Chem. 2020, 28, 110–136. [Google Scholar] [CrossRef]
- Fuentes-Lemus, E.; Hägglund, P.; López-Alarcón, C.; Davies, M.J. Oxidative Crosslinking of Peptides and Proteins: Mechanisms of Formation, Detection, Characterization and Quantification. Molecules 2021, 27, 15. [Google Scholar] [CrossRef]
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [Green Version]
- Christensen, L.P.; Christensen, K.B. The Role of Direct and Indirect Polyphenolic Antioxidants in Protection Against Oxidative Stress. In Polyphenols in Human Health and Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 289–309. [Google Scholar]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Wadhwani, A. Antioxidant Enzymes and Human Health. In Antioxidant Enzyme; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Flohé, L.; Brigelius-Flohé, R. Selenoproteins of the Glutathione Peroxidase Family. In Selenium; Springer: New York, NY, USA, 2011; pp. 167–180. [Google Scholar]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta—Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas-Rodríguez, N.; Pedraza-Chaverri, J. Especies reactivas de oxígeno y sistemas antioxidantes: Aspectos básicos. Educ. Química 2006, 17, 164–173. [Google Scholar]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Johnson, R.J. Uric Acid: The Oxidant-Antioxidant Paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Mayo, J.C.; Tan, D.-X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Moussa, Z.; Judeh, Z.M.A.; Ahmed, S.A. Nonenzymatic Exogenous and Endogenous Antioxidants. In Free Radical Medicine and Biology; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar]
- Bouayed, J.; Bohn, T. Exogenous Antioxidants—Double-Edged Swords in Cellular Redox State: Health Beneficial Effects at Physiologic Doses versus Deleterious Effects at High Doses. Oxid. Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef]
- Neha, K.; Haider, M.R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.S.; Bao, B. Molecular Mechanisms of Zinc as a Pro-Antioxidant Mediator: Clinical Therapeutic Implications. Antioxidants 2019, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxid. Med. Cell. Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [Green Version]
- Morais, J.B.S.; Severo, J.S.; dos Santos, L.R.; de Sousa Melo, S.R.; de Oliveira Santos, R.; de Oliveira, A.R.S.; Cruz, K.J.C.; do Nascimento Marreiro, D. Role of Magnesium in Oxidative Stress in Individuals with Obesity. Biol. Trace Elem. Res. 2017, 176, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, W.; Beyersmann, D. Cadmium-induced apoptosis in C6 glioma cells: Influence of oxidative stress. Biometals 2004, 17, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Tang, M.; Kong, L.; Ying, J.; Wu, T.; Xue, Y.; Pu, Y. Liver Toxicity of Cadmium Telluride Quantum Dots (CdTe QDs) Due to Oxidative Stress In Vitro and In Vivo. Int. J. Mol. Sci. 2015, 16, 23279–23299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, C.; Marcondes, M. Cadmium chloride-induced oxidative stress in skeletal muscle cells in vitro. Free Radic. Biol. Med. 2005, 39, 1378–1384. [Google Scholar] [CrossRef]
- Ashour, T.H.; El-Shemi, A.G. Caffeic acid phenyl ester prevents cadmium intoxication induced disturbances in erythrocyte indices and blood coagulability, hepatorenal dysfunction and oxidative stress in rats. Acta Haematol. Pol. 2014, 45, 272–278. [Google Scholar] [CrossRef]
- El-Boshy, M.E.; Risha, E.F.; Abdelhamid, F.M.; Mubarak, M.S.; Hadda, T. Ben Protective effects of selenium against cadmium induced hematological disturbances, immunosuppressive, oxidative stress and hepatorenal damage in rats. J. Trace Elem. Med. Biol. 2015, 29, 104–110. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Bitto, A.; Pallio, G.; Mannino, F.; Arcoraci, V.; Aliquò, F.; Minutoli, L.; De Ponte, C.; D’andrea, P.; et al. Cadmium-Induced Oxidative Stress Impairs Glycemic Control in Adolescents. Oxid. Med. Cell. Longev. 2017, 2017, 6341671. [Google Scholar] [CrossRef]
- Hormozi, M.; Mirzaei, R.; Nakhaee, A.; Payandeh, A.; Izadi, S.; Haghighi, J.D. Effects of coenzyme Q10 supplementation on oxidative stress and antioxidant enzyme activity in glazers with occupational cadmium exposure: A randomized, double-blind, placebo-controlled crossover clinical trial. Toxicol. Ind. Health 2019, 35, 32–42. [Google Scholar] [CrossRef]
- Alkharashi, N.A.O.; Periasamy, V.S.; Athinarayanan, J.; Alshatwi, A.A. Cadmium triggers mitochondrial oxidative stress in human peripheral blood lymphocytes and monocytes: Analysis using in vitro and system toxicology approaches. J. Trace Elem. Med. Biol. 2017, 42, 117–128. [Google Scholar] [CrossRef]
- Nuran Ercal, B.S.P.; Hande Gurer-Orhan, B.S.P.; Nukhet Aykin-Burns, B.S.P. Toxic Metals and Oxidative Stress Part I: Mechanisms Involved in Me-tal induced Oxidative Damage. Curr. Top. Med. Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef]
