
Physiologically Based Pharmacokinetic Modelling of UGT Substrate Drugs Lamotrigine and Raltegravir during Pregnancy


Abstract
:1. Introduction
1.1. Challenges of Medication Use in Pregnancy
1.2. Physiological Changes during Pregnancy
1.3. Current PBPK Modelling in Pregnancy
1.4. PBPK Modelling of Typical UGT Substrate Drugs Lamotrigine and Raltegravir in Pregnancy
2. Methods
2.1. Clinical PK Data Collection
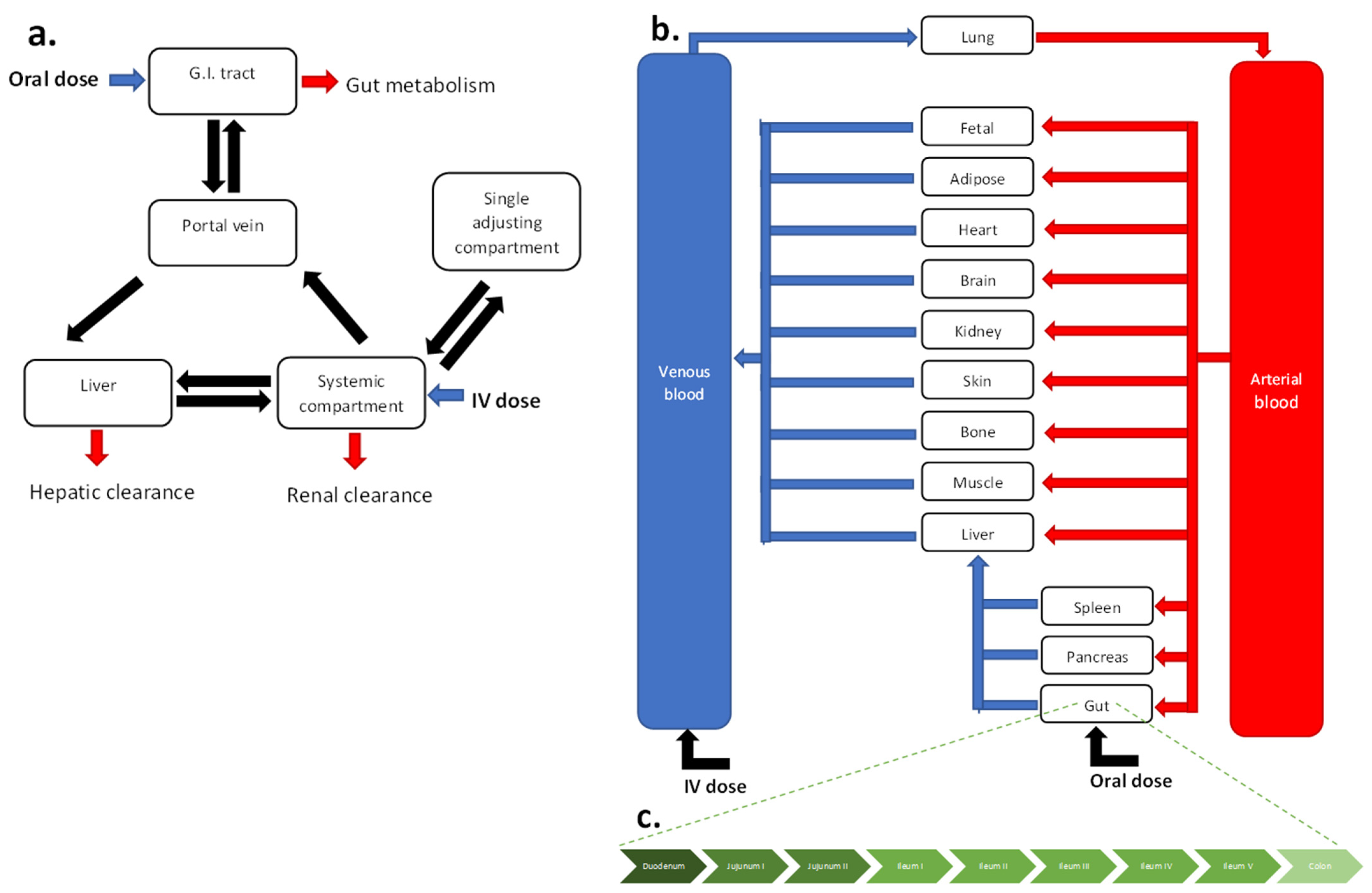
2.2. Adult Model Development
2.3. Adult Model Validation
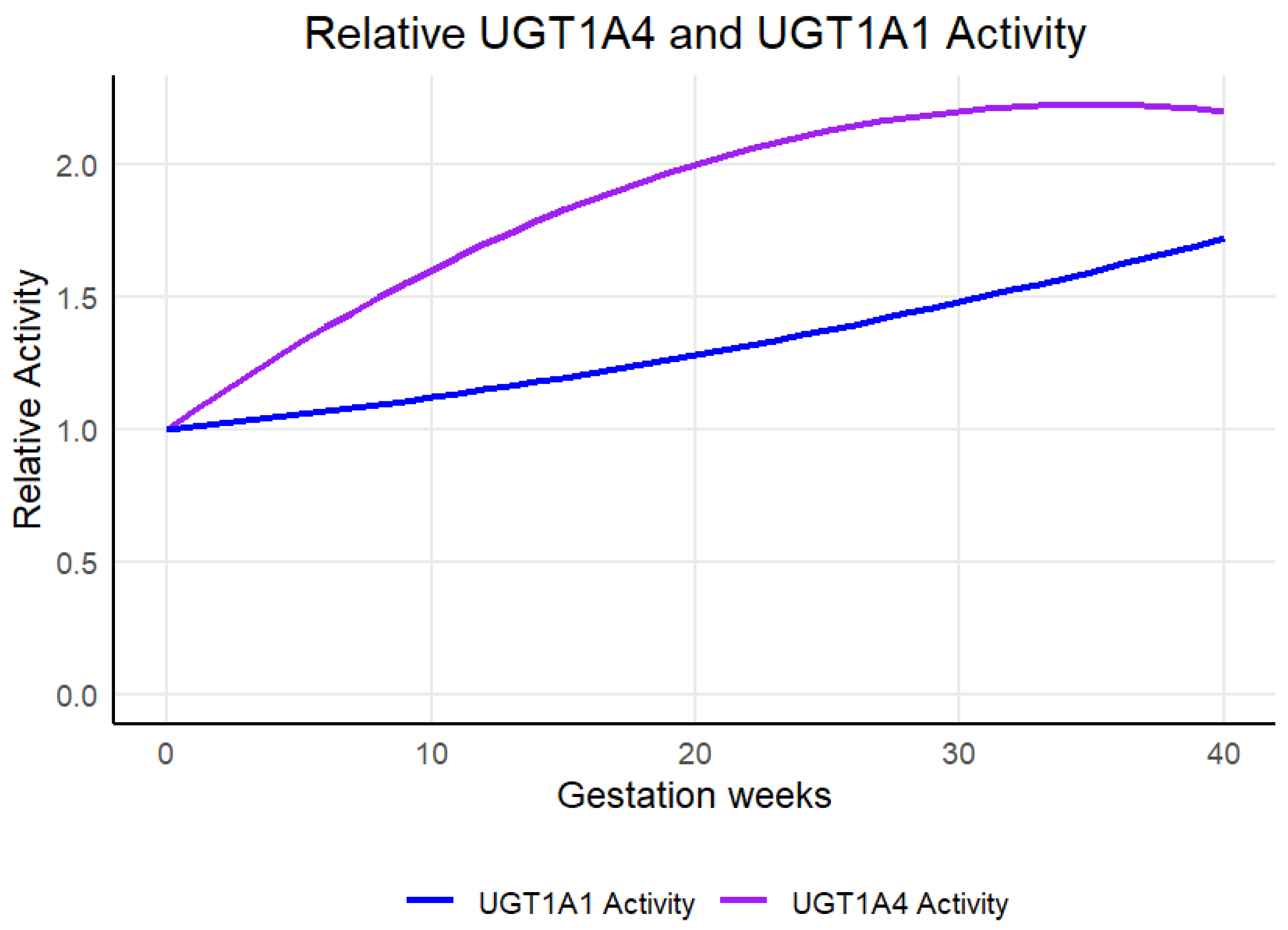
2.4. Pregnancy Population Model Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | B1 | B2 | B3 | B4 |
|---|---|---|---|---|
| UGT1A4 | 0.07 | −0.001 | 0 | 0 |
| UGT1A1 | 0.01 | 0.0002 | 0 | 0 |
2.5. Predction of PK in Pregnancy Population Models
3. Results
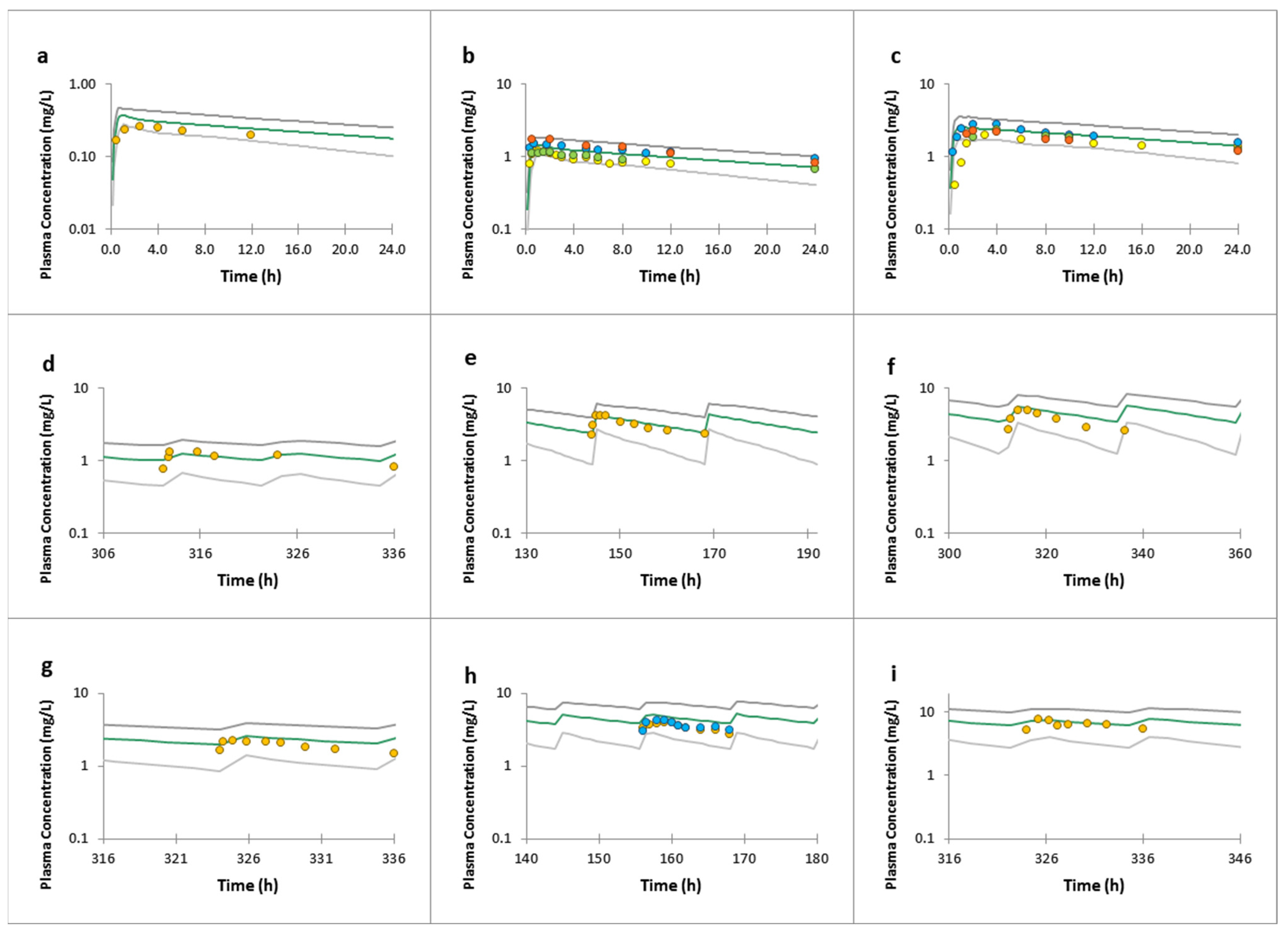
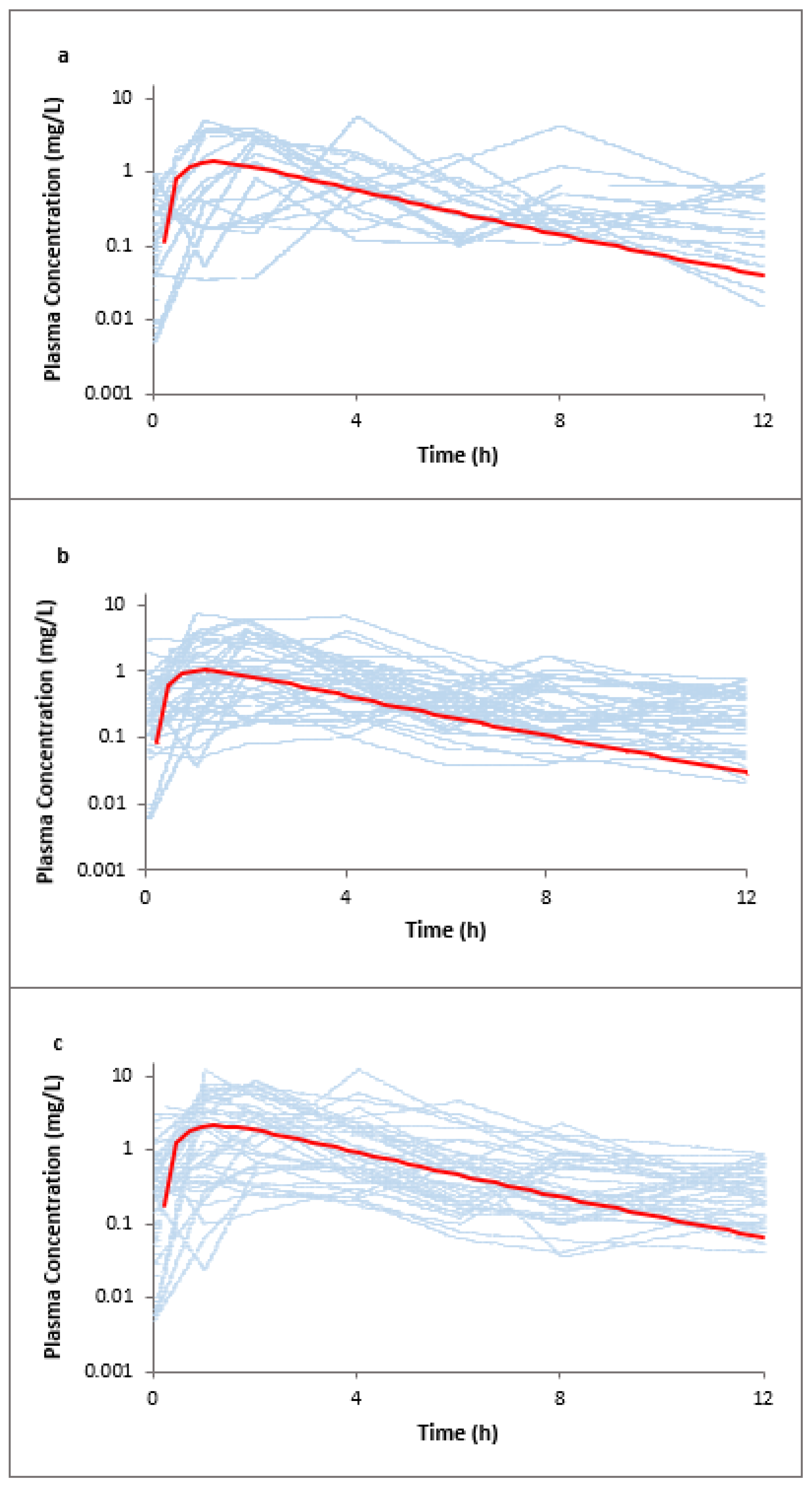
3.1. Lamotrigine Full PBPK Model Validation
3.2. Lamotrigine PK Prediction in Pregnancy
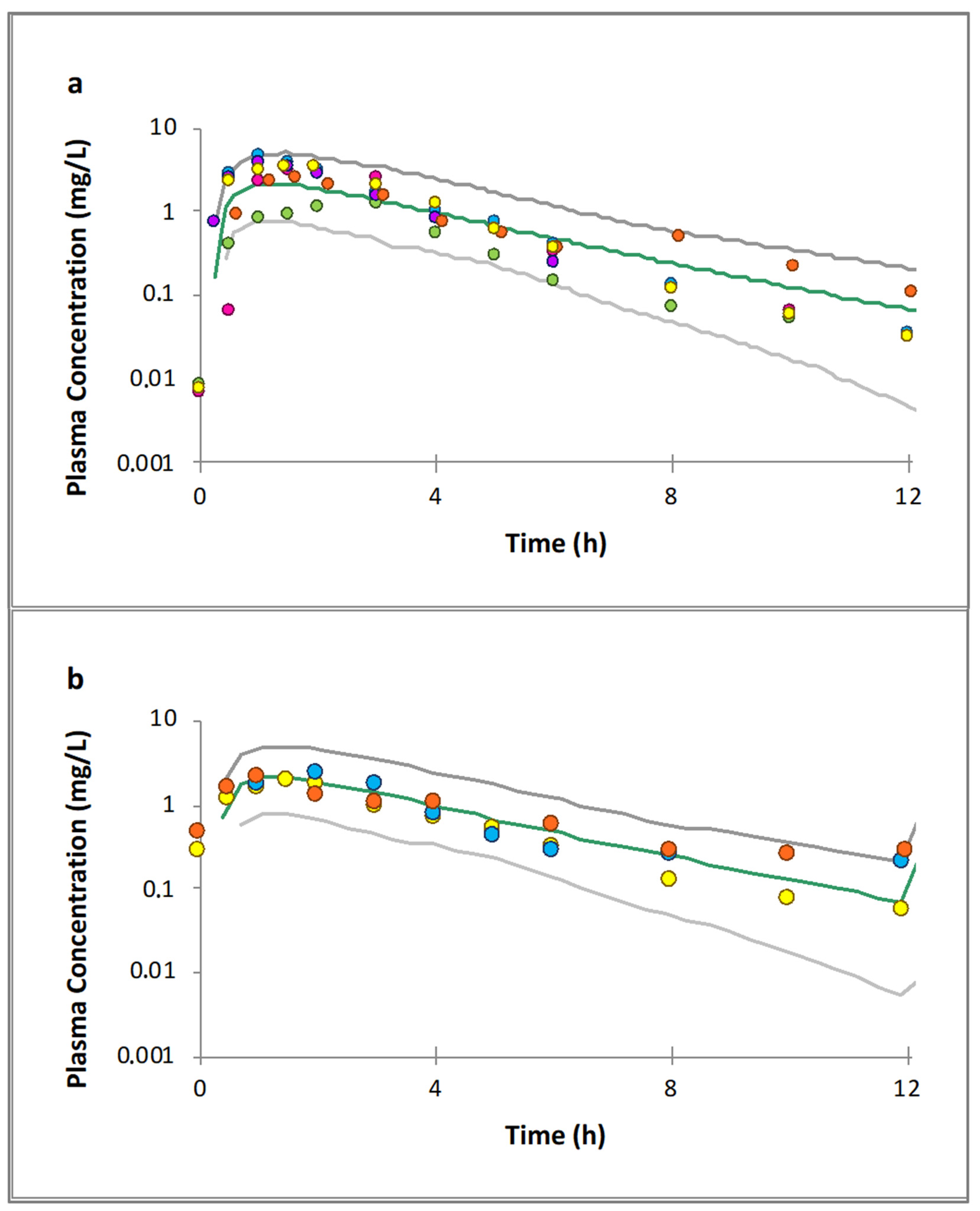
3.3. Raltegravir Full PBPK Model Validation
3.4. Raltegravir PK Prediction in Pregnancy
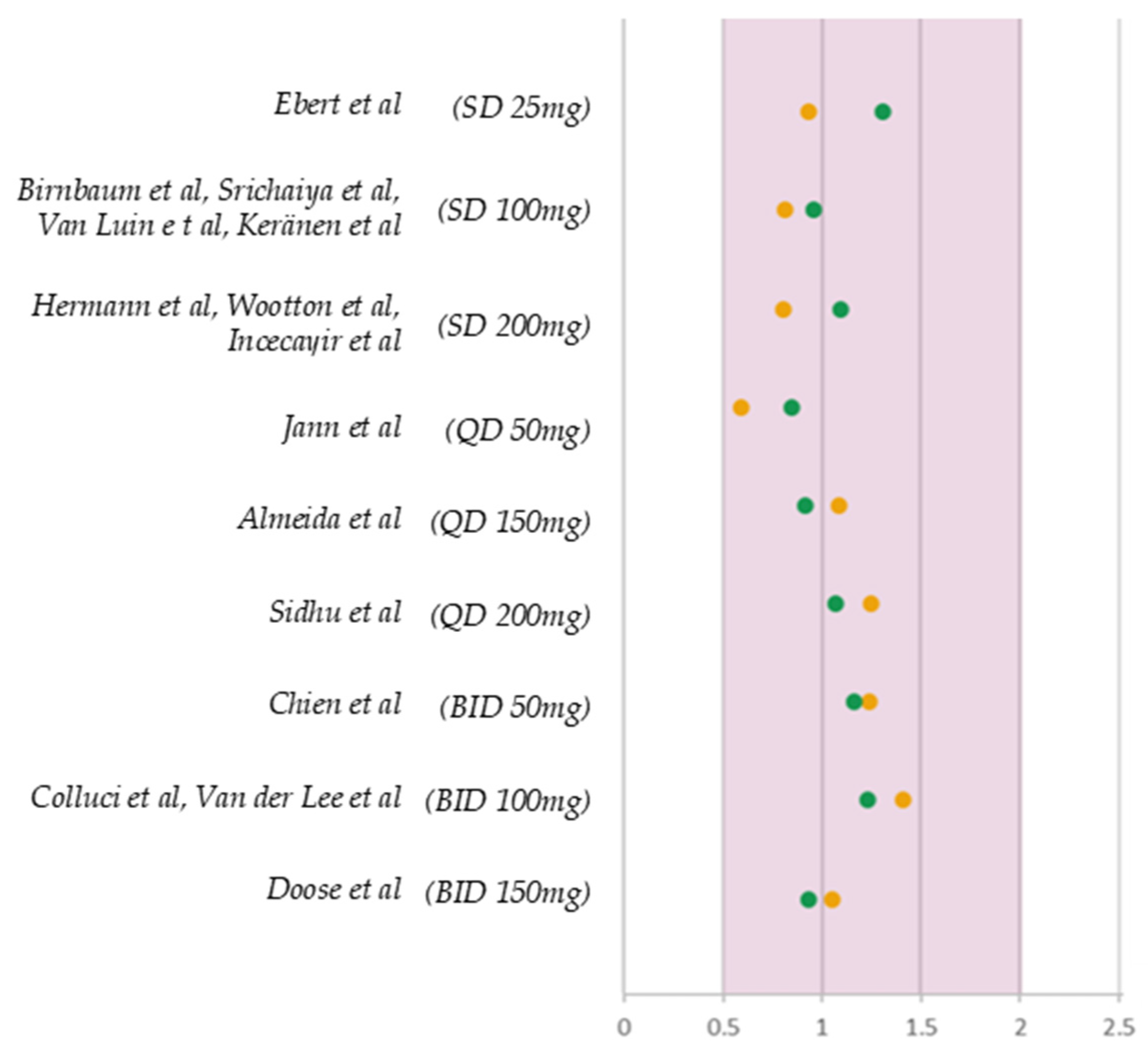
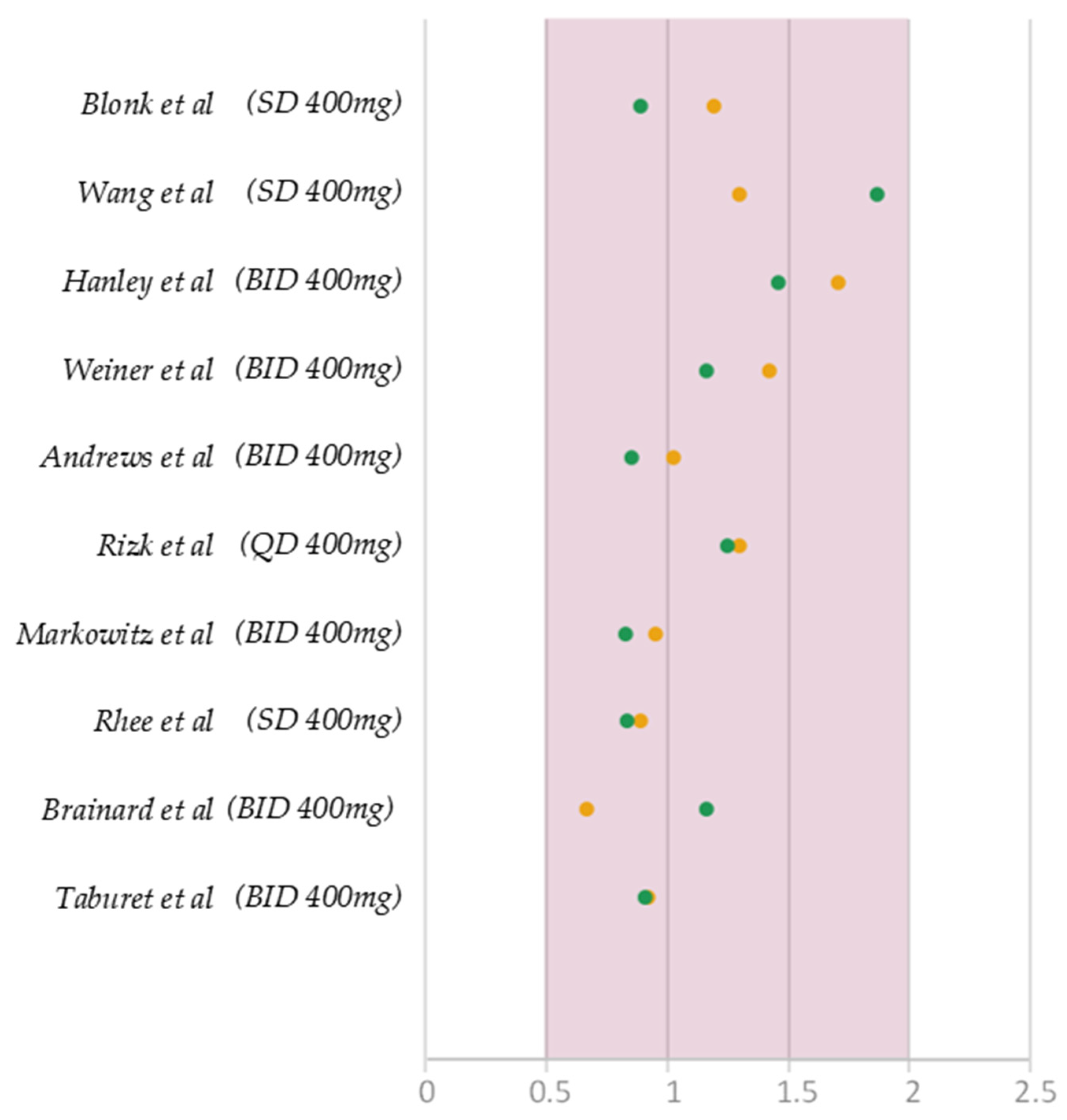
3.5. Model Credibility Evaluation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berezowska, M.; Sharma, P.; Pilla Reddy, V.; Coppola, P. Physiologically Based Pharmacokinetic modelling of drugs in pregnancy: A mini-review on availability and limitations. Fundam. Clin. Pharmacol. 2023, e12967. [Google Scholar] [CrossRef]
- Lupattelli, A.; Spigset, O.; Twigg, M.J.; Zagorodnikova, K.; Mårdby, A.C.; Moretti, M.E.; Drozd, M.; Panchaud, A.; Hämeen-Anttila, K.; Rieutord, A.; et al. Medication use in pregnancy: A cross-sectional, multinational web-based study. BMJ Open 2014, 4, e004365. [Google Scholar] [CrossRef]
- Borda, L.A.; Någård, M.; Boulton, D.W.; Venkataramanan, R.; Coppola, P. A systematic review of pregnancy-related clinical intervention of drug regimens due to pharmacokinetic reasons. Front. Med. 2023, 10, 1241456. [Google Scholar] [CrossRef]
- Coppola, P.; Kerwash, E.; Nooney, J.; Omran, A.; Cole, S. Pharmacokinetic data in pregnancy: A review of available literature data and important considerations in collecting clinical data. Front. Med. 2022, 9, 940644. [Google Scholar] [CrossRef]
- Abduljalil, K.; Furness, P.; Johnson, T.N.; Rostami-Hodjegan, A.; Soltani, H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: A database for parameters required in physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 2012, 51, 365–396. [Google Scholar] [CrossRef]
- Feghali, M.; Venkataramanan, R.; Caritis, S. Pharmacokinetics of drugs in pregnancy. Semin. Perinatol. 2015, 39, 512–519. [Google Scholar] [CrossRef]
- Pariente, G.; Leibson, T.; Carls, A.; Adams-Webber, T.; Ito, S.; Koren, G. Pregnancy-Associated Changes in Pharmacokinetics: A Systematic Review. PLoS Med. 2016, 13, e1002160. [Google Scholar] [CrossRef]
