
Acute Promyelocytic Leukemia (APL): A Review of the Classic and Emerging Target Therapies towards Molecular Heterogeneity

Abstract
:1. Introduction
1.1. Pathogenesis
1.1.1. Leading Cause of Death: Disseminated Intravascular Coagulation
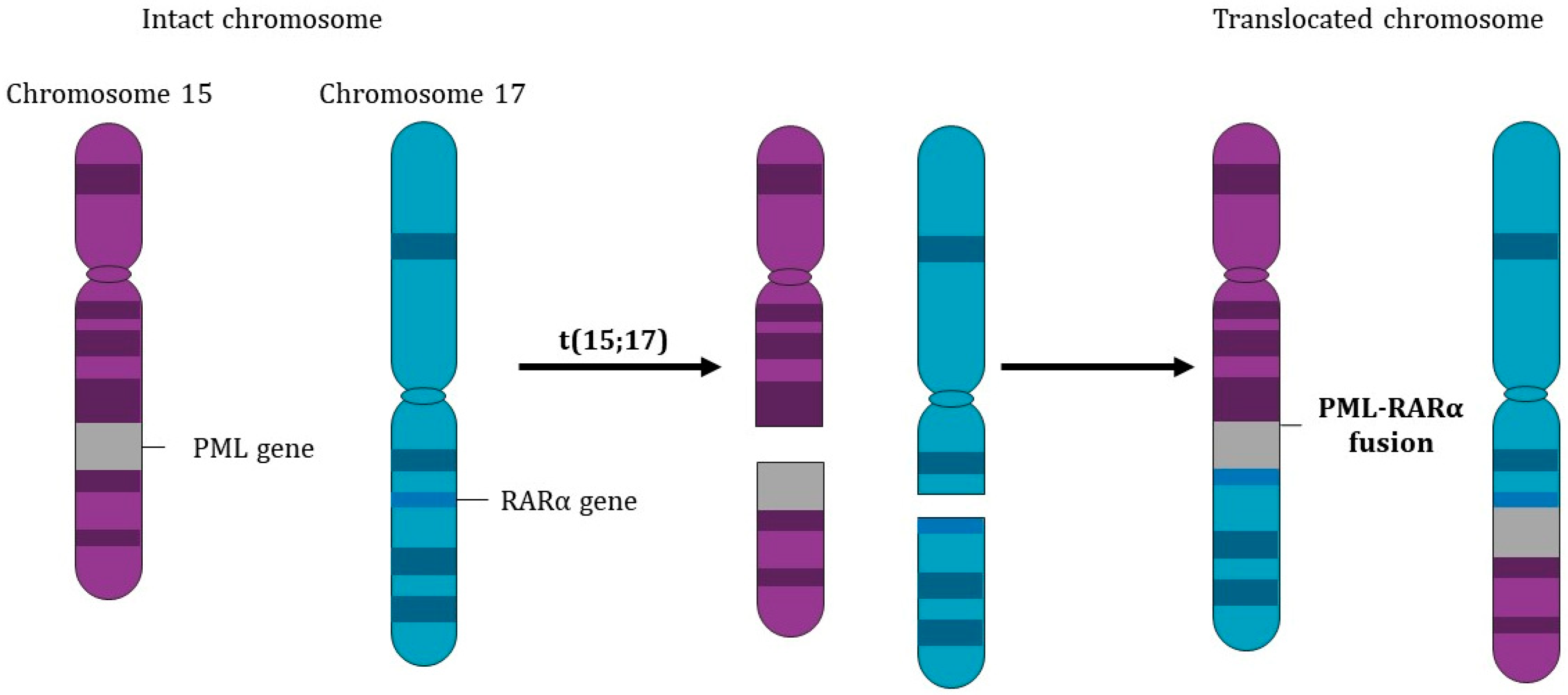
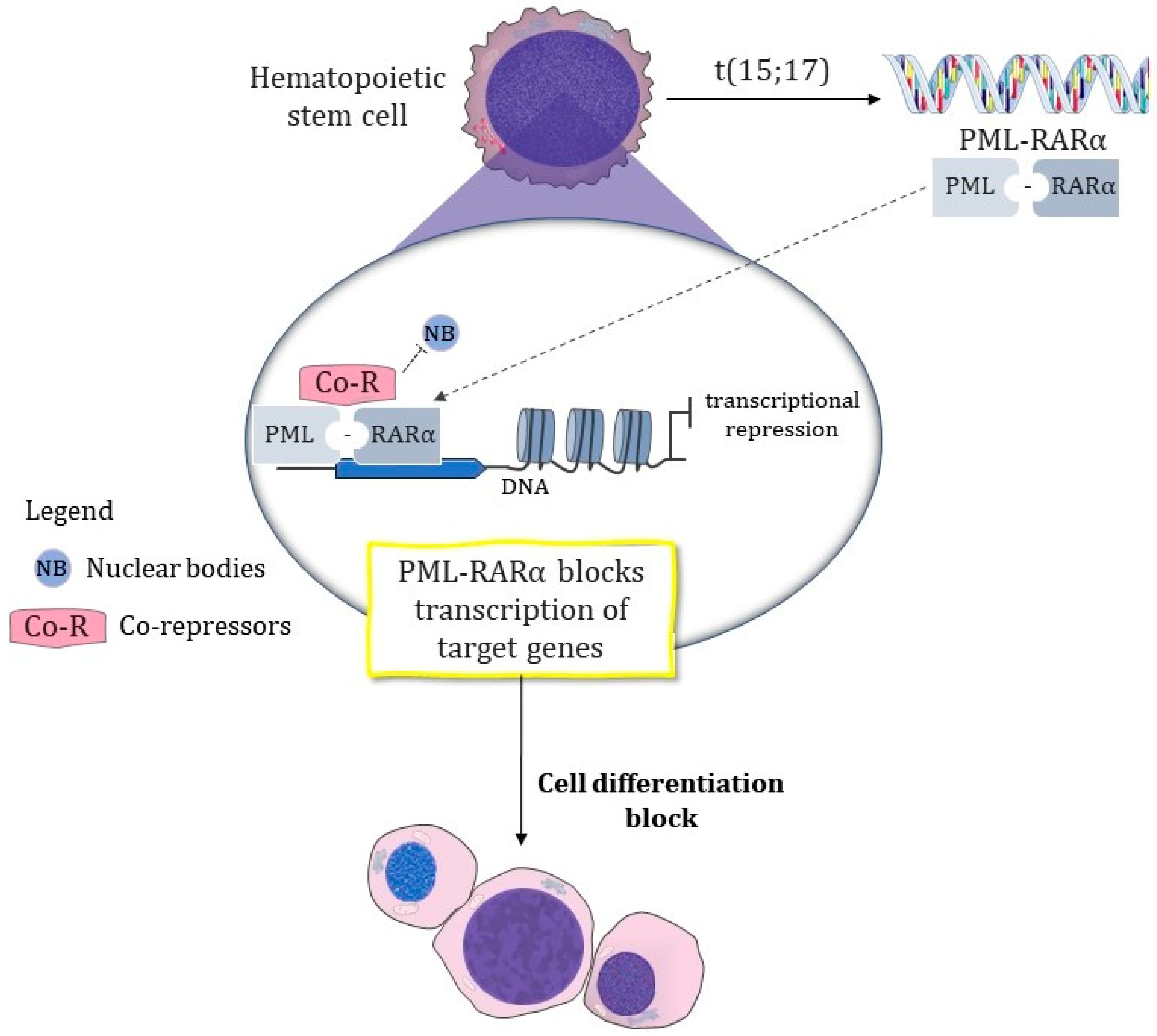
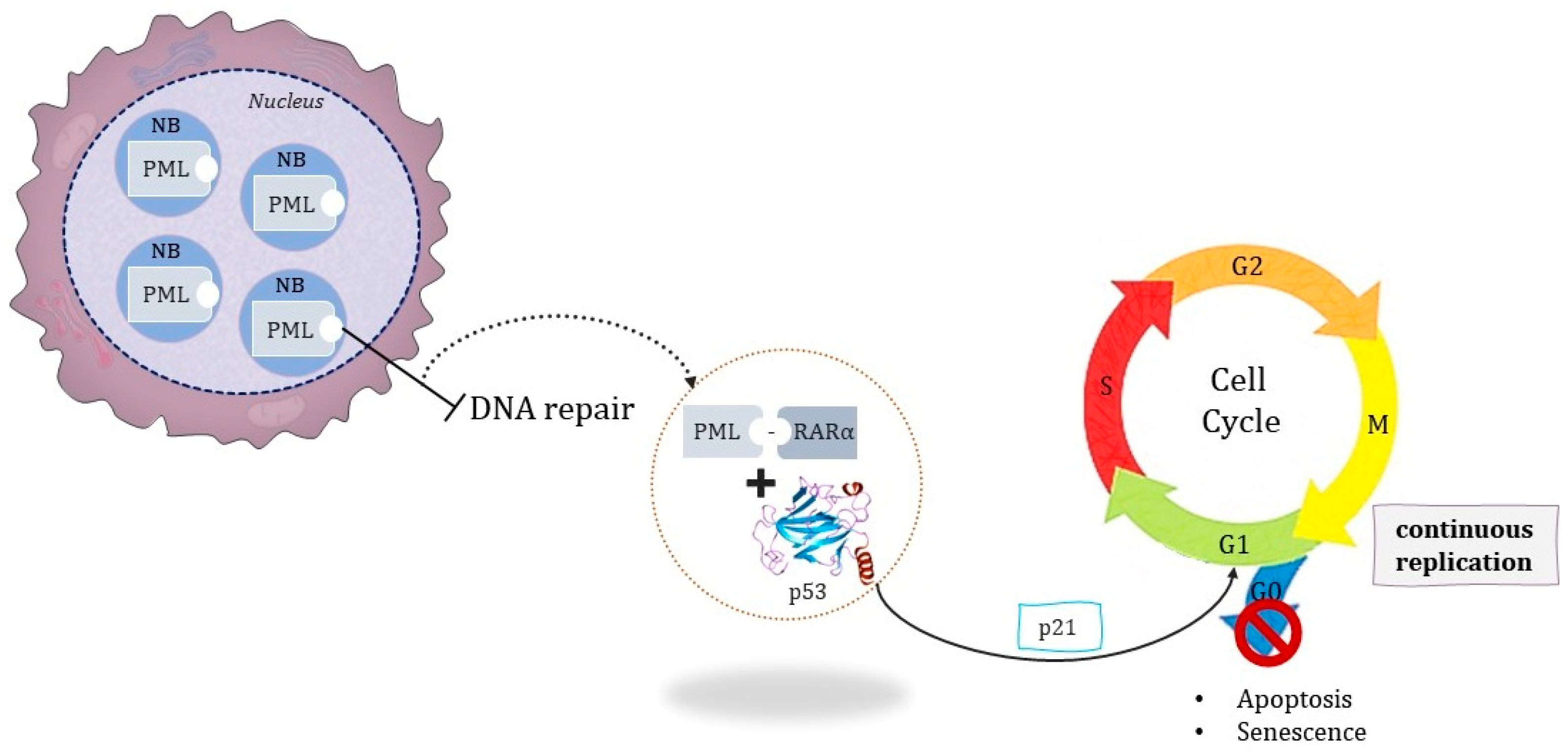
1.1.2. The Pathognomonic Genetic Event
1.1.3. Associated Mutations
2. Target Therapy Has Been the Mainstay Treatment for APL since the 1980s
2.1. Recommended Treatment
2.2. Consolidation Therapy
3. Differentiation Syndrome and Its Problems
4. Other Mutations That Evade Conventional Treatment
5. Possible Treatments for Each Mutation
- The use of cytotoxic drugs, especially topoisomerase inhibitors, alkalinizing agents, and anthracyclines, leads to changes in DNA structure and secondary changes, most of which include treatment-related myelodysplastic syndrome or AML;
- APL is accompanied by other clones in the early stages of the disease but masked by the dominant clones because the application of ATRA, ATO, and chemotherapy eliminates abnormal promyelocytes so that other clones have a better chance of survival;
- As reported in the literature, the prognosis of APL, secondary to treated acute myeloid leukemia, is poor and survival is relatively short. Although patients have achieved complete remission after one cycle of chemotherapy, their long-term survival still needs to be investigated because it is unclear whether the choice of follow-up consolidation therapy should be medium-dose cytarabine-based chemotherapy or allogeneic hematopoietic stem cell transplantation.
