Abstract
Purpose: Pharmaceutical parenteral drug products (PDPs) and orally inhaled nasal drug products (OINDPs) are critical medications for patient care, for which the route of administration is intravenous or oral/nasal inhalation, and the drug products directly infuse into the bloodstream or lungs, but they are categorized as high-risk for leachables. Method: These external foreign chemical substances (leachables) may adversely affect and alter patient safety. Results: These primary container closure systems and manufacturing process equipment mainly comprise rubber elastomers, polypropylene, resin, ink, adhesives, glass, or plastic material. To establish the ID of detected compounds and their quantity in the finished parenteral drug formulation and then to assess the formulation for toxicological safety, broad-scope non-specific analytical screening methods are required that are capable of screening out and quantifying the predicted/unpredicted leachable compounds at the levels that pose anticipated toxicological concerns for human patients. Before the selection of the final primary packaging system for the parenteral drug product, their extractable screening profile/knowledge is required to minimize leachable compounds in the finished drug product formulation and to develop and manufacture a safe product for human patients. The adverse effect or toxicity of leachables proportionally increases with an increase in the dose of the drug product or the duration of therapy because the volume of the drug product administered to a patient in a larger quantity is directly proportional to the concentration of the detected leachable. Conclusion: This document outlines the detailed process/scientific approach for conducting an organic leachable screening profile for parenteral drug products with respect to the chemical nature of leachables, i.e., polarity, propensity, volatility, and techniques.
1. Introduction