- Meah, V.L.; Cockcroft, J.R.; Backx, K.; Shave, R.; Stohr, E.J. Cardiac output and related haemodynamics during pregnancy: A series of meta-analyses. Heart 2016, 102, 518–526. [Google Scholar] [CrossRef]
- Everson, G.T. Gastrointestinal motility in pregnancy. Gastroenterol. Clin. N. Am. 1992, 21, 751–776. [Google Scholar] [CrossRef]
- Aguree, S.; Gernand, A.D. Plasma volume expansion across healthy pregnancy: A systematic review and meta-analysis of longitudinal studies. BMC Pregnancy Childbirth 2019, 19, 508. [Google Scholar] [CrossRef]
- Coppola, P.; Butler, A.; Cole, S.; Kerwash, E. Total and Free Blood and Plasma Concentration Changes in Pregnancy for Medicines Highly Bound to Plasma Proteins: Application of Physiologically Based Pharmacokinetic Modelling to Understand the Impact on Efficacy. Pharmaceutics 2023, 15, 2455. [Google Scholar] [CrossRef]
- Tracy, T.S.; Venkataramanan, R.; Glover, D.D.; Caritis, S.N.; for the National Institute for Child Health and Human Development Network of Maternal-Fetal-Medicine Units. Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am. J. Obs. Gynecol. 2005, 192, 633–639. [Google Scholar] [CrossRef]
- Khatri, R.; Fallon, J.K.; Sykes, C.; Kulick, N.; Rementer, R.J.B.; Miner, T.A.; Schauer, A.P.; Kashuba, A.D.M.; Boggess, K.A.; Brouwer, K.L.R.; et al. Pregnancy-Related Hormones Increase UGT1A1-Mediated Labetalol Metabolism in Human Hepatocytes. Front. Pharmacol. 2021, 12, 655320. [Google Scholar] [CrossRef]
- Pennell, P.B.; Newport, D.J.; Stowe, Z.N.; Helmers, S.L.; Montgomery, J.Q.; Henry, T.R. The impact of pregnancy and childbirth on the metabolism of lamotrigine. Neurology 2004, 62, 292–295. [Google Scholar] [CrossRef]
- Desai, D.K.; Moodley, J.; Naidoo, D.P. Echocardiographic assessment of cardiovascular hemodynamics in normal pregnancy. Obs. Gynecol. 2004, 104, 20–29. [Google Scholar] [CrossRef]
- Davison, J.M.; Dunlop, W. Renal hemodynamics and tubular function normal human pregnancy. Kidney Int. 1980, 18, 152–161. [Google Scholar] [CrossRef]
- Peng, J.; Ladumor, M.K.; Unadkat, J.D. Prediction of Pregnancy-Induced Changes in Secretory and Total Renal Clearance of Drugs Transported by Organic Anion Transporters. Drug Metab. Dispos. 2021, 49, 929–937. [Google Scholar] [CrossRef]
- Bergagnini-Kolev, M.C.; Hebert, M.F.; Easterling, T.R.; Lin, Y.S. Pregnancy Increases the Renal Secretion of N(1)-methylnicotinamide, an Endogenous Probe for Renal Cation Transporters, in Patients Prescribed Metformin. Drug Metab. Dispos. 2017, 45, 325–329. [Google Scholar] [CrossRef]
- Green, D.J.; Park, K.; Bhatt-Mehta, V.; Snyder, D.; Burckart, G.J. Regulatory Considerations for the Mother, Fetus and Neonate in Fetal Pharmacology Modeling. Front. Pediatr. 2021, 9, 698611. [Google Scholar] [CrossRef]
- Nordmark, A. EMA Draft Guideline on the Qualification and Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. Dissolution and Translational Modeling Strategies Enabling Patient-Centric Product Development. 2017. Available online: https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/dissolution/day3_anna-nordmark.pdf (accessed on 3 January 2024).