- Kurochkin, I.O.; Etzkorn, M.; Buchwalter, D.; Leamy, L.; Sokolova, I.M. Top-down control analysis of the cadmium effects on molluscan mitochondria and the mechanisms of cadmium-induced mitochondrial dysfunction. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R21–R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fang, J.; Leonard, S.S.; Krishna Rao, K.M. Cadmium inhibits the electron transfer chain and induces Reactive Oxygen Species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro Júnior, J.E.G.; Moraes, P.Z.; Rodriguez, M.D.; Simões, M.R.; Cibin, F.; Pinton, S.; Barbosa Junior, F.; Peçanha, F.M.; Vassallo, D.V.; Miguel, M.; et al. Cadmium exposure activates NADPH oxidase, renin–angiotensin system and cyclooxygenase 2 pathways in arteries, inducing hypertension and vascular damage. Toxicol. Lett. 2020, 333, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Dorta, D.J.; Leite, S.; DeMarco, K.C.; Prado, I.M.R.; Rodrigues, T.; Mingatto, F.E.; Uyemura, S.A.; Santos, A.C.; Curti, C. A proposed sequence of events for cadmium-induced mitochondrial impairment. J. Inorg. Biochem. 2003, 97, 251–257. [Google Scholar] [CrossRef]
- López, E.; Arce, C.; Oset-Gasque, M.J.; Cañadas, S.; González, M.P. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic. Biol. Med. 2006, 40, 940–951. [Google Scholar] [CrossRef]
- Casalino, E.; Calzaretti, G.; Sblano, C.; Landriscina, C. Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 2002, 179, 37–50. [Google Scholar] [CrossRef]
- Wroñska-Nofer, T.; Wisniewska-Knypl, J.; Dziubaltowska, E.; Wyszyñska, K. Prooxidative and genotoxic effect of transition metals (cadmium, nickel, chromium, and vanadium) in mice. Trace Elem. Electrolytes 1999, 16, 87–92. [Google Scholar]
- Jihen, E.H.; Imed, M.; Fatima, H.; Abdelhamid, K. Protective effects of selenium (Se) and zinc (Zn) on cadmium (Cd) toxicity in the liver of the rat: Effects on the oxidative stress. Ecotoxicol. Environ. Saf. 2009, 72, 1559–1564. [Google Scholar] [CrossRef]
- Newairy, A.A.; El-Sharaky, A.S.; Badreldeen, M.M.; Eweda, S.M.; Sheweita, S.A. The hepatoprotective effects of selenium against cadmium toxicity in rats. Toxicology 2007, 242, 23–30. [Google Scholar] [CrossRef]
- Ognjanović, B.; Marković, S.; Pavlović, S.; Žikić, R.; Štajn, A.; Saičić, Z. Effect of chronic cadmium exposure on antioxidant defense system in some tissues of rats: Protective effect of selenium. Physiol. Res. 2008, 57, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Albasher, G.; Albrahin, T.; Aljarba, N.; Alharbi, R.I.; Alsultan, N.; Alsairi, J.; Rizwana, H. Involvement of redox status and the nuclear-related factor 2 in protecting against cadmium-induced renal injury with Sana Makki (Cassia senna L.) pre-treatment in male rats. An. Acad. Bras. Cienc. 2020, 92, e20191237. [Google Scholar] [CrossRef] [PubMed]
- Almeer, R.S.; AlBasher, G.I.; Alarifi, S.; Alkahtani, S.; Ali, D.; Abdel Moneim, A.E. Royal jelly attenuates cadmium-induced nephrotoxicity in male mice. Sci. Rep. 2019, 9, 5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Zhang, C.; Sun, Y.-C.; Zhang, Q.; Lv, M.-W.; Guo, K.; Li, J.-L. Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci. Total Environ. 2019, 689, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, C.H.; Subastri, A.; Suyavaran, A.; Subbaiah, K.C.V.; Valluru, L.; Thirunavukkarasu, C. Solanum torvum Swartz. fruit attenuates cadmium-induced liver and kidney damage through modulation of oxidative stress and glycosylation. Environ. Sci. Pollut. Res. 2016, 23, 7919–7929. [Google Scholar] [CrossRef]
- Xu, P.; Mo, Z.; Wu, L.; Chen, W.; He, S.; Chen, Y.; Xu, D.; Xiang, J.; Chen, Z.; Lou, X.; et al. Elevated cadmium and 8-hydroxy-2’-deoxyguanosine (8-OHdG) levels in residents living near electroplating industries. Environ. Sci. Pollut. Res. 2021, 28, 34427–34435. [Google Scholar] [CrossRef]
- Xu, X.; Liao, W.; Lin, Y.; Dai, Y.; Shi, Z.; Huo, X. Blood concentrations of lead, cadmium, mercury and their association with biomarkers of DNA oxidative damage in preschool children living in an e-waste recycling area. Environ. Geochem. Health 2018, 40, 1481–1494. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Luo, K.; Liu, Y.; Zhou, M.; Yan, S.; Shi, H.; Cai, Y. The protective effects of selenium on cadmium-induced oxidative stress and apoptosis via mitochondria pathway in mice kidney. Food Chem. Toxicol. 2013, 58, 61–67. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Zhu, J.Y.; Chan, K.M. Effects of cadmium on cell proliferation, apoptosis, and proto-oncogene expression in zebrafish liver cells. Aquat. Toxicol. 2014, 157, 196–206. [Google Scholar] [CrossRef]
- Matović, V.; Buha, A.; Ðukić-Ćosić, D.; Bulat, Z. Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem. Toxicol. 2015, 78, 130–140. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.; Ha, H.C. Environmental epigenetics in metal exposure. Epigenetics 2011, 6, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Corrales, F.J.; Ruiz, F.; Sánchez-Góngora, E.; Mingorance, J.; Carretero, M.V.; Mato, J.M. Specific interaction of methionine adenosyltransferase with free radicals. BioFactors 1998, 8, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Hoover, D.M.; Ludwig, M.L.; Matthews, R.G. The Mechanism of Adenosylmethionine-Dependent Activation of Methionine Synthase: A Rapid Kinetic Analysis of Intermediates in Reductive Methylation of Cob(II)alamin Enzyme. Biochemistry 1998, 37, 12649–12658. [Google Scholar] [CrossRef]
- Pajares, M.A.; Durán, C.; Corrales, F.; Pliego, M.M.; Mato, J.M. Modulation of rat liver S-adenosylmethionine synthetase activity by glutathione. J. Biol. Chem. 1992, 267, 17598–17605. [Google Scholar] [CrossRef]
- Newell-Price, J.; Clark, A.J.L.; King, P. DNA Methylation and Silencing of Gene Expression. Trends Endocrinol. Metab. 2000, 11, 142–148. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Puri, P.; McCann, A.; Bannigan, J.; Thompson, J. Epigenetic Effect of Cadmium on Global De Novo DNA Hypomethylation in the Cadmium-Induced Ventral Body Wall Defect (VBWD) in the Chick Model. Toxicol. Sci. 2011, 120, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Xu, L.; Song, S.; Zhu, C.; Wu, Q.; Zhang, L.; Wu, L. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology 2008, 244, 49–55. [Google Scholar] [CrossRef]