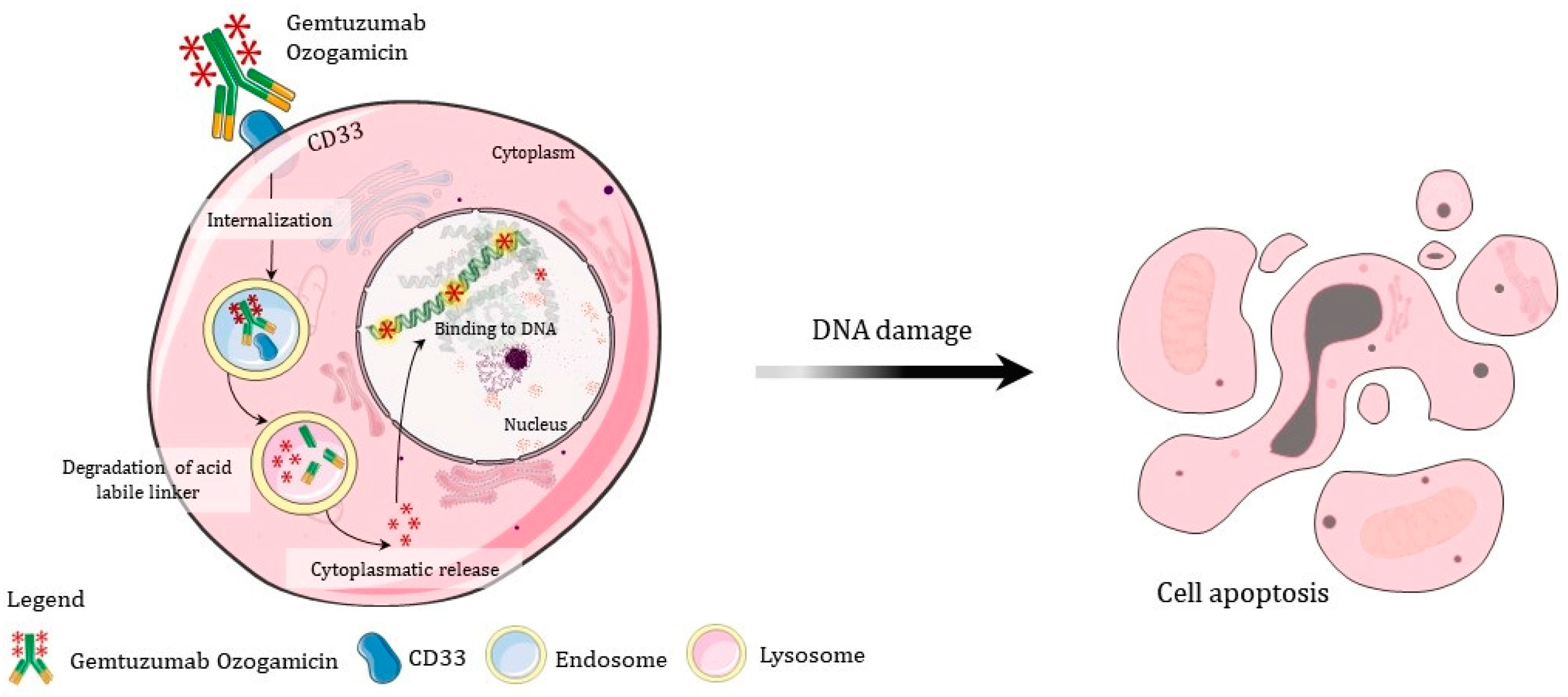
- Gentuzumab is recommended for immature cells with CD33 markers; thus, if it is not a blastic crisis, perhaps other drugs may be more effective;
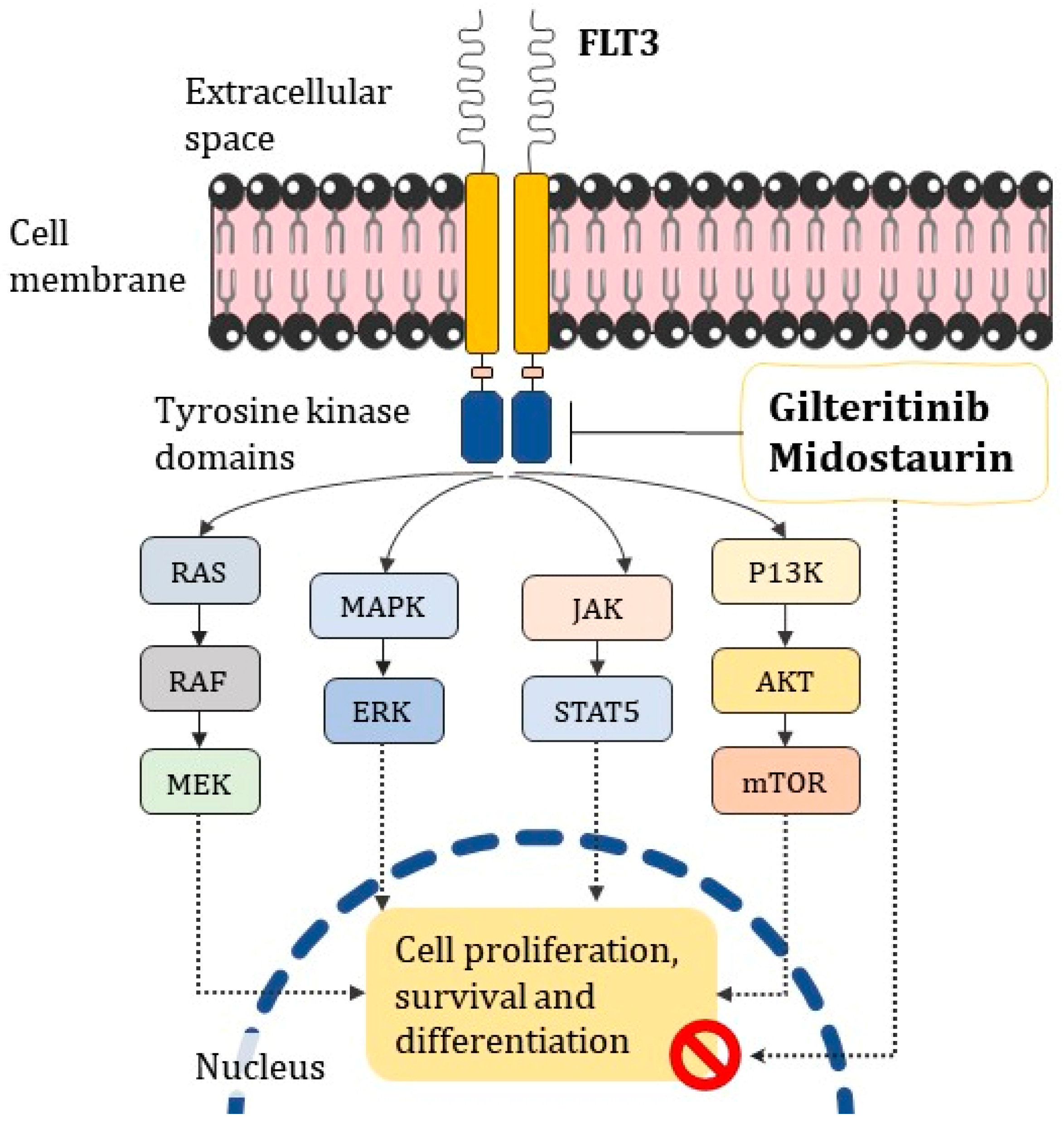
- Midostaurin and Gilteritinib targeting FLT3, which is commonly reported in oncohematological disorders, may help in cases where it is uncertain whether conventional ATRA/ATO treatment is effective;
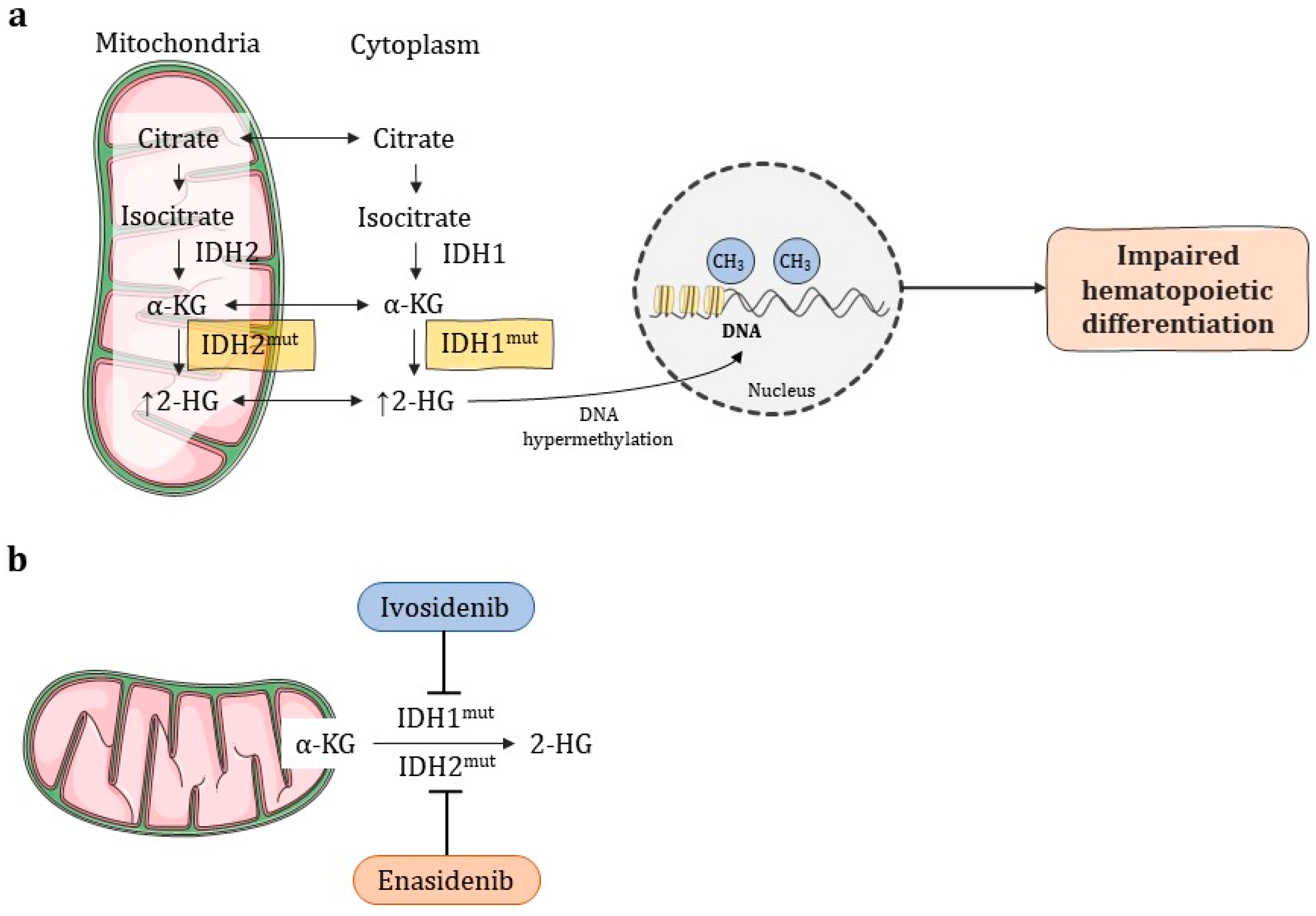
- Enadesinib and Ivodesinib have shown good results for cases in which the previous remission was also impaired;
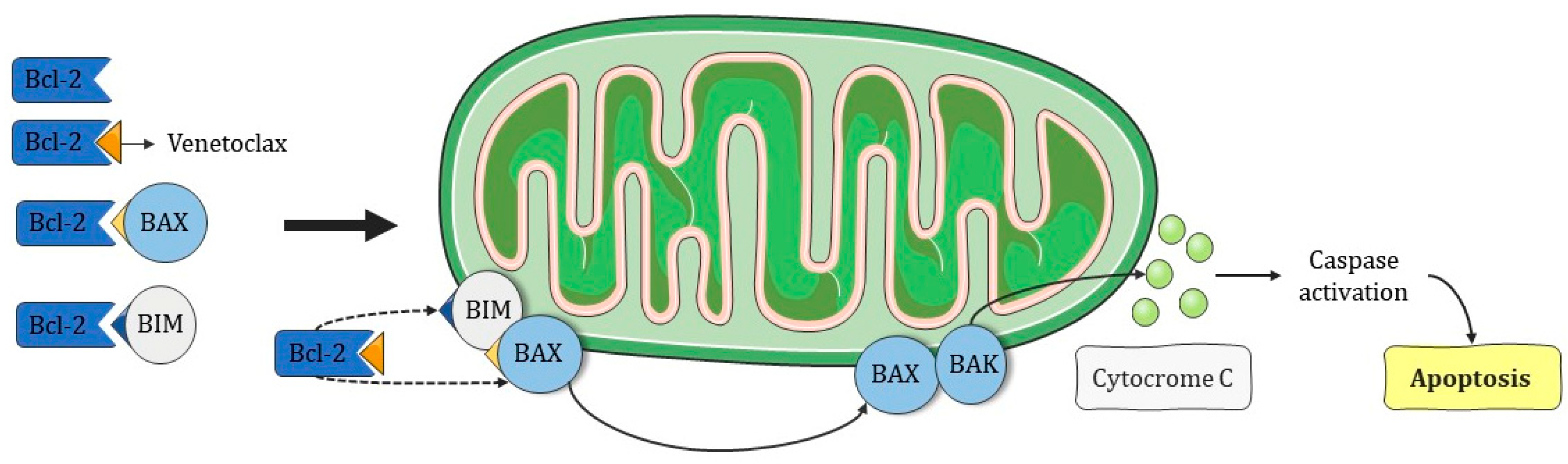
- Venetoclax acts by restoring apoptosis in cells poorly responsive to chemotherapy;
- Since this is such a heterogeneous disease, it is of great importance to outline the appropriate combined therapy of chemotherapy and target therapy to improve survival in APL patients.
6. Emerging and Combined Target Therapies
6.1. Immunotherapies
6.1.1. CAR-T
6.1.2. Cancer Vaccines
7. Hematopoietic Cell Transplantation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, E.S.; Viana, L.G.; Faria, R.M.D.; Santos, S.M.E. Medicina Laboratorial Para o Clínico, 1st ed.; Coopmed: Belo Horizonte, Brazil, 2009; pp. 305–341. [Google Scholar]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 13, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Chale, R.S.; Abad, A.C.; Schally, A.V. Acute promyelocytic leukemia (APL): A review of the literature. Oncotarget 2020, 11, 992–1003. [Google Scholar] [CrossRef]
- Tedesco, J.; Qualtieri, J.; Head, D.; Savani, B.N.; Reddy, N. High prevalence of obesity in acute promyelocytic leukemia (APL): Implications for differentiating agents in APL and metabolic syndrome. Ther. Adv. Hematol. 2011, 2, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Noguera, N.I.; Catalano, G.; Banella, C.; Divona, M.; Faraoni, I.; Ottone, T.; Arcese, W.; Voso, M.T. Acute promyelocytic Leukemia: Update on the mechanisms of leukemogenesis, resistance and on innovative treatment strategies. Cancers 2019, 11, 1591. [Google Scholar] [CrossRef]
- Reis, R.S.; Silva, N.P.; Santos, M.O.; Oliveira, J.F.P.; Thuler, L.C.S.; de Camargo, B.; Pombo-de-Oliveira, M.S. Características mãe-filho ao nascer e leucemias na primeira infância: Um estudo de caso-coorte de base populacional no Brasil. J. Pediatr. 2017, 93, 610–618. [Google Scholar] [CrossRef]
- Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years—From the Most Malignant to the most Curable Form of Acute Leukemia. Oncol. Ther. 2019, 7, 33–65. [Google Scholar] [CrossRef]