- Coppola, P.; Kerwash, E.; Cole, S. Physiologically Based Pharmacokinetics Model in Pregnancy: A Regulatory Perspective on Model Evaluation. Front. Pediatr. 2021, 9, 687978. [Google Scholar] [CrossRef]
- Vajda, F.J.; O’Brien, T.; Lander, C.; Graham, J.; Eadie, M. The efficacy of the newer antiepileptic drugs in controlling seizures in pregnancy. Epilepsia 2014, 55, 1229–1234. [Google Scholar] [CrossRef]
- Ebert, U.; Thong, N.Q.; Oertel, R.; Kirch, W. Effects of rifampicin and cimetidine on pharmacokinetics and pharmacodynamics of lamotrigine in healthy subjects. Eur. J. Clin. Pharmacol. 2000, 56, 299–304. [Google Scholar] [CrossRef]
- Birnbaum, A.K.; Kriel, R.L.; Im, Y.; Remmel, R.P. Relative bioavailability of lamotrigine chewable dispersible tablets administered rectally. Pharmacotherapy 2001, 21, 158–162. [Google Scholar] [CrossRef]
- Srichaiya, A.; Longchoopol, C.; Oo-Puthinan, S.; Sayasathid, J.; Sripalakit, P.; Viyoch, J. Bioequivalence of generic lamotrigine 100-mg tablets in healthy Thai male volunteers: A randomized, single-dose, two-period, two-sequence crossover study. Clin. Ther. 2008, 30, 1844–1851. [Google Scholar] [CrossRef]
- van Luin, M.; Colbers, A.; Wissen, C.P.W.G.M.V.; van Ewijk-Beneken-Kolmer, E.W.J.; van der Kolk, M.; Hoitsma, A.; da Silva, H.G.; Burger, D.M. The effect of raltegravir on the glucuronidation of lamotrigine. J. Clin. Pharmacol. 2009, 49, 1220–1227. [Google Scholar] [CrossRef]
- Keränen, T.; Sorri, A.; Moilanen, E.; Ylitalo, P. Effects of charcoal on the absorption and elimination of the antiepileptic drugs lamotrigine and oxcarbazepine. Arzneimittelforschung 2010, 60, 421–426. [Google Scholar] [CrossRef]
- Hermann, R.; Knebel, N.G.; Niebch, G.; Richards, L.; Borlak, J.; Locher, M. Pharmacokinetic interaction between retigabine and lamotrigine in healthy subjects. Eur. J. Clin. Pharmacol. 2003, 58, 795–802. [Google Scholar] [CrossRef]
- Wootton, R.; Soul-Lawton, J.; Rolan, P.E.; Sheung, C.T.; Cooper, J.D.; Posner, J. Comparison of the pharmacokinetics of lamotrigine in patients with chronic renal failure and healthy volunteers. Br. J. Clin. Pharmacol. 1997, 43, 23–27. [Google Scholar] [CrossRef]
- Incecayir, T.; Agabeyoglu, I.; Gucuyener, K. Comparison of plasma and saliva concentrations of lamotrigine in healthy volunteers. Arzneimittelforschung 2007, 57, 517–521. [Google Scholar] [CrossRef]
- Jann, M.W.; Hon, Y.Y.; Shamsi, S.A.; Zheng, J.; Awad, E.A.; Spratlin, V. Lack of pharmacokinetic interaction between lamotrigine and olanzapine in healthy volunteers. Pharmacotherapy 2006, 26, 627–633. [Google Scholar] [CrossRef]
- Almeida, L.; Nunes, T.; Sicard, E.; Rocha, J.-F.; Falcão, A.; Brunet, J.-S.; Lefebvre, M.; Soares-Da-Silva, P. Pharmacokinetic interaction study between eslicarbazepine acetate and lamotrigine in healthy subjects. Acta Neurol. Scand. 2010, 121, 257–264. [Google Scholar] [CrossRef]
- Sidhu, J.; Job, S.; Bullman, J.; Francis, E.; Abbott, R.; Ascher, J.; Theis, J.G.W. Pharmacokinetics and tolerability of lamotrigine and olanzapine coadministered to healthy subjects. Br. J. Clin. Pharmacol. 2006, 61, 420–426. [Google Scholar] [CrossRef]