- Hossain, M.B.; Vahter, M.; Concha, G.; Broberg, K. Low-Level Environmental Cadmium Exposure Is Associated with DNA Hypomethylation in Argentinean Women. Environ. Health Perspect. 2012, 120, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Yagi, T.; Sawayama, H.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Baba, H. Long Interspersed Element-1 Methylation Level as a Prognostic Biomarker in Gastrointestinal Cancers. Digestion 2018, 97, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Yin, H.; Zhang, B.; Zhu, W. Effect of cadmium on TET enzymes and DNA methylation changes in human embryonic kidney cell. Zhonghua Yu Fang Yi Xue Za Zhi 2015, 49, 822–827. [Google Scholar] [PubMed]
- Sanders, A.; Smeester, L.; Rojas, D.; DeBussycher, T.; Wu, M.; Wright, F.; Zhou, Y.-H.; Laine, J.; Rager, J.; Swamy, G.; et al. Cadmium exposure and the epigenome: Exposure-associated patterns of DNA methylation in leukocytes from mother-baby pairs. Epigenetics 2014, 9, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.J.; Yu, S.Y.; Kim, S.H.; Lee, J.S.; Hwang, S.Y. Prenatal Exposure to Heavy Metals Affects Gestational Age by Altering DNA Methylation Patterns. Nanomaterials 2021, 11, 2871. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lei, Y.; Wang, C. Analysis of Aberrant Methylation in DNA Repair Genes During Malignant Transformation of Human Bronchial Epithelial Cells Induced by Cadmium. Toxicol. Sci. 2012, 125, 412–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Ye, S.; Pan, Y.; Bao, Y.; Chen, H.; Shao, C. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutat. Res. Toxicol. Environ. Mutagen. 2013, 757, 125–131. [Google Scholar] [CrossRef]
- Vilahur, N.; Vahter, M.; Broberg, K. The Epigenetic Effects of Prenatal Cadmium Exposure. Curr. Environ. Health Rep. 2015, 2, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Cribiu, P.; Chaumot, A.; Geffard, O.; Ravanat, J.-L.; Bastide, T.; Delorme, N.; Quéau, H.; Caillat, S.; Devaux, A.; Bony, S. Natural variability and modulation by environmental stressors of global genomic cytosine methylation levels in a freshwater crustacean, Gammarus fossarum. Aquat. Toxicol. 2018, 205, 11–18. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y.; Shao, C.; Tan, Y.; Cai, L. Cadmium and Its Epigenetic Effects. Curr. Med. Chem. 2012, 19, 2611–2620. [Google Scholar] [CrossRef]
- Huang, D.; Zhang, Y.; Qi, Y.; Chen, C.; Ji, W. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol. Lett. 2008, 179, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Šrut, M.; Drechsel, V.; Höckner, M. Low levels of Cd induce persisting epigenetic modifications and acclimation mechanisms in the earthworm Lumbricus terrestris. PLoS ONE 2017, 12, e0176047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, J.; Zhang, Y.; Wang, X.; Wang, M.; Su, P. Differential Expression Profiles and Potential Intergenerational Functions of tRNA-Derived Small RNAs in Mice After Cadmium Exposure. Front. Cell Dev. Biol. 2022, 9, 791784. [Google Scholar] [CrossRef] [PubMed]
- Ronco, A.M.; Llaguno, E.; Epuñan, M.J.; Llanos, M.N. Effect of cadmium on cortisol production and 11β-hydroxysteroid dehydrogenase 2 expression by cultured human choriocarcinoma cells (JEG-3). Toxicol. Vitr. 2010, 24, 1532–1537. [Google Scholar] [CrossRef]
- Gliga, A.R.; Malin Igra, A.; Hellberg, A.; Engström, K.; Raqib, R.; Rahman, A.; Vahter, M.; Kippler, M.; Broberg, K. Maternal exposure to cadmium during pregnancy is associated with changes in DNA methylation that are persistent at 9 years of age. Environ. Int. 2022, 163, 107188. [Google Scholar] [CrossRef]
- Vidal, A.C.; Semenova, V.; Darrah, T.; Vengosh, A.; Huang, Z.; King, K.; Nye, M.D.; Fry, R.; Skaar, D.; Maguire, R.; et al. Maternal cadmium, iron and zinc levels, DNA methylation and birth weight. BMC Pharmacol. Toxicol. 2015, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Everson, T.M.; Armstrong, D.A.; Jackson, B.P.; Green, B.B.; Karagas, M.R.; Marsit, C.J. Maternal cadmium, placental PCDHAC1, and fetal development. Reprod. Toxicol. 2016, 65, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, A.F.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Afsharinejad, Z.; Burbacher, T.M.; Siscovick, D.S.; Williams, M.A.; Enquobahrie, D.A. Infant sex-specific placental cadmium and DNA methylation associations. Environ. Res. 2015, 138, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Hasani Nourian, Y.; Beh-Pajooh, A.; Aliomrani, M.; Amini, M.; Sahraian, M.A.; Hosseini, R.; Mohammadi, S.; Ghahremani, M.H. Changes in DNA methylation in APOE and ACKR3 genes in multiple sclerosis patients and the relationship with their heavy metal blood levels. Neurotoxicology 2021, 87, 182–187. [Google Scholar] [CrossRef]
- Smith, M.M. Histone structure and function. Curr. Opin. Cell Biol. 1991, 3, 429–437. [Google Scholar] [CrossRef]
- Biterge, B.; Schneider, R. Histone variants: Key players of chromatin. Cell Tissue Res. 2014, 356, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.; Ponte, I.; Suau, P. Interplay between histone H1 structure and function. Biochim. Biophys. Acta—Gene Regul. Mech. 2016, 1859, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Inducible gene expression: Diverse regulatory mechanisms. Nat. Rev. Genet. 2010, 11, 426–437. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, C. Histone modifying enzymes: Novel disease biomarkers and assay development. Expert Rev. Mol. Diagn. 2016, 16, 297–306. [Google Scholar] [CrossRef]