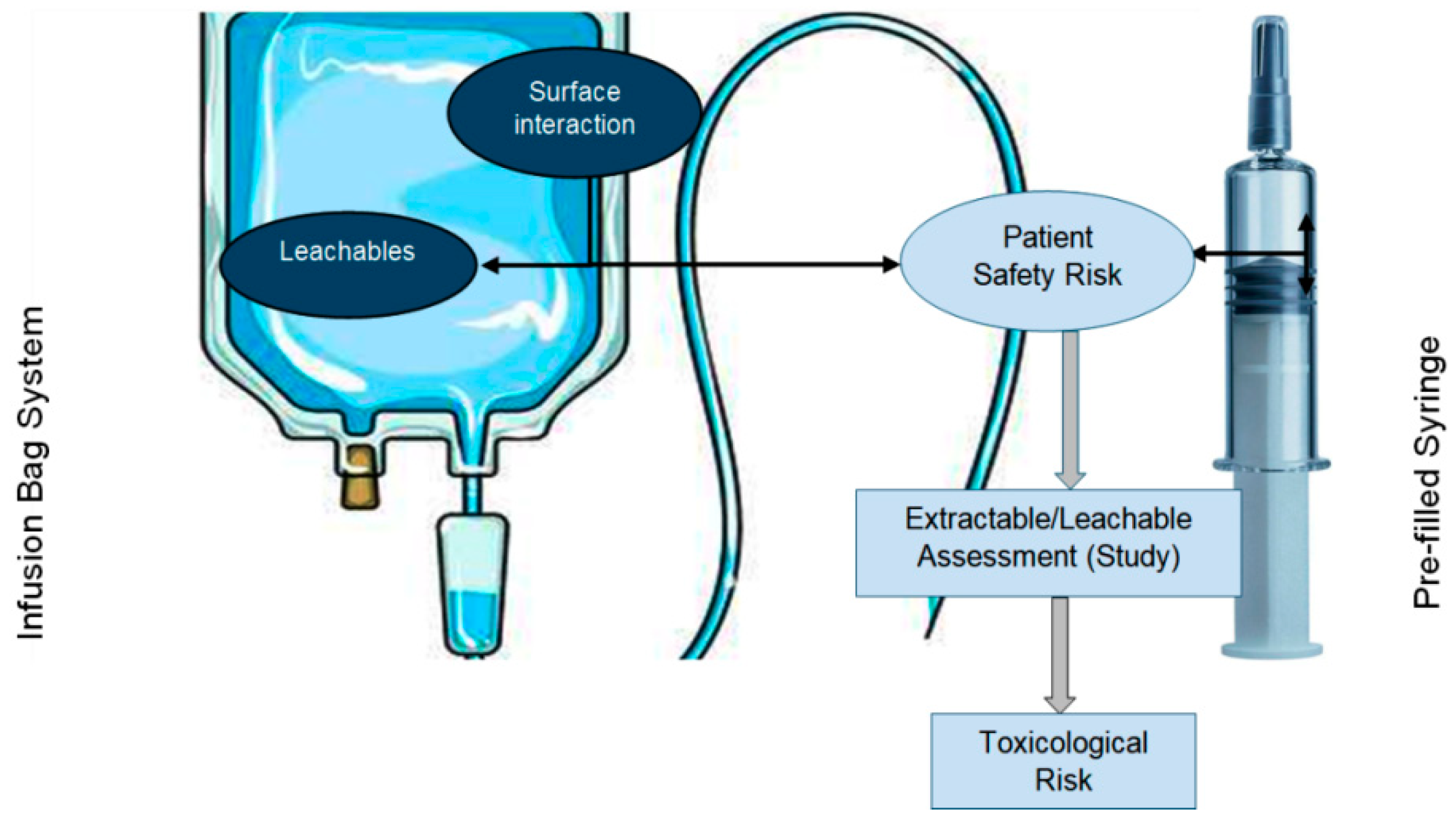
Leachables are a class of chemical compounds that can transfer or migrate from manufacturing process equipment, packaging (Figure 1), or medication delivery systems to a finished liquid formulation due to direct contact during normal storage conditions during the product’s whole shelf life [1]. In drug products, leachables may arise from components that are used to package drug products, delivery systems, and manufacturing process systems/equipment and may compromise the quality of drug products and directly impact or alter patient safety. When studying the stability and storage of drug product samples in terms of leachables, the orientation of the packaging system should be in a position where very-high-risk materials like plastic, rubber, etc., avoid contact with the drug product as much as possible (e.g., a vial in an inverted orientation and a pre-filled syringe in a horizontal orientation). Because of the liquid form of drug products, there is a higher chance that the liquid product will interact with the packaging system and that some chemical moieties will leach out into the drug product, which may affect patient safety due to the packaging system being made from plastic or rubber materials. USP general chapter <1664>categorizes risk based on the formulation and the route of administration. Extractable are chemical compounds that are purposefully extracted from manufacturing process equipment or materials, packaging container closure systems (CCSs), delivery systems, or medical devices in the presence of model solvent systems under controlled laboratory extraction conditions, including controlled-temperature and controlled-duration conditions, in order to characterize the material of construction of these systems, which helps in selecting good-quality packaging systems or manufacturing processing materials, thus reducing the risk of leachables [2].