- Kwaan, H.C. The unique hemostatic dysfunction in acute promyelocytic leukemia. Semin. Thromb. Hemost. 2014, 40, 332–336. [Google Scholar]
- Jácomo, R.H.; Figueiredo-Pontes, L.L.; Rego, E.M. Do paradigma molecular ao impacto no prognóstico: Uma visão da leucemia promielocítica aguda. Rev. Assoc. Med. Bras. 2008, 54, 82–89. [Google Scholar] [CrossRef]
- Kwaan, H.C.; Weiss, I.; Tallman, M.S. The Role of Abnormal Hemostasis and Fibrinolysis in Morbidity and Mortality of Acute Promyelocytic Leukemia. Semin Thromb Hemost. 2019, 45, 612–621. [Google Scholar] [CrossRef]
- de Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell. 2017, 32, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Biol. 2016, 6, 1–16. [Google Scholar]
- Kreis, N.-N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.A.; Djavaheri-Mergny, M. Autophagy: New insights into mechanisms of action and resistance of treatment in acute promyelocytic leukemia. Int. J. Mol. Sci. 2019, 20, 3559. [Google Scholar] [CrossRef]
- Garay, C.M.; Sánchez, K.C.; Lagunes, L.L.F.; Olivares, M.J.; Rivas, A.M.; Torres, B.E.V.; Aguilar, H.F.; Enríquez, J.C.N.; Hernández, E.J.; Méndez, V.C.B.; et al. Profiling FLT3 Mutations in Mexican Acute Myeloid Leukemia Pediatric Patients: Impact on Overall Survival. Front. Pediatr. 2020, 8, 586. [Google Scholar] [CrossRef]
- Gale, R.E.; Hills, R.; Pizzey, A.R.; Kottaridis, P.D.; Swirsky, D.; Gilkes, A.F.; Nugent, E.; Mills, K.I.; Wheatley, K.; Solomon, E.; et al. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 2005, 106, 3768–3776. [Google Scholar] [CrossRef]
- Beitinjaneh, A.; Jang, S.; Roukoz, H.; Majhail, N.S. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: A systematic review. Leuk. Res. 2010, 34, 831–836. [Google Scholar] [CrossRef]
- McCulloch, D.; Brown, C.; Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. Onco. Targets. Ther. 2017, 10, 1585–1601. [Google Scholar] [CrossRef]
- Esnault, C.; Rahmé, R.; Rice, K.L.; Berthier, C.; Gaillard, C.; Quentin, S.; Maubert, A.L.; Kogan, S.; de Thé, H. FLT3-ITD impedes retinoic acid, but not arsenic, responses in murine acute promyelocytic leukemias. Blood 2019, 133, 1495–1506. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kim, H.-J.; Kwak, D.-H.; Park, S.-S.; Jeon, Y.-W.; Lee, S.E.; Cho, B.S.; Eom, K.S.; Kim, Y.J.; Lee, S.; et al. High WT1 expression is an early predictor for relapse in patients with acute promyelocytic leukemia in first remission with negative PML-RARa after anthracycline-based chemotherapy: A single-center cohort study. Clin. Res. Inf. Serv. 2017, 10, 30. [Google Scholar] [CrossRef]