- Gräff, J.; Tsai, L.-H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Gujral, P.; Mahajan, V.; Lissaman, A.C.; Ponnampalam, A.P. Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reprod. Biol. Endocrinol. 2020, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhao, J.; Li, D.; Chen, Y.; Xiao, Y.; Fan, A.; Chen, X.-T.; Wang, H.-L. Combined exposure of lead and cadmium leads to the aggravated neurotoxicity through regulating the expression of histone deacetylase 2. Chemosphere 2020, 252, 126589. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, Z.; Yang, F.; Zhu, M.; Cao, J.; Chen, J.; Lin, Y.; Guo, S.; Li, J.; Liu, Z. Cadmium disturbs epigenetic modification and induces DNA damage in mouse preimplantation embryos. Ecotoxicol. Environ. Saf. 2021, 219, 112306. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Murray, K. The Occurrence of iε-N-Methyl Lysine in Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef]
- Byvoet, P.; Shepherd, G.R.; Hardin, J.M.; Noland, B.J. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972, 148, 558–567. [Google Scholar] [CrossRef]
- Fischle, W.; Franz, H.; Jacobs, S.A.; Allis, C.D.; Khorasanizadeh, S. Specificity of the Chromodomain Y Chromosome Family of Chromodomains for Lysine-methylated ARK(S/T) Motifs. J. Biol. Chem. 2008, 283, 19626–19635. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Schneider, R.; Kouzarides, T. Histone Methylation. Cell 2002, 109, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Singh, D.; Nishi, K.; Khambata, K.; Balasinor, N.H. Introduction to epigenetics: Basic concepts and advancements in the field. In Epigenetics and Reproductive Health; Elsevier: Amsterdam, The Netherlands, 2020; pp. xxv–xliv. [Google Scholar]
- Barcia-Sanjurjo, I.; Vázquez-Cedeira, M.; Barcia, R.; Lazo, P.A. Sensitivity of the kinase activity of human vaccinia-related kinase proteins to toxic metals. JBIC J. Biol. Inorg. Chem. 2013, 18, 473–482. [Google Scholar] [CrossRef] [PubMed]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Zardo, G.; Ciolfi, A.; Vian, L.; Starnes, L.M.; Billi, M.; Racanicchi, S.; Maresca, C.; Fazi, F.; Travaglini, L.; Noguera, N.; et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 2012, 119, 4034–4046. [Google Scholar] [CrossRef] [Green Version]
- Miguel, V.; Lamas, S.; Espinosa-Diez, C. Role of non-coding-RNAs in response to environmental stressors and consequences on human health. Redox Biol. 2020, 37, 101580. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.T.; Olson, E.N. MicroRNAs in Stress Signaling and Human Disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Yosim, A.; Fry, R.C. Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead, and mercury: Strategies and challenges. Front. Genet. 2014, 5, 201. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.; Li, H.; Goel, A. Piwi-interacting RNAs (piRNAs) and cancer: Emerging biological concepts and potential clinical implications. Biochim. Biophys. Acta—Rev. Cancer 2019, 1871, 160–169. [Google Scholar] [CrossRef]
- Pérez-Alvarado, J.; Moreno-Ortiz, J.M. piRNAs, un nuevo campo de biomarcadores en cáncer. Rev. BIOMÉDICA 2017, 28, 117–122. [Google Scholar] [CrossRef]
- Sarkar, A.; Maji, R.K.; Saha, S.; Ghosh, Z. piRNAQuest: Searching the piRNAome for silencers. BMC Genom. 2014, 15, 555. [Google Scholar] [CrossRef] [Green Version]
- Ketting, R.F.; Fischer, S.E.J.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H.A. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.; Rager, J.E. Epigenetics: An overview of CpG methylation, chromatin remodeling, and regulatory/noncoding RNAs. In Environmental Epigenetics in Toxicology and Public Health; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3–32. [Google Scholar]
- Dorsett, Y.; Tuschl, T. siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef]
- Li, S.; Wang, H.; Qi, Y.; Tu, J.; Bai, Y.; Tian, T.; Huang, N.; Wang, Y.; Xiong, F.; Lu, Z.; et al. Assessment of nanomaterial cytotoxicity with SOLiD sequencing-based microRNA expression profiling. Biomaterials 2011, 32, 9021–9030. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Luo, L.; Li, Y.; Liu, J.; Zhang, W. Effect of cadmium on kitl pre-mRNA alternative splicing in murine ovarian granulosa cells and its associated regulation by miRNAs. J. Appl. Toxicol. 2018, 38, 227–239. [Google Scholar] [CrossRef]
- Brooks, S.A.; Martin, E.; Smeester, L.; Grace, M.R.; Boggess, K.; Fry, R.C. miRNAs as common regulators of the transforming growth factor (TGF)-β pathway in the preeclamptic placenta and cadmium-treated trophoblasts: Links between the environment, the epigenome and preeclampsia. Food Chem. Toxicol. 2016, 98, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Satarug, S. Dietary Cadmium Intake and Its Effects on Kidneys. Toxics 2018, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Fay, M.; Alt, L.; Ryba, D.; Salamah, R.; Peach, R.; Papaeliou, A.; Zawadzka, S.; Weiss, A.; Patel, N.; Rahman, A.; et al. Cadmium Nephrotoxicity Is Associated with Altered MicroRNA Expression in the Rat Renal Cortex. Toxics 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, K.L.; Gerlach, C.V.; Craciun, F.L.; Ramachandran, K.; Bijol, V.; Kissick, H.T.; Vaidya, V.S. Application of small RNA sequencing to identify microRNAs in acute kidney injury and fibrosis. Toxicol. Appl. Pharmacol. 2016, 312, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Ngalame, N.N.O.; Waalkes, M.P.; Tokar, E.J. Silencing KRAS Overexpression in Cadmium-Transformed Prostate Epithelial Cells Mitigates Malignant Phenotype. Chem. Res. Toxicol. 2016, 29, 1458–1467. [Google Scholar] [CrossRef] [Green Version]