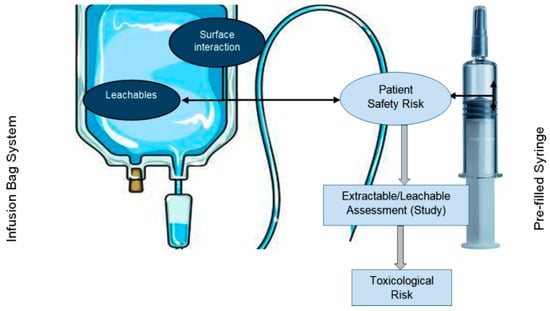
Figure 1.
Material drug product interaction leading to leachables and patient safety risks.
2. Materials and Methods
2.1. Sources and Methodology
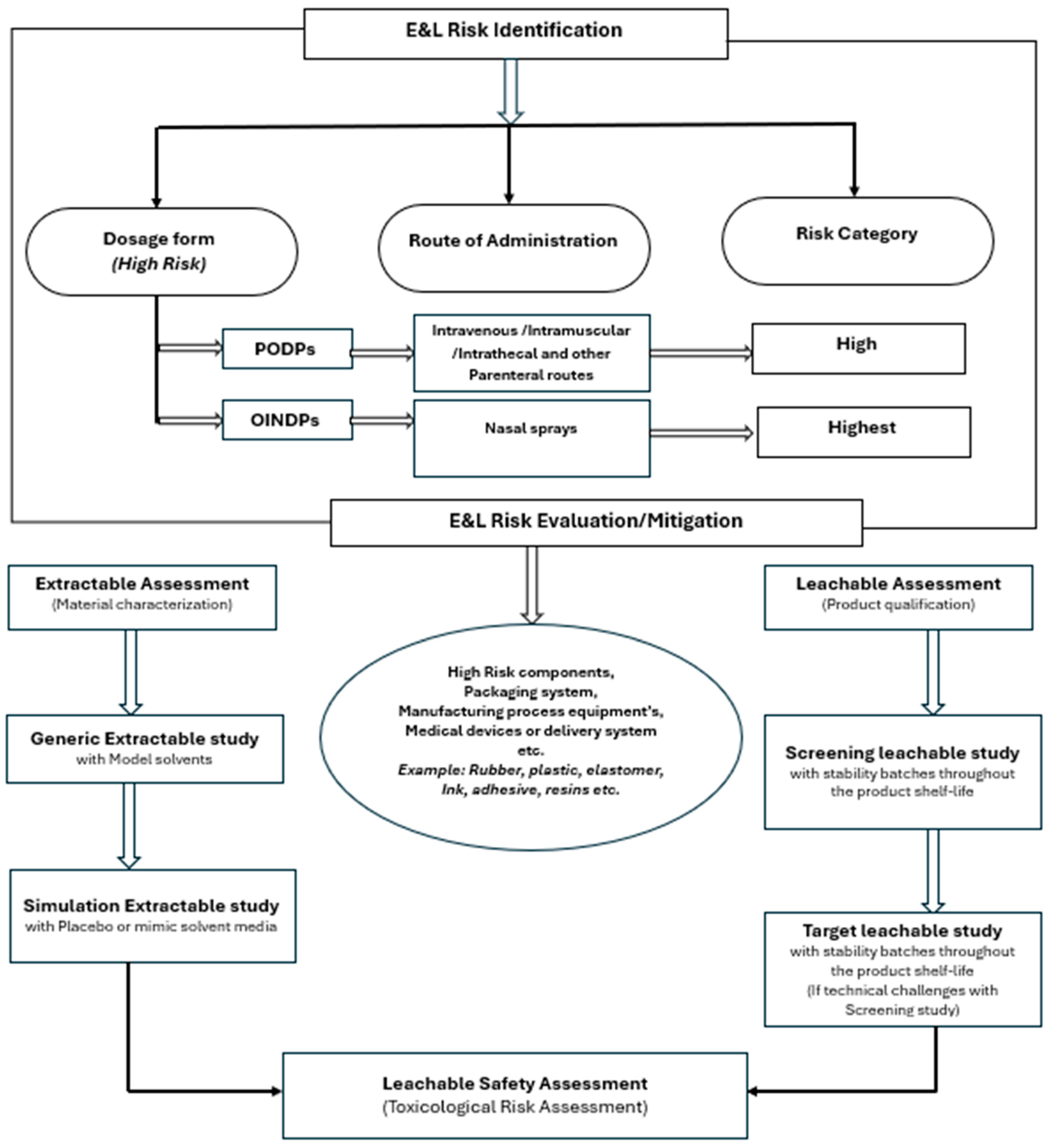
References and research-related data were collected via the U.S. Pharmacopeial Convention and different open- or controlled-access sites and regulatory guidance documents. The search terms were as follows: leachable, OINDPs, container closure systems, and pharmaceutical toxicity. All the relevant data were compiled in a table, and the methods used to evaluate the leachable screening studies for pharmaceutical and parenteral drug products were underpinned by scientific research (Scheme 1).
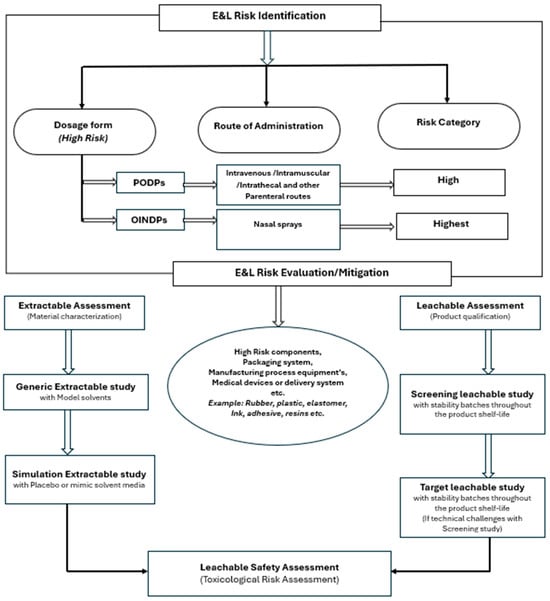
Scheme 1.
Schematic diagram of risk identification/evaluation and mitigation for extractable and leachable compounds for pharmaceutical drug products.
2.2. Leachable Study Design
The following were the types of study designs that were employed: studies that performed a leachable screening study of the finished liquid product or formulation and studies that assessed the toxicology of products if any chemical moiety leached out in the product above the threshold that could affect patient safety.
2.3. Leachable Screening Study
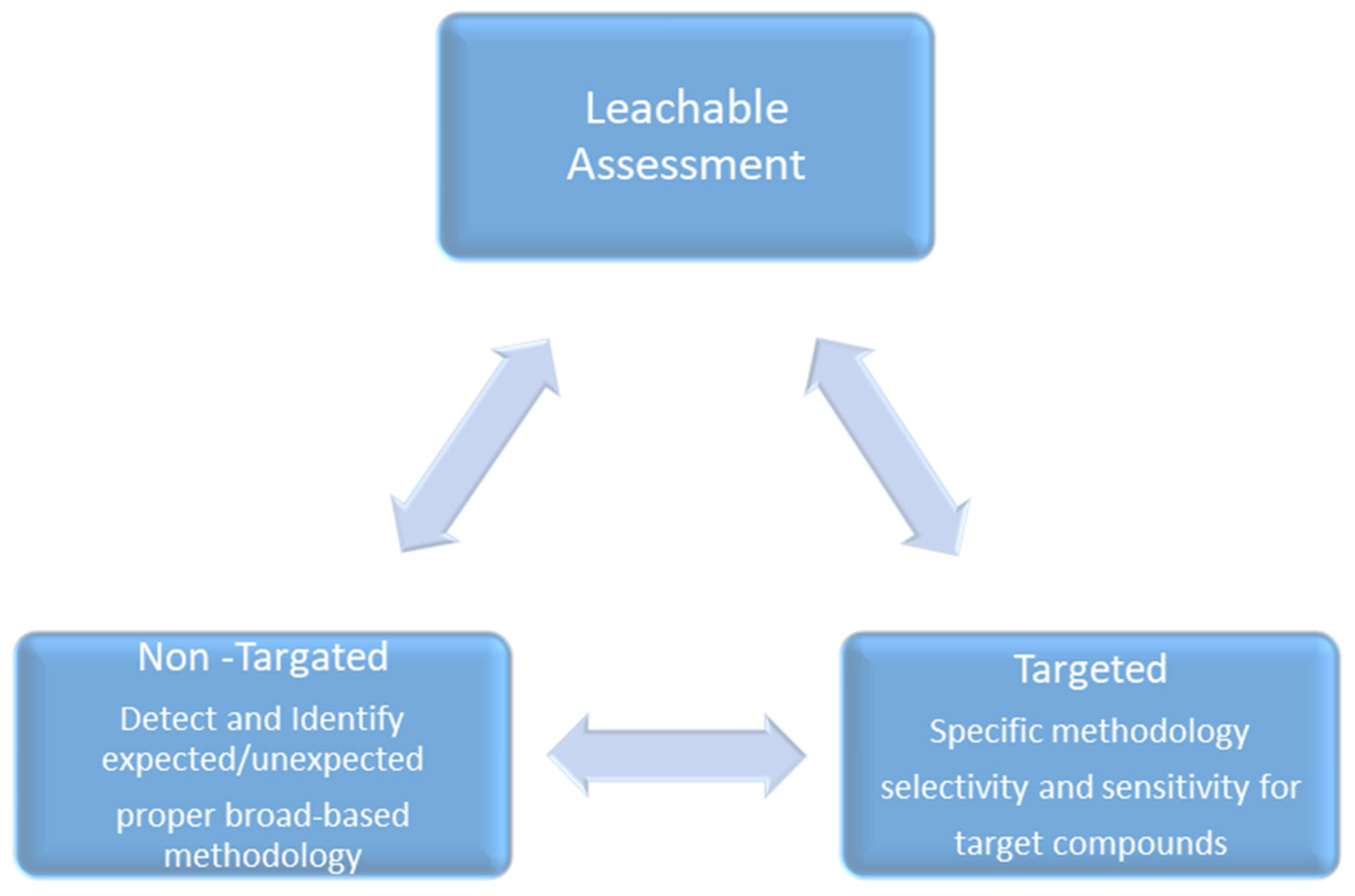
To address the patient safety risk caused by any expected/unpredicted leachable from a container closure system, manufacturing process equipment, or delivery system, a leachable screening profile is the best scientifically justified approach for screening out and quantifying all expected/unexpected leachables from a finished drug product that may not have previously been detected as extractable. The leachable screening study approach is capable of detecting and quantifying all leachable moieties that may leach out from a material, like a packaging system, manufacturing system, or medical device, into the finished drug product, even in trace amounts, which helps in preventing the underestimation of patient safety risk (Figure 2).