- Chien, S.; Yao, C.; Mertens, A.; Verhaeghe, T.; Solanki, B.; Doose, D.R.; Novak, G.; Bialer, M. An interaction study between the new antiepileptic and CNS drug carisbamate (RWJ-333369) and lamotrigine and valproic acid. Epilepsia 2007, 48, 1328–1338. [Google Scholar] [CrossRef]
- Colucci, R.; Glue, P.; Holt, B.; Banfield, C.; Reidenberg, P.; Meehan, J.W.; Pai, S.; Nomeir, A.; Lim, J.; Lin, C.-C.; et al. Effect of felbamate on the pharmacokinetics of lamotrigine. J. Clin. Pharmacol. 1996, 36, 634–638. [Google Scholar] [CrossRef]
- van der Lee, M.J.; Dawood, L.; ter Hofstede, H.J.; de Graaff-Teulen, M.J.; van Ewijk-Beneken Kolmer, E.W.; Caliskan-Yassen, N.; Koopmans, P.P.; PharmD, D.M.B. Lopinavir/ritonavir reduces lamotrigine plasma concentrations in healthy subjects. Clin. Pharmacol. Ther. 2006, 80, 159–168. [Google Scholar] [CrossRef]
- Doose, D.R.; Brodie, M.J.; Wilson, E.A.; Chadwick, D.; Oxbury, J.; Berry, D.J.; Schwabe, S.; Bialer, M. Topiramate and lamotrigine pharmacokinetics during repetitive monotherapy and combination therapy in epilepsy patients. Epilepsia 2003, 44, 917–922. [Google Scholar] [CrossRef]
- Reimers, A.; Helde, G.; Brathen, G.; Brodtkorb, E. Lamotrigine and its N2-glucuronide during pregnancy: The significance of renal clearance and estradiol. Epilepsy Res. 2011, 94, 198–205. [Google Scholar] [CrossRef]
- Reisinger, T.L.; Newman, M.; Loring, D.W.; Pennell, P.B.; Meador, K.J. Antiepileptic drug clearance and seizure frequency during pregnancy in women with epilepsy. Epilepsy Behav. 2013, 29, 13–18. [Google Scholar] [CrossRef]
- Pennell, P.B.; Peng, L.; Newport, D.J.; Ritchie, J.C.; Koganti, A.; Holley, D.K.; Newman, M.; Stowe, Z.N. Lamotrigine in pregnancy: Clearance, therapeutic drug monitoring, and seizure frequency. Neurology 2008, 70 Pt 2, 2130–2136. [Google Scholar] [CrossRef]
- Ding, Y.; Tan, X.; Zhang, S.; Guo, Y. Pharmacokinetic changes and therapeutic drug monitoring of lamotrigine during pregnancy. Brain Behav. 2019, 9, e01315. [Google Scholar] [CrossRef]
- Fotopoulou, C.; Kretz, R.; Bauer, S.; Schefold, J.; Schmitz, B.; Dudenhausen, J.W.; Henrich, W. Prospectively assessed changes in lamotrigine-concentration in women with epilepsy during pregnancy, lactation and the neonatal period. Epilepsy Res. 2009, 85, 60–64. [Google Scholar] [CrossRef]
- Wenning, L.A.; Hanley, W.D.; Brainard, D.M.; Petry, A.S.; Ghosh, K.; Jin, B.; Mangin, E.; Marbury, T.C.; Berg, J.K.; Chodakewitz, J.A.; et al. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 2009, 53, 2852–2856. [Google Scholar] [CrossRef]
- Iwamoto, M.; Wenning, L.A.; Petry, A.S.; Laethem, M.; De Smet, M.; Kost, J.T.; Breidinger, S.A.; Mangin, E.C.; Azrolan, N.; Greenberg, H.E.; et al. Minimal effects of ritonavir and efavirenz on the pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 2008, 52, 4338–4343. [Google Scholar] [CrossRef]