- Urani, C.; Melchioretto, P.; Fabbri, M.; Bowe, G.; Maserati, E.; Gribaldo, L. Cadmium Impairs p53 Activity in HepG2 Cells. ISRN Toxicol. 2014, 2014, 976428. [Google Scholar] [CrossRef] [Green Version]
- Batinac, T.; Gruber, F.; Lipozencić, J.; Zamolo-Koncar, G.; Stasić, A.; Brajac, I. Protein p53--structure, function, and possible therapeutic implications. Acta Dermatovenerol. Croat. 2003, 11, 225–230. [Google Scholar]
- Liu, Q.; Zheng, C.; Shen, H.; Zhou, Z.; Lei, Y. MicroRNAs-mRNAs Expression Profile and Their Potential Role in Malignant Transformation of Human Bronchial Epithelial Cells Induced by Cadmium. Biomed Res. Int. 2015, 2015, 902025. [Google Scholar] [CrossRef]
- Moawad, A.M.; Hassan, F.M.; Sabry Abdelfattah, D.; Basyoni, H.A.M. Long non-coding RNA ENST00000414355 as a biomarker of cadmium exposure regulates DNA damage and apoptosis. Toxicol. Ind. Health 2021, 37, 745–751. [Google Scholar] [CrossRef]
- Lin, H.-P.; Rea, M.; Wang, Z.; Yang, C. Down-regulation of lncRNA MEG3 promotes chronic low dose cadmium exposure-induced cell transformation and cancer stem cell-like property. Toxicol. Appl. Pharmacol. 2021, 430, 115724. [Google Scholar] [CrossRef]
- McCall, J.L.; Varney, M.E.; Rice, E.; Dziadowicz, S.A.; Hall, C.; Blethen, K.E.; Hu, G.; Barnett, J.B.; Martinez, I. Prenatal Cadmium Exposure Alters Proliferation in Mouse CD4+ T Cells via LncRNA Snhg7. Front. Immunol. 2022, 12, 720635. [Google Scholar] [CrossRef]
- Valinluck, V. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Hou, N.; Xiao, L.; Xu, Y.; Xu, H.; Li, F. Dynamic monitoring of oxidative DNA double-strand break and repair in cardiomyocytes. Cardiovasc. Pathol. 2016, 25, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkena, K.V.; Young, C.Y.F.; Tindall, D.J. Oxidative Stress and DNA Methylation in Prostate Cancer. Obstet. Gynecol. Int. 2010, 2010, 302051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyr, A.R.; Domann, F.E. The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Wachsman, J.T. DNA methylation and the association between genetic and epigenetic changes: Relation to carcinogenesis. Mutat. Res. Mol. Mech. Mutagen. 1997, 375, 1–8. [Google Scholar] [CrossRef]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [CrossRef]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Formation of 8-hydroxyguanine moiety in deoxyribonucleic acid on.gamma.-irradiation in aqueous solution. Biochemistry 1985, 24, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Turk, P.W.; Laayoun, A.; Smith, S.S.; Weitzman, S.A. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 1995, 16, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Kitkumthorn, N.; Mutirangura, A.; Chongsrisawat, V.; Poovorawan, Y.; Honsawek, S. Global methylation, oxidative stress and relative telomere length in biliary atresia patients. Sci. Rep. 2016, 6, 26969. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar] [CrossRef] [Green Version]
- Chia, N.; Wang, L.; Lu, X.; Senut, M.-C.; Brenner, C.A.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Yin, L.; Dong, F.; Zheng, M.; Zhao, Y.; Fu, S.; Zhang, W.; Chen, X. Effects of long-term cadmium exposure on growth, antioxidant defense and DNA methylation in juvenile Nile tilapia (Oreochromis niloticus). Aquat. Toxicol. 2021, 241, 106014. [Google Scholar] [CrossRef]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Qu, W.; Ke, H.; Pi, J.; Broderick, D.; French, J.E.; Webber, M.M.; Waalkes, M.P. Acquisition of Apoptotic Resistance in Cadmium-Transformed Human Prostate Epithelial Cells: Bcl-2 Overexpression Blocks the Activation of JNK Signal Transduction Pathway. Environ. Health Perspect. 2007, 115, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Li, J.; Lv, X.; Yu, L.; Wu, K.; Yang, Y. Metal contamination, bioaccumulation, ROS generation, and epigenotoxicity influences on zebrafish exposed to river water polluted by mining activities. J. Hazard. Mater. 2021, 405, 124150. [Google Scholar] [CrossRef]
- Cerda, S.; Weitzman, S. Influence of oxygen radical injury on DNA methylation. Mutat. Res. Mutat. Res. 1997, 386, 141–152. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Maciejewski, J.P.; Mukherji, M. A mechanistic overview of TET-mediated 5-methylcytosine oxidation. Biochem. Biophys. Res. Commun. 2013, 436, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.M.; Gustafson, C.B.; Young, J.I.; Züchner, S.; Wang, G. Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate. Biochem. Biophys. Res. Commun. 2013, 439, 522–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao-Suzuki, M.; Takeda, S.; Sakai, G.; Waalkes, M.P.; Sugihara, N.; Takiguchi, M. Cadmium-stimulated invasion of rat liver cells during malignant transformation: Evidence of the involvement of oxidative stress/TET1-sensitive machinery. Toxicology 2021, 447, 152631. [Google Scholar] [CrossRef]