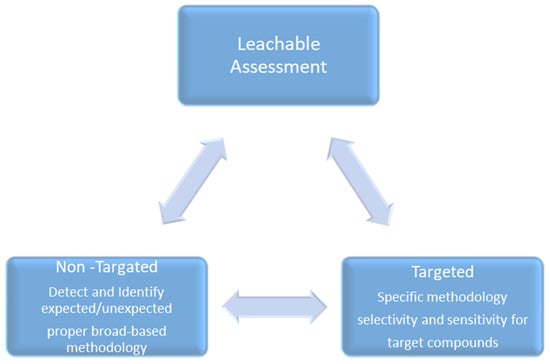
Figure 2.
Holistic leachable-risk-based approaches.
2.4. Target Leachable Study
The target leachable analytical method gives only precise information about the expected selected target compounds that are potentially detected in an extractable study. This approach helps when drug products have complex matrices, such as oil-based emulsions and glycol-based or non-aqueous formulations. Then, a leachable study can be designed for only the selected target chemical compound that is detected in a respective container closure extractable study.
2.5. Leachable Strategy
To address the patient safety risk concerning leachables from parenteral or orally inhaled nasal drug products, the following are stepwise approaches that are used to screen out the drug product and to conclude that the product is safe for human patients and that there is no risk from the product manufacturing process and/or packaging system:
- ⮚
- Derivation of the analytical evaluation threshold (AET) concerning the drug product’s maximum daily dose and the safety concern threshold (SCT) with respect to the route of administration.
- ⮚
- Analytical instrumentation and methodologies for the determination of the highly volatile nature, semi-volatile nature, and non-volatile nature of organic leachable species.
- ⮚
- Leachable screening method development and verification using published references like ICH.
- ⮚
- Leachable screening study on the finished drug product using registration stability batches (batches manufactured with the same process, such as those intended for commercial use).
- ⮚
- Data interpretation and reporting of observed leachable compounds.
- ⮚
- Toxicological risk assessment for the detected leachables if they are found at or above the safety or analytical threshold.