- Iwamoto, M.; Wenning, L.; Petry, A.; Laethem, M.; De Smet, M.; Kost, J.; Merschman, S.; Strohmaier, K.; Ramael, S.; Lasseter, K.; et al. Safety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjects. Clin. Pharmacol. Ther. 2008, 83, 293–299. [Google Scholar] [CrossRef]
- Wang, L.; Soon, G.H.; Seng, K.-Y.; Li, J.; Lee, E.; Yong, E.-L.; Goh, B.-C.; Flexner, C.; Lee, L. Pharmacokinetic modeling of plasma and intracellular concentrations of raltegravir in healthy volunteers. Antimicrob. Agents Chemother. 2011, 55, 4090–4095. [Google Scholar] [CrossRef]
- Blonk, M.; Colbers, A.; Poirters, A.; Schouwenberg, B.; Burger, D. Effect of ginkgo biloba on the pharmacokinetics of raltegravir in healthy volunteers. Antimicrob. Agents Chemother. 2012, 56, 5070–5075. [Google Scholar] [CrossRef]
- Taburet, A.-M.; Sauvageon, H.; Grinsztejn, B.; Assuied, A.; Veloso, V.; Pilotto, J.H.; De Castro, N.; Grondin, C.; Fagard, C.; Molina, J.-M. Pharmacokinetics of raltegravir in HIV-Infected Patients on Rifampicin-Based Antitubercular Therapy. Clin. Infect. Dis. 2015, 61, 1328–1335. [Google Scholar] [CrossRef]
- Hanley, W.D.; Wenning, L.A.; Moreau, A.; Kost, J.T.; Mangin, E.; Shamp, T.; Stone, J.A.; Gottesdiener, K.M.; Wagner, J.A.; Iwamoto, M. Effect of tipranavir-ritonavir on pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 2009, 53, 2752–2755. [Google Scholar] [CrossRef]
- Weiner, M.; Egelund, E.F.; Engle, M.; Kiser, M.; Prihoda, T.J.; Gelfond, J.A.L.; Mac Kenzie, W.; Peloquin, C.A. Pharmacokinetic interaction of rifapentine and raltegravir in healthy volunteers. J. Antimicrob. Chemother. 2014, 69, 1079–1085. [Google Scholar] [CrossRef]
- Rhee, E.G.; Rizk, M.L.; Brainard, D.M.; Gendrano, I.N., 3rd; Jin, B.; Wenning, L.A.; Wagner, J.A.; Iwamoto, M. A pharmacokinetic comparison of adult and paediatric formulations of raltegravir in healthy adults. Antivir. Ther. 2014, 19, 619–624. [Google Scholar] [CrossRef]
- Rizk, M.L.; Hang, Y.; Luo, W.-L.; Su, J.; Zhao, J.; Campbell, H.; Nguyen, B.-Y.T.; Sklar, P.; Eron, J.J.; Wenning, L. Pharmacokinetics and pharmacodynamics of once-daily versus twice-daily raltegravir in treatment-naive HIV-infected patients. Antimicrob. Agents Chemother. 2012, 56, 3101–3106. [Google Scholar] [CrossRef]
- Markowitz, M.; O Morales-Ramirez, J.; Nguyen, B.-Y.; Kovacs, C.M.; Steigbigel, R.T.; A Cooper, D.; Liporace, R.; Schwartz, R.; Isaacs, R.; Gilde, L.R.; et al. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 2006, 43, 509–515. [Google Scholar] [CrossRef]
- Andrews, E.; Glue, P.; Fang, J.; Crownover, P.; Tressler, R.; Damle, B. Assessment of the pharmacokinetics of co-administered maraviroc and raltegravir. Br. J. Clin. Pharmacol. 2010, 69, 51–57. [Google Scholar] [CrossRef]
- Brainard, D.M.; Friedman, E.J.; Jin, B.; Breidinger, S.A.; Tillan, M.D.; Wenning, L.A.; Stone, J.A.; Chodakewitz, J.A.; Wagner, J.A.; Iwamoto, M. Effect of low-, moderate-, and high-fat meals on raltegravir pharmacokinetics. J. Clin. Pharmacol. 2011, 51, 422–427. [Google Scholar] [CrossRef]