- Mitrovic, M.; Kostic, T.; Virijevic, M.; Karan-Djurasevic, T.; Vukovic, N.S.; Pavlovic, S.; Tosic, N. The influence of Wilms’ tumor 1 gene expression level on prognosis and risk stratification of acute promyelocytic leukemia patients. Int. J. Lab. Hematol. 2020, 42, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Zhang, Y.; Chang, J.; Zheng, S.; Metayer, C.; Zhang, L.; Smith, M.T.; Ma, X.; Selvin, S.; Buffler, P.A.; et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia 2005, 19, 415–419. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, D.; Candoni, A. The Role of Wilms’ Tumor Gene (WT1) Expression as a Marker of Minimal Residual Disease in Acute Myeloid Leukemia. J. Clin. Med. 2022, 11, 3306. [Google Scholar] [CrossRef] [PubMed]
- Ivanschitz, L.; Thé, H.D.; Le Bras, M. PML, SUMOylation, and Senescence. Front Oncol. 2013, 3, 171. [Google Scholar] [CrossRef]
- Rahul, E.; Goel, H.; Chopra, A.; Ranjan, A.; Gupta, A.K.; Meena, J.P.; Bakhshi, S.; Misra, A.; Hussain, S.; Viswanathan, G.K.; et al. An updated account on molecular heterogeneity of acute leukemia. Am. J. Blood Res. 2021, 11, 22–43, ISSN: 2160-1992/AJBR0123439. [Google Scholar] [PubMed]
- Chen, S.J.; Zhou, G.B.; Zhang, X.W.; Mao, J.H.; Thé, H.D.; Chen, Z. From an old remedy to a magic bullet: Molecular mechanisms underlying the therapeutic effects of arsenic in fighting leucemia. Blood 2011, 117, 6425–6437. [Google Scholar] [CrossRef]
- Ministério da saúde: Instituto nacional de câncer José Alencar Gomes Da Silva, (INCA). Estimativa de Incidência de Câncer no Brasil, 1st ed.; Ministério da Saúde: Rio de Janeiro, Brazil, 2020; ISBN 978-85-7318-389-4. [Google Scholar]
- Stahl, M.; Tallman, M.S. Differentiation syndrome in acute promyelocytic leukaemia. Br. J. Haematol. 2019, 187, 157–162. [Google Scholar] [CrossRef]
- Rabellino, A.; Scaglioni, P.P. PML degradation: Multiple ways to eliminate PML. Front. Oncol. 2013, 3, 60. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network®. Acute Myeloid Leukemia. NCCN Guidel. Patients® Acute Myeloid Leuk. 2022, 1, 60–69. [Google Scholar]
- Gbadamosi, M.; Meshinchi, S.; Lamba, J.K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Futur. Oncol. 2018, 14, 3199–3213. [Google Scholar] [CrossRef] [PubMed]