3. Results
3.1. Derivation of the Analytical Evaluation Threshold (AET) for Drug Product Total Maximum Daily Dose
Definition: The analytical evaluation threshold (AET) is the concentration of an organic extractable or leachable that is less than that which is unlikely to present any significant safety concern [3,4]. The AET is derived from the allowable maximum daily dose (MDD) of the drug formulation and the lowest toxicological threshold that addresses the risk of sensitization/irritation, mutagenicity, and general toxicity. The guidance given in ICH M7 states that a TTC (threshold of toxicological concern) of 120 µg/day is appropriate for addressing the risk of genotoxic compounds in drug products that are dosed for less than 1 month [5]. This is higher than the qualification threshold (QT), which is recommended in the safety guidance by issued PQRI for parenteral drug products to address the risk of sensitizers/irritants. As such, consistent with the statements in the PQRI PDP guidance document for acutely dosed drug products, the AET should be calculated based on an appropriate toxicological threshold of 5 µg/day. Additionally, in ICH M7, it is stated that if the duration of therapy is more than 10 years or the product is taken for the patient’s whole life, the TTC-based acceptable intake for multiple mutagenic impurities of 5 µg/day is appropriate. The main challenge for a leachable study is to achieve a very low AET level for large-volume parenteral drug products (LVPs), as the AET depends on the dosing regimen of the product. Scientifically, an alternate approach to achieve the AET is to enrich the sample concentration to the desired AET, but a limitation of this method comes from the sample folding and the matrix concentration increasing, which may create further instrumental challenges for sample application to high-end hyphenated techniques. To overcome these challenges, the drug product sample can be extracted with a suitable solvent with a proven recovery.
3.2. AET Calculation
The estimated leachable AET can be determined as follows, in which a 200 mg MDD, 10 mg/mL product strength, and 10 mL fill volume of CCS is assumed:
UF = Uncertainty factor
3.3. Instrumentation and Methodologies
For leachable screening studies, broad-scope non-specific analytical screening methods with very high sensitivity at or below the AET level are required to screen out all the expected and/or unexpected leachable chemical compounds from the finished drug product. These broad-scope non-specific analytical screening methods have been proven to be able to detect and quantify a diverse range of chemical compounds at or below the AET level.
The following broad-scope non-specific analytical screening methods can be used to establish volatile, semi-volatile, and non-volatile leachable screening profiles for parenteral drug products.
These screening methods are semi-quantitative, as we do not know what leachable compounds may leach out in the drug product from the packaging, delivery system, and/or manufacturing process materials (Table 1). The quantitation can be carried out in either of the following ways:

Table 1.
Pros and cons of leachable screening and target studies.
External system suitability standards can be obtained from existing knowledge of the packaging, delivery system, and manufacturing contact material extractable data (potential extractable), and a group of selected compounds can collectively be used as the system suitability standard for the method (without the sample matrix), and the same can be used to calculate the leachable quantity in the drug product after determining the method’s suitability for those standards in the drug product (recovery) and the use recovery factor during the final calculation of the leachables for the external method.
3.4. The Internal Standard Method
In this method, an internal standard (IS) is spiked into the drug product matrix, and the response factor (RF) is the area of the IS in the matrix and the concentration of the IS, and this RF is then used to calculate the quantity of any leachables.
Volatile organic compounds can be screened out by head space gas chromatography and mass spectrometry, and semi-volatile organic compounds can be screened out by gas chromatography–mass spectrometry using an electron impact (EI) ionization source because it has a high electronic energy (70 eV), which breaks organic molecules into ions, and these ions then transfer towards the MS detector and give a signal related to the analyte. The mass range for volatile and semi-volatile compounds can be selected from 40 m/z to 700 m/z. This range covers all the ranges of volatile compounds to semi-volatile. A mass range below 40 m/zi is not recommended scientifically, as there is a lot of interference from atmospheric gases like nitrogen, carbon dioxide, and oxygen.
The screening of non-volatile organic compounds is carried out by ultra-high-performance liquid chromatography with a mass spectrometer (UPLCMS) (Table 2). Ion sources can be selected based on the scientific justification for non-polar organic compounds, and atmospheric-pressure chemical ionization (APCI) and electro spray ionization (ESI) can be used for polar-to-mid-polar organic compounds. The mass range can be selected from 100 m/z to 1300 m/z, which covers the full range of polar-to-non-polar compounds.
Table 2.
Screening leachable study techniques.
3.5. Leachable Screening Method Development and Verification
Patient safety risks from leachables that are transferred from packaging systems, delivery systems, and manufacturing process equipment [6,7,8,9] into the finished drug product are established using a broad-scope non-specific analytical screening method. The terms broad-scope and non-specific mean that the leachable method is not specific to expected or predicted compounds. This method is also applicable to detecting and quantifying leachable compounds that were not predicted or were not detected previously in an extractable study.