- Watts, D.H.; Stek, A.; Best, B.M.; Wang, J.; Capparelli, E.V.; Cressey, T.R.; Aweeka, F.; Lizak, P.; Kreitchmann, R.; Burchett, S.K.; et al. Raltegravir pharmacokinetics during pregnancy. J. Acquir. Immune Defic. Syndr. 2014, 67, 375–381. [Google Scholar] [CrossRef]
- Blonk, M.I.; Colbers, A.P.; Hidalgo-Tenorio, C.; Kabeya, K.; Weizsäcker, K.; Haberl, A.E.; Moltó, J.; Hawkins, D.A.; van der Ende, M.E.; Gingelmaier, A.; et al. Raltegravir in HIV-1-Infected Pregnant Women: Pharmacokinetics, Safety, and Efficacy. Clin. Infect. Dis. 2015, 61, 809–816. [Google Scholar] [CrossRef]
- Zheng, Y.; Hirt, D.; Delmas, S.; Lui, G.; Benaboud, S.; Lechedanec, J.; Tréluyer, J.-M.; Chenevier-Gobeaux, C.; Arezes, E.; Gelley, A.; et al. Effect of Pregnancy on Unbound raltegravir Concentrations in the ANRS 160 RalFe Trial. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef]
- Polepally, A.R.; Pennell, P.B.; Brundage, R.C.; Stowe, Z.N.; Newport, D.J.; Viguera, A.C.; Ritchie, J.C.; Birnbaum, A.K. Model-Based lamotrigine clearance changes during pregancy: Clinical implication. Ann. Clin. Transl. Neurol. 2014, 1, 99–106. [Google Scholar] [CrossRef]
- Conner, T.M.; Reed, R.C.; Zhang, T. A Physiologically Based Pharmacokinetic Model for Optimally Profiling lamotrigine Disposition and Drug-Drug Interactions. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 389–408. [Google Scholar] [CrossRef]
- Chen, H.; Yang, K.; Choi, S.; Fischer, J.H.; Jeong, H. Up-regulation of UDP-glucuronosyltransferase (UGT) 1A4 by 17beta-estradiol: A potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab. Dispos. 2009, 37, 1841–1847. [Google Scholar] [CrossRef]
- Chaphekar, N.; Dodeja, P.; Shaik, I.H.; Caritis, S.; Venkataramanan, R. Maternal-Fetal Pharmacology of Drugs: A Review of Current Status of the Application of Physiologically Based Pharmacokinetic Models. Front. Pediatr. 2021, 9, 733823. [Google Scholar] [CrossRef]
- Chen, S.; Yueh, M.F.; Evans, R.M.; Tukey, R.H. Pregnane-x-receptor controls hepatic glucuronidation during pregnancy and neonatal development in humanized UGT1 mice. Hepatology 2012, 56, 658–667. [Google Scholar] [CrossRef]
- Sychterz, C.; Galetin, A.; Taskar, K.S. When special populations intersect with drug-drug interactions: Application of physiologically-based pharmacokinetic modeling in pregnant populations. Biopharm. Drug Dispos. 2021, 42, 160–177. [Google Scholar] [CrossRef]
- Petrenaite, V.; Öhman, I.; Ekström, L.; Sæbye, D.; Hansen, T.F.; Tomson, T.; Sabers, A. UGT polymorphisms and lamotrigine clearance during pregnancy. Epilepsy Res. 2018, 140, 199–208. [Google Scholar] [CrossRef]
- Wang, M.-L.; Tao, Y.-Y.; Sun, X.-Y.; Guo, Y.; Wang, Z.-Y.; Cao, Y.-F.; Zhao, L. Estrogen profile- and pharmacogenetics-based lamotrigine dosing regimen optimization: Recommendations for pregnant women with epilepsy. Pharmacol. Res. 2021, 169, 105610. [Google Scholar] [CrossRef]
- Wegner, I.; Wilhelm, A.J.; Lambrechts, D.A.J.E.; Sander, J.W.; Lindhout, D. Effect of oral contraceptives on lamotrigine levels depends on comedication. Acta Neurol. Scand. 2014, 129, 393–398. [Google Scholar] [CrossRef]