- Vrijens, K.; Trippas, A.-J.; Lefebvre, W.; Vanpoucke, C.; Penders, J.; Janssen, B.G.; Nawrot, T.S. Association of Prenatal Exposure to Ambient Air Pollution With Circulating Histone Levels in Maternal Cord Blood. JAMA Netw. Open 2020, 3, e205156. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Romá-Mateo, C.; Pallardó, F.V. Oxidative post-translational modifications in histones. BioFactors 2019, 45, 641–650. [Google Scholar] [CrossRef]
- Agudelo, M.; Gandhi, N.; Saiyed, Z.; Pichili, V.; Thangavel, S.; Khatavkar, P.; Yndart-Arias, A.; Nair, M. Effects of Alcohol on Histone Deacetylase 2 (HDAC2) and the Neuroprotective Role of Trichostatin A (TSA). Alcohol. Clin. Exp. Res. 2011, 35, 1550–1556. [Google Scholar] [CrossRef]
- Hwang, J.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Castonguay, Z.; Auger, C.; Thomas, S.C.; Chahma, M.; Appanna, V.D. Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes. Biochem. Biophys. Res. Commun. 2014, 454, 172–177. [Google Scholar] [CrossRef]
- Hu, S.; Liu, H.; Ha, Y.; Luo, X.; Motamedi, M.; Gupta, M.P.; Ma, J.-X.; Tilton, R.G.; Zhang, W. Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic. Biol. Med. 2015, 79, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.; Fitzpatrick, F.A. Redox Signaling, Alkylation (Carbonylation) of Conserved Cysteines Inactivates Class I Histone Deacetylases 1, 2, and 3 and Antagonizes Their Transcriptional Repressor Function. J. Biol. Chem. 2010, 285, 17417–17424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodie, F.M.; Marwick, J.A.; Anderson, C.S.; Szulakowski, P.; Biswas, S.K.; Bauter, M.R.; Kilty, I.; Rahman, I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004, 18, 1897–1899. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Smathers, R.L.; Roede, J.R.; Shearn, C.T.; Reigan, P.; Petersen, D.R. 4-Hydroxynonenal Inhibits SIRT3 via Thiol-Specific Modification. Chem. Res. Toxicol. 2011, 24, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żabka, A.; Winnicki, K.; Polit, J.T.; Wróblewski, M.; Maszewski, J. Cadmium (II)-Induced Oxidative Stress Results in Replication Stress and Epigenetic Modifications in Root Meristem Cell Nuclei of Vicia faba. Cells 2021, 10, 640. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Flashman, E.; Mecinović, J.; Kramer, H.B.; Kessler, B.M.; Frapart, Y.M.; Boucher, J.-L.; Clifton, I.J.; McDonough, M.A.; Schofield, C.J. Studies on the Reaction of Nitric Oxide with the Hypoxia-Inducible Factor Prolyl Hydroxylase Domain 2 (EGLN1). J. Mol. Biol. 2011, 410, 268–279. [Google Scholar] [CrossRef]
- Hickok, J.R.; Vasudevan, D.; Antholine, W.E.; Thomas, D.D. Nitric Oxide Modifies Global Histone Methylation by Inhibiting Jumonji C Domain-containing Demethylases. J. Biol. Chem. 2013, 288, 16004–16015. [Google Scholar] [CrossRef] [Green Version]
- Hitchler, M.J.; Domann, F.E. Redox regulation of the epigenetic landscape in Cancer: A role for metabolic reprogramming in remodeling the epigenome. Free Radic. Biol. Med. 2012, 53, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Wang, B.; Liu, Q.; Nenoi, M.; Onoda, M. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014, 156, 85–95. [Google Scholar] [CrossRef]
- Rusnak, F.; Reiter, T. Sensing electrons: Protein phosphatase redox regulation. Trends Biochem. Sci. 2000, 25, 527–529. [Google Scholar] [CrossRef]
- KKhan, M.A.; Dixit, K.; Moinuddin; Arif, Z.; Alam, K. Studies on peroxynitrite-modified H1 histone: Implications in systemic lupus erythematosus. Biochimie 2014, 97, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Dixit, K.; Khan, M.A.; Moinuddin; Arif, Z.; Alam, K. Physicochemical studies on peroxynitrite-modified H3 histone. Int. J. Biol. Macromol. 2010, 46, 20–26. [Google Scholar] [CrossRef]
- Alvarez-Olmedo, D.G.; Biaggio, V.S.; Koumbadinga, G.A.; Gómez, N.N.; Shi, C.; Ciocca, D.R.; Batulan, Z.; Fanelli, M.A.; O’Brien, E.R. Recombinant heat shock protein 27 (HSP27/HSPB1) protects against cadmium-induced oxidative stress and toxicity in human cervical cancer cells. Cell Stress Chaperones 2017, 22, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, J.J.; Rose, K.L.; Beavers, W.N.; Hill, S.; Tallman, K.A.; Tansey, W.P.; Marnett, L.J. Stable Histone Adduction by 4-Oxo-2-nonenal: A Potential Link between Oxidative Stress and Epigenetics. J. Am. Chem. Soc. 2014, 136, 11864–11866. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.; Petroze, R.; Castegna, A.; Ding, Q.; Keller, J.N.; Markesbery, W.R.; Lovell, M.A.; Butterfield, D.A. 4-Hydroxynonenal oxidatively modifies histones: Implications for Alzheimer’s disease. Neurosci. Lett. 2004, 356, 155–158. [Google Scholar] [CrossRef]
- Lan, J.; Huang, Z.; Han, J.; Shao, J.; Huang, C. Redox regulation of microRNAs in cancer. Cancer Lett. 2018, 418, 250–259. [Google Scholar] [CrossRef]
- He, J.; Jiang, B.-H. Interplay Between Reactive Oxygen Species and MicroRNAs in Cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; McKinney, G.J.; Nichols, K.M.; Colbourne, J.K.; Sepúlveda, M.S. Novel Cadmium Responsive MicroRNAs in Daphnia pulex. Environ. Sci. Technol. 2015, 49, 14605–14613. [Google Scholar] [CrossRef]
- Barr, I.; Smith, A.T.; Chen, Y.; Senturia, R.; Burstyn, J.N.; Guo, F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc. Natl. Acad. Sci. USA 2012, 109, 1919–1924. [Google Scholar] [CrossRef] [Green Version]
- Jaksik, R.; Lalik, A.; Skonieczna, M.; Cieslar-Pobuda, A.; Student, S.; Rzeszowska-Wolny, J. MicroRNAs and reactive oxygen species: Are they in the same regulatory circuit? Mutat. Res. Toxicol. Environ. Mutagen. 2014, 764–765, 64–71. [Google Scholar] [CrossRef]