The profiles of volatile organic leachable compounds by HS/GC-MS, semi-volatile leachable compounds by direct-injection GC/MS, and non-volatile organic leachable compounds by UPLCMS (Table 2), the following steps are used:
Separate generic screening methods (not product-specific) for volatile, semi-volatile, and non-volatile organic compounds are developed for solvents with various polarities and propensities.
The proper pH range for the aqueous solvent media is selected to mimic the aqueous-based drug product chemical profile. Leachable screening methods are developed and verified by using these mimic solvent media and non-specific screening methods to generate a leachable screening profile of the aqueous-based drug product (Table 3) [10].
Table 3.
Selection of mimic solvent media for generic method development.
Drug products contain some extended organic phases, and organic solvent media with different polarities can be used to simulate the chemical properties of organic-based drug products, and the leachable screening method is developed and verified by using these mimic solvent media.
The following are the proposed solvent media using a generic leachable method for development and verification covering aqueous and/or organic-based drug products, which have varying chemical natures and properties (e.g., pH, polarity).
The choice of the system suitability standard depends on the chemicals or their compositions used to manufacture drug product packaging systems, delivery systems, or manufacturing materials like tubing and filters [11,12,13,14,15,16]. Standard mixes are used to analyze the leachable screening method’s suitability to facilitate consistent analytical method performance through the sample sequence, and they are also used to calculate the amount of leachables present in the drug product, respectively. The leachable screening methods are verified at the AET or below the AET level by using standard mixes to prove that these methods are fit to propose screening out if any expected and/or unexpected chemical compounds from the CCS or manufacturing processing materials leach out in the drug product.
During the development of the system, suitability standards spike into the mimic solvent media at as low as possible a concentration to detect the response, adjust the sample preparation, and optimize the method parameters to obtain as low as possible a response and separate each standard from others.
After the screening, the method developed and the system suitability parameters concerning the techniques, solvent media, and method are verified using the following minimum parameters:
- ⮚
- Specificity;
- ⮚
- Limit of detection and limit of quantitation;
- ⮚
- Linearity;
- ⮚
- Method precision;
- ⮚
- Method accuracy.
3.6. Leachable Screening Study
After the verification of generic screening methods at or below the AET level concerning the solvent media techniques, they can be used directly for the study of the leachables in the finished paracentral drug product with a respective product matrix and polarity. For example, if the product is acidic, then a water-based media-specific generic method at pH 2.5 can be used directly by performing a minimum method compatibility/suitability (accuracy) test with a control drug product (same formulation prepared without the contact of polymeric parts and filled using a method other than that with the final packaging system) to analyze the accuracy at the desired AET level or below the AET level followed by a leachable screening study (Table 4).
Table 4.
Leachable screening study minimum requirements.