| Absorption | Increased gastric pH ↑ |
| Increased cardiac output ↑ | |
| Decreased intestinal motility ↓ | |
| Nausea/Vomiting | |
| Distribution | Increased plasma volume ↑ |
| Increased body water and fat ↑ | |
| Decreased plasma protein concentration ↓ | |
| Metabolism | Increased CYP3A4 activity ↑ |
| Increased CYP2B6 ↑ | |
| Increased CYP2D6 ↑ | |
| Increased CYP2E1 ↑ | |
| Increased CYP2C8 ↑ | |
| Increased CYP2C9 ↑ | |
| Decreased CYP2C19 ↓ | |
| Decreased CYP1A2 ↓ | |
| Increased UGT1A1 ↑ | |
| Increased UGT1A4 ↑ | |
| Increased UGT2B7 ↑ | |
| Elimination | Increased glomerular filtration rate ↑ |
| Increased renal P-gp, OATs, OCTs and MATE transport ↑ |
| Drug | Population | Start at
Gestational Week | Dosing Schedule | Dose Level (mg) | Clinical Trial Reference | n | Data Used for Validation |
|---|---|---|---|---|---|---|---|
| Lamotrigine | Non-pregnant | - | SD | 25 | Ebert et al. [23] | 10 | Graphical and numerical |
| 100 | Birnbaum et al. [24] | 12 | |||||
| Srichaiya et al. [25] | 24 | ||||||
| Van Luin et al. [26] | 24 | ||||||
| Keränen et al. [27] | 6 | ||||||
| 200 | Hermann et al. [28] | 15 | |||||
| Wootton et al. [29] | 12 | ||||||
| Incecayir et al. [30] | 14 | ||||||
| QD | 50 | Jann et al. [31] | 14 | ||||
| 150 | Almeida et al. [32] | 32 | |||||
| 200 | Sidhu et al. [33] | 12 | |||||
| BID | 50 | Chien et al. [34] | 24 | ||||
| 100 | Colluci et al. [35] | 21 | |||||
| Van der Lee et al. [36] | 8 | ||||||
| 150 | Doose et al. [37] | 13 | |||||
| Pregnant | 0 | BID | 400 | Reimers et al. [38] | 19 | Numerical | |
| 36 | |||||||
| 0 | 406.54 | Reisinger et al. [39] | 69 | ||||
| 10 | 426.81 | Pennell et al. [40] | 14 | ||||
| 20 | 564.90 | Ding et al. [41] | 12 | ||||
| 36 | 664.49 | Fotopoulou et al. [42] | 9 | ||||
| 0 | 350 | Fotopoulou et al. [43] | 9 | ||||
| 40 | 600 | ||||||
| Raltegravir | Non-pregnant | - | SD | 400 | Wenning et al. [44] | 10 | Graphical |
| Iwamoto a et al. [45] | 10 | ||||||
| Iwamoto b et al. [46] | 6 | ||||||
| Wang et al. [47] | 6 | Graphical and numerical | |||||
| Blonk et al. [48] | 18 | ||||||
| BID | Taburet et al. [48] | 21 | |||||
| Hanley et al. [49] | 15 | ||||||
| Weiner et al. [50] | 21 | ||||||
| SD | Rhee et al. [51] | 10 | Numerical | ||||
| QD | Rizk et al. [52] | 22 | |||||
| BD | Markowitz et al. [53] | 8 | |||||
| Andrews et al. [54] | 18 | ||||||
| Brainard et al. [55] | 20 | ||||||
| Pregnant | 0 | BID | 400 | Watts et al. [56] | 16 | Graphical and numerical | |
| 21 | 41 | ||||||
| 34 | 38 | ||||||
| 0 | Blonk et al. [57] | 22 | Numerical | ||||
| 34 | |||||||
| 0 | Zheng et al. [58] | 43 | |||||
| 34 |
| Parameter | Lamotrigine | Raltegravir |
|---|---|---|
| Phys Chem And Blood Biding | ||
| Molecular weight (g/mol) | 256.09 | 444.42 |
| LogO:W | 1.19 | 1.07 |
| Molecule type | Monoprotic base | Monoprotic acid |
| pKa | 5.5 | 6.7 |
| B/P | 1 | 0.62 |
| Fu | 0.45 | 0.17 |
| Absorption | ||
| fa | 0.99915 (Predicted) | 1 |
| Fugut | 1 (User input) | 1 |