- Abaza, Y.; Kantarjian, H.; Manero, G.; Estey, E.; Borthakur, G.; Jabbour, E.; Faderl, S.; O’Brien, S.; Wierda, W.; Pierce, S.; et al. Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood 2017, 129, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, G.; Liu, X.; Song, Y.; Xie, J.; Li, G.; Ren, J.; Wang, H.; Mou, J.; Dai, J.; et al. Sorafenib inhibited cell growth through the MEK/ERK signaling pathway in acute promyelocytic leukemia cells. Oncol. Lett. 2018, 15, 5620–5626. [Google Scholar] [PubMed]
- Kenedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Kucukyurt, S.; Eskazan, A.E. New drugs approved for acute myeloid leukaemia in 2018. Br. J. Clin. Pharmacol. 2019, 85, 2689–2693. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Wang, W.; Li, X.; Tan, Y.; Zhang, X.; Qian, W. Treatment of Central Nervous System Relapse in Acute Promyelocytic Leukemia by Venetoclax: A Case Report. Front. Oncol. 2021, 11, 693670. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients withmutant-IDH2acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef]
- Mugoni, V.; Panella, R.; Cheloni, G.; Chen, M.; Pozdnyakova, O.; Stroopinsky, D.; Guarnerio, J.; Monteleone, E.; Lee, J.D.; Mendez, L.; et al. Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res. 2019, 29, 446–459. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Reyhanoglu, G.; Hugues, B.; King, K.E.; Cambridge, R. Differentiation Syndrome, a Side Effect from the Therapy of Acute Promyelocytic Leukemia. Cureus 2020, 12, 12042. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.W.; Su, Y.Z.; Tao, H.F. All-trans Retinoic Acid, Arsenic Trioxide, and Anthracycline-based Chemotherapy improves outcome in newly diagnosed acute promyelocytic leukemia regardless of FLT3-ITD mutation status. Curr. Med. Sci. 2021, 41, 491–497. [Google Scholar] [CrossRef]
- Mazzarella, L.; Botteri, E.; Matthews, A.; Gatti, E.; Salvatore, D.D.; Bagnardi, V.; Breccia, M.; Montesinos, P.; Bernal, T.; Gil, C.; et al. Obesity is a risk factor for acute promyelocytic leukemia: Evidence from population and cross-sectional studies and correlation with FLT3 mutations and polyunsaturated fatty acid metabolism. Haematology 2020, 105, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene. Cancers 2021, 12, 624. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat. Commun. 2018, 9, 2047. [Google Scholar] [CrossRef]
- FDA Approves Olutasidenib for Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation. News Release. FDA. 1 December 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olutasidenib-relapsed-or-refractory-acute-myeloid-leukemia-susceptible-idh1-mutation (accessed on 1 December 2022).