In the study of the system’s accuracy, the suitability and internal standards are spiked into the control drug product at or below the AET level, which may be up to 50% below the AET level, considering the method uncertainty (UF). After determining the method’s suitability concerning the drug product, a leachable screening study is performed on the finished drug product formulation to establish the patient safety risk due to the leachable throughout the product’s shelf-life using the following minimum requirements.
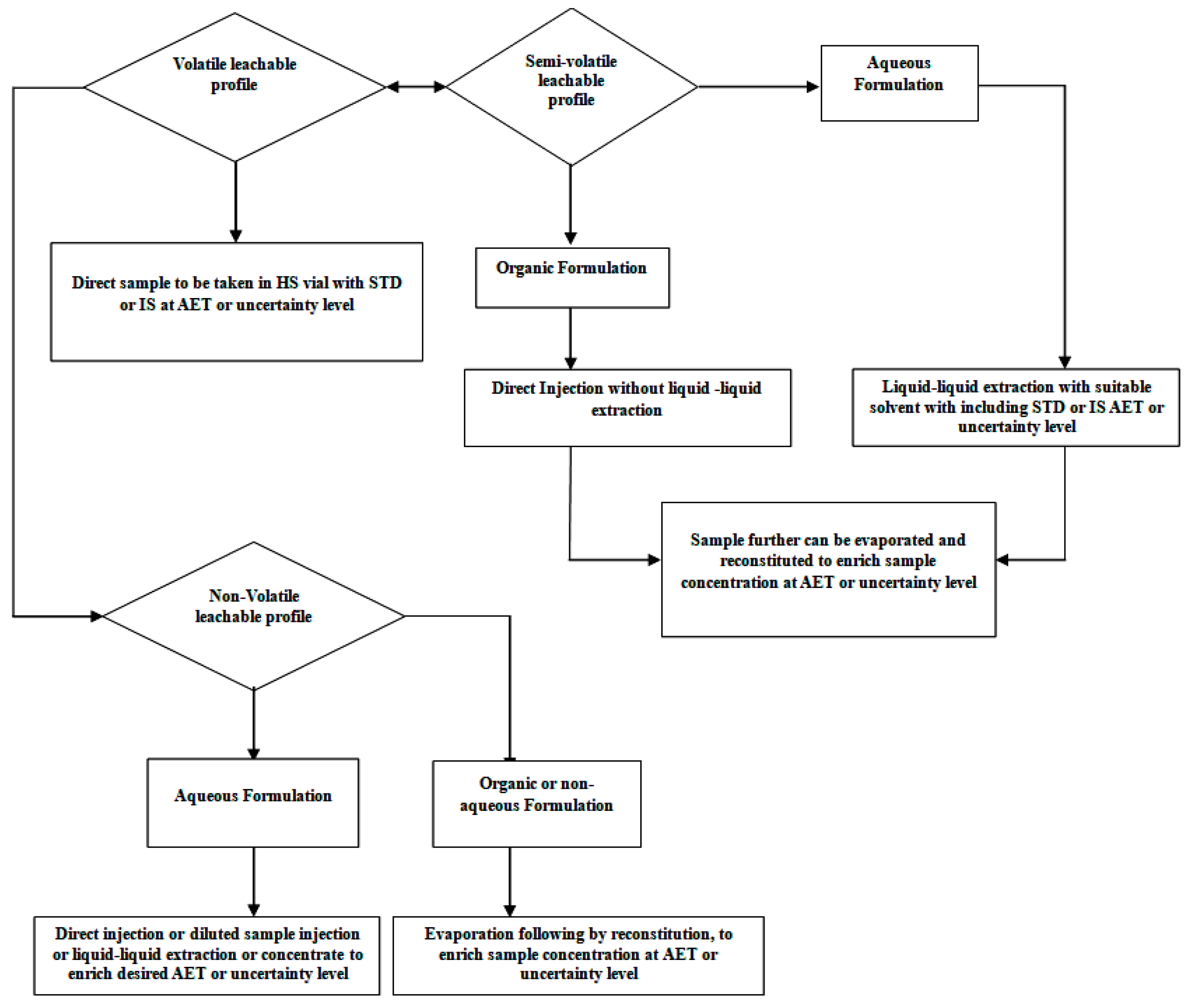
For screening a leachable study, drug product samples and standards (internal or external) are prepared at a concentration of the AET level or below the AET level, considering the uncertainty factor of the analytical method. To diminish peaks from the drug product formulation matrix and to identify leachables that leach out of the packaging system, delivery system, and manufacturing process equipment, a lab-prepared control drug product sample (without contact with manufacturing polymeric parts like the tubing and filter and filled using a method other than that used in the final packaging system) is prepared and analyzed in the same sequence. A leachable sample can be prepared with a similar procedure that was adopted during the method’s verification for generic solvent media. There are the following recommendations for sample preparation for volatile, semi-volatile, and non-volatile organic compounds (Figure 3).
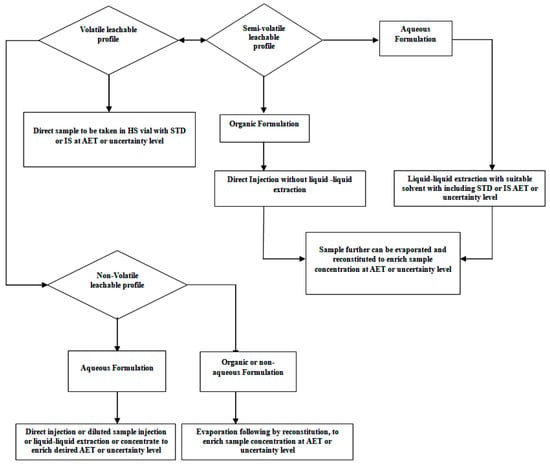
Figure 3.
Process flow for sample preparation and introduction into the system.
3.7. Data Interpretation
The leachable screening study of the finished drug product presents analytical challenges, especially in terms of data integration or interpretation and the structural identification of unknown compounds. The integration/interpretation of analytical instrumental data for the control and stability batches (finished product) is conventionally performed manually, and the process can be very lengthy and time-consuming. Software-based analytical data integration/interpretation greatly reduces this time-consuming challenge [17,18]. Mass Profiler Professional (MPP), a software application, performs differential analysis and provides a means to readily visualize the distribution of compounds across the drug product stability samples and control samples.
The identification of unknown compounds encountered during leachable screening analysis using GC/MS with electron ionization (EI) and LCMS with ESI/and or APCI ionization requires a degree of specialized knowledge. The use of EI often results in a mass spectrum that does not contain a distinct molecular ion, and its identification is dependent on matching characteristic fragmentation patterns with a breakdown of molecular bonding. In leachable screening analysis, matching scores of fragmentation patterns of m/z ions can be relatively poor, where compounds are present in trace amounts or interfered with by strong chemical background noise in the baseline. Therefore, not all compounds may be identified unequivocally based on their m/z fragmentation pattern alone. Generally, hyphenated techniques come with an inbuilt library, and the analyst can match the fragmentation pattern of unknown compounds with library-suggested compounds to predict the spectra and identify the unknown compounds. These libraries come with updates on a regular basis. For example, GCMS technology comes with an inbuilt NIST mass library, which is a database created based on a universal EI pattern with 70 EV.