| ka(1/h) | 3.3889 (Predicted) | 0.4 |
| Lag time (h) | 0.3 | |
| Peff,man (10−4 cm/s) (Predicted) | 7.761 | 2.0864 |
| Caco-2 (10−6 cm/s) 6.5:7:4 P&A | 73.7 | 9.2 |
| Distribution | ||
| Full PBPK | ||
| Vss (L/kg) (Predicted—Method 2) | 1.0575 | 0.1452 |
| Kp scalar | 2.2 | 1 |
| Elimination | ||
| Enzyme kinetics | ||
| Enzyme | UGT1A4 | UGT1A1 |
| CLint (pmol/min/pmol of isoform) | 1.48 | |
| Vmax (pmol/min/pmol of isoform) | 153 | |
| Km (µM) | 550 | |
| Fumic | 1 | 1 |
| ISEF | 0.077 | 1 |
| Enzyme | UGT1A3 | |
| Vmax (pmol/min/pmol of isoform) | 17 | |
| Km (µM) | 700 | |
| Fumic | 1 | |
| ISEF | 0.077 | |
| CLR (L/h) | 0.2 | 3.3 |
| Baseline | 1st Trimester | 2nd Trimester | 3rd Trimester | 3rd Trim/Baseline %Change | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pred. | Obsv. | Pred. | Obsv. | Pred. | Obsv. | Pred. | Obsv | Pred. | Obsv. | |
| Cmax (mg/L) [38] | 9.97 | 14.5 ± 9.2 | - | - | - | - | 4.95 | 5.3 ± 4.3 | −63% | −50% |
| AUC (mg/Lxh) [38] | 187.2 | 137.3 ± 66.4 | - | - | - | - | 79.4 | 43.4 ± 34.2 | −68% | −58% |
| CL (mg/(mg/L)) [39,40,41,42] | 40.6 | 50.75 | 82.66 | 78.89 | 115.05 | 120.19 | 144.45 | 124.20 | +256% | +164–224% |
| 39 | 77 | 92 | 103 | |||||||
| 52.9 | 88.5 | 132.5 | 171.2 | |||||||
| 40.3 | 73.9 | 122.2 | 120.5 | |||||||
| Baseline | 2nd Trimester | 3rd Trimester | 2nd Trim/Baseline %Change | 3rd Trim/Baseline %Change | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pred. | Obsv. | Pred. | Obsv. | Pred. | Obsv. | Pred. | Obsv | Pred. | Obsv. | |
| Cmax (mg/L) | 2.23 | 3.04 | 1.42 | 2.25 | 1.06 | 1.77 | −36% | −25% | −52% | −42% |
| 1.76 | 1.43 | |||||||||
| AUC (mg/L h) | 8.65 | 11.6 | 5.36 | 6.6 | 4.00 | 5.4 | −38% | −43% | −53% | −53% |
| 7.11 | 5 | |||||||||
| 6.77 | 3.92 | |||||||||
| 9.29 | 5.36 | |||||||||
| CL (L/h) | 61.06 | 34.8 | 100.81 | 60 | 139.21 | 74.8 | +65% | +72% | +128% | +115% |
| 56.2 | 80.1 | |||||||||
| 53 | 102 | |||||||||
| Lamotrigine | Raltegravir | |||
|---|---|---|---|---|
| AFE | AAFE | AFE | AAFE | |
| Cmax a | 1.03 | 1.07 | 1.05 | 1.17 |
| AUC a | 0.99 | 1.13 | 1.06 | 1.18 |
| CL p | 1.02 | 1.11 | - | - |
| AUC p | - | - | 0.82 | 1.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berezowska, M.; Coppola, P.; Pilla Reddy, V.; Sharma, P. Physiologically Based Pharmacokinetic Modelling of UGT Substrate Drugs Lamotrigine and Raltegravir during Pregnancy. Future Pharmacol. 2024, 4, 317-335. https://doi.org/10.3390/futurepharmacol4020018
Berezowska M, Coppola P, Pilla Reddy V, Sharma P. Physiologically Based Pharmacokinetic Modelling of UGT Substrate Drugs Lamotrigine and Raltegravir during Pregnancy. Future Pharmacology. 2024; 4(2):317-335. https://doi.org/10.3390/futurepharmacol4020018
Chicago/Turabian StyleBerezowska, Monika, Paola Coppola, Venkatesh Pilla Reddy, and Pradeep Sharma. 2024. "Physiologically Based Pharmacokinetic Modelling of UGT Substrate Drugs Lamotrigine and Raltegravir during Pregnancy" Future Pharmacology 4, no. 2: 317-335. https://doi.org/10.3390/futurepharmacol4020018