- Hongtao, L. Emerging agents and regimens for AML. J. Hematol. Oncol. 2021, 14, 49. [Google Scholar]
- U.S. Food and Drug Administration. FDA Approves Ivosidenib in Combination with Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-combination-azacitidine-newly-diagnosed-acute-myeloid-leukemia (accessed on 30 November 2022).
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, O.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef]
- Janssen, M.; Schmidt, C.; Bruch, P.M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax synergizes with Gilteritinib in FLT3 wildtype high-risk Acute Myeloid Leukemia by suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef]
- Wang, E.S.; Montesinos, P.; Minden, M.D.; Lee, J.H.; Heuser, M.; Naoe, T.; Chou, W.C.; Laribi, K.; Esteve, J.; Altman, J.K.; et al. Phase 3 trial of gilteritinib plus azacitidine vs azacitidine for newly diagnosed FLT3mut+ AML ineligible for intensive chemotherapy. Blood 2022, 140, 1845–1857. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Frankfurt, O.; Schuh, A.C.; Döhner, H.; Martinelli, G.; Patel, P.A.; Raffoux, E.; et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination With Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 57–65. [Google Scholar] [CrossRef] [PubMed]
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02129101 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03625505 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03048344 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT04655391 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03386513 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03328078 (accessed on 5 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02124174 (accessed on 5 December 2022).
- Aureli, A.; Marziani, B.; Sconocchia, T.; Del Principe, M.I.; Buzzatti, E.; Pasqualone, G.; Venditti, A.; Sconocchia, G. Immunotherapy as a Turning Point in the Treatment of Acute Myeloid Leukemia. Cancers 2021, 13, 6246. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.; Kayser, S. Immune-Based Therapeutic Strategies for Acute Myeloid Leukemia. Cancers 2022, 14, 105. [Google Scholar] [CrossRef]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.-E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef]
- Maucher, M.; Srour, M.; Danhof, S.; Einsele, H.; Hudecek, M.; Yakoub-Agha, I. Current Limitations and Perspectives of Chimeric Antigen Receptor-T-Cells in Acute Myeloid Leukemia. Cancers 2021, 13, 6157. [Google Scholar] [CrossRef]
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/study/NCT04097301 (accessed on 10 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/study/NCT03473457 (accessed on 10 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/study/NCT03126864 (accessed on 10 December 2022).
- NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/study/NCT03672851 (accessed on 10 December 2022).
- Barbullushi, K.; Rampi, N.; Serpenti, F.; Sciumè, M.; Fabris, S.; De Roberto, P.; Fracchiolla, N.S. Vaccination Therapy for Acute Myeloid Leukemia: Where Do We Stand? Cancers 2022, 14, 2994. [Google Scholar] [CrossRef]
- Van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.J.A.M.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef]
- Khoury, H.J.; Collins, R.H.; Blum, W., Jr.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D.; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Ganzel, C.; Mathews, V.; Alimoghaddam, K.; Ghavamzadeh, A.; Kuk, D.; Devlin, S.; Wang, H.; Zhang, M.J.; Weisdorf, D.; Douer, D.; et al. Autologous transplant remains the preferred therapy for relapsed APL in CR2. Bone Marrow Transpl. 2016, 51, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Yanada, M.; Matsuda, K.; Ishii, H.; Fukuda, T.; Ozeki, K.; Ota, S.; Tashiro, H.; Uchida, N.; Kako, S.; Doki, N.; et al. Allogeneic Hematopoietic Cell Transplantation for Patients with Relapsed Acute Promyelocytic Leukemia. Transpl. Cell Ther. 2022, 28, 847.e1–847.e8. [Google Scholar] [CrossRef] [PubMed]
- Holter Chakrabarty, J.L.; Rubinger, M.; Le-Rademacher, J.; Wang, H.L.; Grigg, A.; Selby, G.B.; Szer, J.; Rowe, J.M.; Weisdorf, D.J.; Tallman, M.S. Autologous is superior to allogeneic hematopoietic cell transplantation for acute promyelocytic leukemia in second complete remission. Biol. Blood Marrow Transpl. 2014, 20, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.A.; Sánchez-Ortega, I.; Corbacioglu, S.; Basak, G.W.; Chabannon, C.; de la Camara, R.; Dolstra, H.; Duarte, R.F.; Glass, B.; Greco, R.; et al. Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: Current practice in Europe, 2022. Bone Marrow Transpl. 2022, 57, 1217–1239. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRUGS | DOSE | MOMENT | PERIOD | |