- Menezo, Y.J.R.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, J.; Tao, Y.-F.; He, J.; Xu, P.; Bao, J.-W.; Sun, Y.-L. miR-122 promotes hepatic antioxidant defense of genetically improved farmed tilapia (GIFT, Oreochromis niloticus) exposed to cadmium by directly targeting a metallothionein gene. Aquat. Toxicol. 2017, 182, 39–48. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, X.; Yin, L.; Xu, L.; Xu, Y.; Qi, Y.; Han, X.; Song, S.; Zhao, Y.; Lin, Y.; et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br. J. Pharmacol. 2017, 174, 2512–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Li, C.; Xu, M.; Liu, Y.; Cong, M.; Liu, S. LncRNA MT1DP Aggravates Cadmium-Induced Oxidative Stress by Repressing the Function of Nrf2 and is Dependent on Interaction with miR-365. Adv. Sci. 2018, 5, 1800087. [Google Scholar] [CrossRef]
- Yu, C.; Yang, C.; Song, X.; Li, J.; Peng, H.; Qiu, M.; Yang, L.; Du, H.; Jiang, X.; Liu, Y. Long Non-coding RNA Expression Profile in Broiler Liver with Cadmium-Induced Oxidative Damage. Biol. Trace Elem. Res. 2021, 199, 3053–3061. [Google Scholar] [CrossRef]
- Huang, S. Microarray profiling of long noncoding RNAs and mRNA expression in the kidney of rats damaged by cadmium. Am. J. Biomed. Sci. Res. 2020, 10, 168–180. [Google Scholar] [CrossRef]
- Qu, T.; Mou, Y.; Dai, J.; Zhang, X.; Li, M.; Gu, S.; He, Z. Changes and relationship of N6-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicol. Lett. 2021, 343, 56–66. [Google Scholar] [CrossRef]
- Smith, S.W. The Role of Chelation in the Treatment of Other Metal Poisonings. J. Med. Toxicol. 2013, 9, 355–369. [Google Scholar] [CrossRef] [Green Version]
- van Lith, R.; Ameer, G.A. Antioxidant Polymers as Biomaterial. In Oxidative Stress and Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 251–296. [Google Scholar]
- Zhang, D.; Zhang, T.; Liu, J.; Chen, J.; Li, Y.; Ning, G.; Huo, N.; Tian, W.; Ma, H. Zn Supplement-Antagonized Cadmium-Induced Cytotoxicity in Macrophages In Vitro: Involvement of Cadmium Bioaccumulation and Metallothioneins Regulation. J. Agric. Food Chem. 2019, 67, 4611–4622. [Google Scholar] [CrossRef]
- Amara, S.; Abdelmelek, H.; Garrel, C.; Guiraud, P.; Douki, T.; Ravanat, J.-L.; Favier, A.; Sakly, M.; Ben Rhouma, K. Preventive Effect of Zinc Against Cadmium-induced Oxidative Stress in the Rat Testis. J. Reprod. Dev. 2008, 54, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formigari, A.; Alberton, P.; Cantale, V.; Nadal, V.D.; Feltrin, M.; Ferronato, S.; Santon, A.; Schiavon, L.; Irato, P. Relationship Between Metal Transcription Factor-1 and Zinc in Resistance to Metals Producing Free Radicals. Curr. Chem. Biol. 2008, 2, 256–266. [Google Scholar] [CrossRef]
- Nordberg, M.N.G. Toxicological aspects of metallothionein. Cell. Mol. Biol. 2000, 46, 451–463. [Google Scholar] [PubMed]
- Brzóska, M.M.; Borowska, S.; Tomczyk, M. Antioxidants as a Potential Preventive and Therapeutic Strategy for Cadmium. Curr. Drug Targets 2016, 17, 1350–1384. [Google Scholar] [CrossRef] [PubMed]
- Abib, R.T.; Peres, K.C.; Barbosa, A.M.; Peres, T.V.; Bernardes, A.; Zimmermann, L.M.; Quincozes-Santos, A.; Fiedler, H.D.; Leal, R.B.; Farina, M.; et al. Epigallocatechin-3-gallate protects rat brain mitochondria against cadmium-induced damage. Food Chem. Toxicol. 2011, 49, 2618–2623. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.-Z.; Zhang, Y.-X.; Zhu, R.-M.; Li, Y.-L.; Jiang, H.-M.; Li, R.-B.; Chen, Q.-X.; Wang, Q.; Tang, L.-Y.; Ren, Z.-F. Identification of epigenetic modifications mediating the antagonistic effect of selenium against cadmium-induced breast carcinogenesis. Environ. Sci. Pollut. Res. 2022, 29, 22056–22068. [Google Scholar] [CrossRef]
- Alshammari, G.M.; Al-Qahtani, W.H.; AlFaris, N.A.; Alzahrani, N.S.; Alkhateeb, M.A.; Yahya, M.A. Quercetin prevents cadmium chloride-induced hepatic steatosis and fibrosis by downregulating the transcription of miR-21. BioFactors 2021, 47, 489–505. [Google Scholar] [CrossRef]
- Wang, W.; Liu, G.; Jiang, X.; Wu, G. Resveratrol ameliorates toxic effects of cadmium on placental development in mouse placenta and human trophoblast cells. Birth Defects Res. 2021, 113, 1470–1483. [Google Scholar] [CrossRef]
- Chen, S.; Luo, T.; Yu, Q.; Dong, W.; Zhang, H.; Zou, H. Isoorientin plays an important role in alleviating Cadmium-induced DNA damage and G0/G1 cell cycle arrest. Ecotoxicol. Environ. Saf. 2020, 187, 109851. [Google Scholar] [CrossRef]
- Li, X.; Yao, Z.; Yang, D.; Jiang, X.; Sun, J.; Tian, L.; Hu, J.; Wu, B.; Bai, W. Cyanidin-3-O-glucoside restores spermatogenic dysfunction in cadmium-exposed pubertal mice via histone ubiquitination and mitigating oxidative damage. J. Hazard. Mater. 2020, 387, 121706. [Google Scholar] [CrossRef]
- Kiełczykowska, M.; Kocot, J.; Paździor, M.; Musik, I. Selenium—A fascinating antioxidant of protective properties. Adv. Clin. Exp. Med. 2018, 27, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [Green Version]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Shi, P.; Ge, J.-J. miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc. Disord. 2017, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Shih, W.; Chang, C.; Chen, H.; Fan, K. Antioxidant activity and leukemia initiation prevention in vitro and in vivo by N-acetyl-L-cysteine. Oncol. Lett. 2018, 16, 2046–2052. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Kosmeder, J.W.; Pezzuto, J.M. Biological Effects of Resveratrol. Antioxid. Redox Signal. 2001, 3, 1041–1064. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Cantó, C. The molecular targets of resveratrol. Biochim. Biophys. Acta—Mol. Basis Dis. 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Lançon, A.; Kaminski, J.; Tili, E.; Michaille, J.-J.; Latruffe, N. Control of MicroRNA Expression as a New Way for Resveratrol To Deliver Its Beneficial Effects. J. Agric. Food Chem. 2012, 60, 8783–8789. [Google Scholar] [CrossRef]