|---|---|---|---|---|
| A | ATRA | 45 mg/m2 daily (in 2 divided doses) | Daily until remission | |
| ATO | 0.15 mg/kg IV I | |||
| B | ATRA | 45 mg/m2 daily (in 2 divided doses) | Daily until remission | |
| ATO | 0.3 mg/kg IV I | Days 1,2,3,4,5 | Week 1 | |
| ATO | 0.25 mg/kg | Twice weekly | Weeks 2–8 |
| DRUGS | DOSE | MOMENT | PERIOD | |
|---|---|---|---|---|
| A | ATRA + IDARUBICIN | 6–12 mg/m2 | Days 2, 4, 6, 8 | |
| ATO | 0.15 mg/kg | Days 09–36 | 2 h IV I | |
| B | ATRA + ATO | 0.15 mg/kg/d IV I | ||
| SINGLE DOSE OF GO | 9 mg/m2 | Day 1, 2, 3, or 4 | ||
| C | ATRA + ATO | 0.3 mg/kg IV I | Days 1, 2, 3, 4, 5 | Week 1 |
| ATRA + ATO | 0.25 mg/kg | Twice weekly | Weeks 2–8 | |
| SINGLE DOSE OF GO | 6 mg/m2 | Day 1, 2, 3, or 4 | ||
| D | ATRA + DAUNORUBICIN | 50 mg/m2 IV I | Days 3, 4, 5, 6 | |
| CYTARABINE | 200 mg/m2 IV I | Days 3, 4, 5, 6, 7, 8, 9 | ||
| E | ATRA + DAUNORUBICIN | 60 mg/m2 | 3 days | |
| CYTARABINE | 200 mg/m2 IV I | 7 days | ||
| F | ATRA+ IDARUBICIN | 12 mg/m2 | Days 2, 4, 6, 8 |
| Indication | Combination |
|---|---|
| IDH1/2 mutation | Ivosidenib +AZA [50] or AZA + Ivosidenib [56] or AZA+ Enasidenib [57] |
| Newly diagnosed, ineligible for intense chemotherapy | AZA+ Venetoclax + Decitabine [51] |
| FLT3 high-risk | Venetoclax + Gilteritinib [52] |
| FLT3 mutation in older patients | Gilteritinib + AZA [53] |
| TP53 mutation | Eprenetapopt +AZA [54] |
| Post-allogenic HCT | Eprenetapopt +AZA [55] |
| Clinical Trial Identifier | Phase | Clinical Test | Related Conditions | Reference |
|---|---|---|---|---|
| NCT02129101 | I/Ib | To evaluate the tolerable dose and efficacy of Azacitidine and Sonidegib or Decitabine | Acute myeloid leukemia Acute promyelocytic leukemia | [58] |
| NCT03625505 | I | To evaluate the safety and efficacy of Venetoclax in combination with Gilteritinib | Relapsed or refractory (R/R) acute myeloid leukemia Acute myeloid leukemia Acute promyelocytic leukemia | [59] |
| NCT03048344 | I | To determine the recommended dose of arsenic trioxide capsule formulation ORH 2014 | Acute myeloid leukemia Acute promyelocytic leukemia Chronic myelomonocytic leukemia Mantle cell lymphoma Myelodysplastic syndromes Myelodysplastic/myeloproliferative neoplasm | [60] |
| NCT04655391 | Ib | To evaluate the best dose and effect of Glasdegib with Venetoclax and Decitabine; Gilteritinib, Bosutinib, Ivosidenib, or Enasidenib | Relapse after HCT Acute myeloid leukemia Acute promyelocytic leukemia | [61] |
| NCT03386513 | I/II | To evaluate the antileukemic activity of IMGN632 when administered as a monotherapy to patients with CD123+ | Acute lymphoblastic leukemia Acute myeloid leukemia Acute promyelocytic leukemia Blastic plasmacytoid Dendritic cell neoplasm Chronic myeloid leukemia Chronic myelomonocytic leukemia Myelodysplastic syndromes Myeloproliferative neoplasm | [62] |
| NCT03328078 | I/II | To evaluate oral administration of CA-4948 (reversible inhibitor of Interleukin-1) | Adult patients with relapsed/refractory hematologic malignancies Acute myeloid leukemia Acute promyelocytic leukemia B-cell non-Hodgkin lymphoma | [63] |
| NCT02124174 | II | To evaluate Vidaza and valproic acid post allogeneic transplant | High-risk AML post allogeneic transplant Acute myeloid leukemia Acute myeloid leukemia with t(8;21), (q22; q22.1), RUNX1-RUNX1T1 Acute promyelocytic leukemia Core binding factor Acute myeloid leukemia Myelodysplastic syndromes | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Almeida, T.D.; Evangelista, F.C.G.; Sabino, A.d.P. Acute Promyelocytic Leukemia (APL): A Review of the Classic and Emerging Target Therapies towards Molecular Heterogeneity. Future Pharmacol. 2023, 3, 162-179. https://doi.org/10.3390/futurepharmacol3010012
de Almeida TD, Evangelista FCG, Sabino AdP. Acute Promyelocytic Leukemia (APL): A Review of the Classic and Emerging Target Therapies towards Molecular Heterogeneity. Future Pharmacology. 2023; 3(1):162-179. https://doi.org/10.3390/futurepharmacol3010012
Chicago/Turabian Stylede Almeida, Tâmara Dauare, Fernanda Cristina Gontijo Evangelista, and Adriano de Paula Sabino. 2023. "Acute Promyelocytic Leukemia (APL): A Review of the Classic and Emerging Target Therapies towards Molecular Heterogeneity" Future Pharmacology 3, no. 1: 162-179. https://doi.org/10.3390/futurepharmacol3010012