- Latruffe, N.; Lançon, A.; Frazzi, R.; Aires, V.; Delmas, D.; Michaille, J.-J.; Djouadi, F.; Bastin, J.; Cherkaoui-Malki, M. Exploring new ways of regulation by resveratrol involving miRNAs, with emphasis on inflammation. Ann. N. Y. Acad. Sci. 2015, 1348, 97–106. [Google Scholar] [CrossRef]
- Britton, R.G.; Kovoor, C.; Brown, K. Direct molecular targets of resveratrol: Identifying key interactions to unlock complex mechanisms. Ann. N. Y. Acad. Sci. 2015, 1348, 124–133. [Google Scholar] [CrossRef]
- Fernandes, G.; Silva, G.; Pavan, A.; Chiba, D.; Chin, C.; Dos Santos, J. Epigenetic Regulatory Mechanisms Induced by Resveratrol. Nutrients 2017, 9, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.-Y.; Wong, Z.C.-F.; Wang, C.; Fung, A.H.-Y.; Wong, E.O.-Y.; Chan, G.K.-L.; Dong, T.T.-X.; Chen, Y.; Tsim, K.W.-K. Isoorientin derived from Gentiana veitchiorum Hemsl. flowers inhibits melanogenesis by down-regulating MITF-induced tyrosinase expression. Phytomedicine 2019, 57, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, J.; Jiang, X.; Sun, J.; Tian, L.; Jiao, R.; Tang, Y.; Bai, W. Cyanidin-3-O-glucoside protects against cadmium-induced dysfunction of sex hormone secretion via the regulation of hypothalamus-pituitary-gonadal axis in male pubertal mice. Food Chem. Toxicol. 2019, 129, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [Green Version]
- Tao, L. Effect of Trichloroethylene and Its Metabolites, Dichloroacetic Acid and Trichloroacetic Acid, on the Methylation and Expression of c-Jun and c-Myc Protooncogenes in Mouse Liver: Prevention by Methionine. Toxicol. Sci. 2000, 54, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Liang, Y.; Dai, Q.; Tian, J.; Chen, J.; Lei, B.; Yang, Z.; Cai, Z. Application of machine learning algorithms to screen potential biomarkers under cadmium exposure based on human urine metabolic profiles. Chin. Chem. Lett. 2022, in press. [Google Scholar] [CrossRef]
- Nzengue, Y.; Steiman, R.; Garrel, C.; Lefèbvre, E.; Guiraud, P. Oxidative stress and DNA damage induced by cadmium in the human keratinocyte HaCaT cell line: Role of glutathione in the resistance to cadmium. Toxicology 2008, 243, 193–206. [Google Scholar] [CrossRef]
- Hirota, K.; Matsuoka, M. N-acetylcysteine restores the cadmium toxicity of Caenorhabditis elegans. BioMetals 2021, 34, 1207–1216. [Google Scholar] [CrossRef]
- Luchese, C.; Zeni, G.; Rocha, J.B.T.; Nogueira, C.W.; Santos, F.W. Cadmium inhibits δ-aminolevulinate dehydratase from rat lung in vitro: Interaction with chelating and antioxidant agents. Chem. Biol. Interact. 2007, 165, 127–137. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Model | Cd Compound | Relevant Effects of the Antioxidant | Appointment |
|---|---|---|---|---|
| Selenium | MCF-7 cells | CdCl2 | Epigenetic regulation of genes affected by Cd: APBA2, KIAA0895, DHX35, CPEB3, SVIL, MYLK, ZFYVE28, ABLIM2, GRB10, and PCDH9↓ Carcinogenesis ↓ PI3K/Akt | [260] |
| Quercetin | Rats | CdCl2 | ↓ miRNA-21 ↑ Nrf2 ↑ GSH ↑ SOD ↓ MDA ↓ ALT ↓ AST ↓ ROS ↓ IL6↓ TNF-α ↓ Total cholesterol ↓ Triglycerides | [261] |
| N-acetyl-l-cysteine | TRL1215 Cells | CdCl2 | ↑ TET1 ↑ ApoE ↓ MT2A | [217] |
| Resveratrol | CD-1 mice and JEG-3 cells | CdCl2 | ↓ DNMT activity ↓ DNMT3B expression ↓ Apoptosis ↓ TNF-α ↓ IFN-γ ↓ CCM-1 ↓ MIP-2 ↓ KC ↑ SIRT1 ↓ PI3K/Akt | [262] |
| Isoorientine | NRK-52E cells and primary cultures (rPT) | CdCl2 | ↓ p-H2AX ↓ ROS ↓ 8-OHdG | [263] |
| Cyanidin-3-O-glucoside | Pubescent mice | CdCl2 | ↑ Spermatogenesis ↓Histone H2A ↓ Histone H2B ↑ Ubiquitination of H2A ↑ SOD ↑ GSH ↑ GSH/GSSG ↓ MDA ↓ p-JNK/JNK ↑ p-ERK/ERK ↓ p-p38/p38 ↓ Caspase 3 ↓ Bax ↓ Bad ↑ bcl-2 | [264] |
| Methionine | K562 cells | CdCl2 | ↑ Global DNA methylation ↓ ROS ↓ 8-OHdG | [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Cruz, E.Y.; Arancibia-Hernández, Y.L.; Loyola-Mondragón, D.Y.; Pedraza-Chaverri, J. Oxidative Stress and Its Role in Cd-Induced Epigenetic Modifications: Use of Antioxidants as a Possible Preventive Strategy. Oxygen 2022, 2, 177-210. https://doi.org/10.3390/oxygen2020015
Hernández-Cruz EY, Arancibia-Hernández YL, Loyola-Mondragón DY, Pedraza-Chaverri J. Oxidative Stress and Its Role in Cd-Induced Epigenetic Modifications: Use of Antioxidants as a Possible Preventive Strategy. Oxygen. 2022; 2(2):177-210. https://doi.org/10.3390/oxygen2020015
Chicago/Turabian StyleHernández-Cruz, Estefani Yaquelin, Yalith Lyzet Arancibia-Hernández, Deyanira Yael Loyola-Mondragón, and José Pedraza-Chaverri. 2022. "Oxidative Stress and Its Role in Cd-Induced Epigenetic Modifications: Use of Antioxidants as a Possible Preventive Strategy" Oxygen 2, no. 2: 177-210. https://doi.org/10.3390/oxygen2020015
APA StyleHernández-Cruz, E. Y., Arancibia-Hernández, Y. L., Loyola-Mondragón, D. Y., & Pedraza-Chaverri, J. (2022). Oxidative Stress and Its Role in Cd-Induced Epigenetic Modifications: Use of Antioxidants as a Possible Preventive Strategy. Oxygen, 2(2), 177-210. https://doi.org/10.3390/oxygen2020015