After analyzing the samples, the leachable screening profile integrates or processes only the peaks that are detected at or above the AET or reporting threshold (uncertainty level). It is a requirement to identify all the unknown compounds that are detected at or above the AET level. The software-based identification of unknown compounds can be achieved with remarkable accuracy based on the match factor with library compounds as follows:
- ⮚
- Confirmed Identification (CD): The unknown compounds can be confirmed if the spectra of the unknown compounds match with the spectra of library compounds by more than 90%, and they can also be confirmed with the respective standard retention time and fragmentation pattern.
- ⮚
- Confident Identification (CI): The unknown compounds can be confidently identified if the spectra of the unknown compounds match the spectra of library compounds by more than 80%.
- ⮚
- Tentative Identification (TI): The unknown compounds can be tentatively identified if the spectra of the unknown compounds match the spectra of library compounds by more than 50%.
- ⮚
- Unknown Identification (Unk): The unknown compounds can not be identified if the spectra of the unknown compounds match the spectra of library compounds by less than 50%.
3.8. Toxicological Assessment
Once interpretation and calculation are carried out for the leachables, a toxicological risk assessment (TRA) should be carried out for all the leachable compounds that are determined at or above the AET or SCT levels. The goal of a toxicological risk assessment for leachable compounds is to evaluate the potential patient safety risks associated with each leachable substance present in a drug product. This assessment involves a detailed analysis of the specific amount of each leachable compound that leaches into the product in terms of the dosage and exposure associated with patient use.
The safety risk assessment of identified leachables from packaging systems, delivery systems, or manufacturing-process-related equipment is a complicated procedure [15]. The number of detected and labeled leachables can be large (e.g., more than 20). It is important to note that a lack of comprehensive data on chronic exposure to many leachables may necessitate the use of conservative assumptions or extrapolation methods to estimate potential risks. Additionally, ongoing research and monitoring of leachables in pharmaceutical products can help to fill gaps in the knowledge and improve the safety of these products.
The qualification of detected leachables and extractable in parenteral and ophthalmic drug products can benefit from the concepts outlined in the OINDP (orally inhaled and nasally inhaled drug product) best practices. However, modifications are necessary to account for the unique characteristics of these routes of administration, drug product characteristics, and the potential for systemic exposure.
The PQRI PODP Toxicology sub-team, which consisted of specialists from the USFDA, Canada health agency, and different pharmaceutical industries, evaluated the OINDP best practices and identified areas where modifications were necessary to ensure their applicability to parenteral and ophthalmic drug products. They also provided guidance on the implementation of these modified best practices for the requirement of leachables in these pharmaceutical drug formulations.
In summary, the PQRI Toxicology sub-team concluded that the suggestions from the appropriate practices could be useful for the assessment of leachables in injectable drug formulations, with modifications to account for differences in the routes of administration, drug product characteristics, and potential for systemic exposure. This conclusion was based on an evaluation of the OINDP best practices by the PQRI PODP toxicology sub-team, which consisted of specialists from different regulatory and pharmaceutical industries [16,19].
- ⮚
- A safety methodology was developed for orally inhaled nasal formulations, and it can be applied to extractable and leachable compounds for the qualification of injectable formulations.
- ⮚
- Based on predominant aqueous-based PDP formulations, an SCT of 1.5 μg/day can be used to calculate an AET.
- ⮚
- The qualification threshold (QT) originally established for organic impurities in orally inhaled and nasal drug products (OINDPs) has been reassessed for its applicability to parenteral drug products (PDPs). This reevaluation aimed to determine whether the existing QT is suitable for PDPs, considering the unique characteristics of these products.
An essential part of leachable safety assessment is the evaluation of the toxicology safety of chemical compounds that may transfer from any of the primary packaging/secondary systems, delivery systems, or manufacturing process equipment into the drug product. For any organic molecules (leachables), a rise in MDD is associated with an increase in the effect on the patient. For example, suppose 1 µg/mL or ppm of a chemical compound (leachable) leaches out from any of the streams, either the manufacturing or packaging systems, into the drug product and that the product’s maximum daily dose or daily volume to the patient is 10 mL; then, the daily patient exposure to that leachable is 10 µg/day (1 µg/mL leachable content × 10 mL maximum daily volume of the drug product to the patient); if the daily dose (MDD) increases, then the leachable exposure also increases, which may affect patient safety. When patients (humans) are tested and clinical animal data are available, a safe level of exposure can be determined for chemicals that exhibit undesirable toxicity. This involves identifying a dosage amount less than that at which harmful effects are not estimated. The following key considerations should be kept in mind during a safety evaluation:
- ⮚
- Available data: Patient (human) and/or animal clinical or toxicity data are available for the chemical in question.
- ⮚
- Toxicology profile: The chemical exhibits undesirable toxicity.
- ⮚
- Dose-response relationship: A dose concentration at a level where there is no harmful effect is estimated or determined.
If the allowable daily dose of the observed leachable compound is greater than the limit of 1.5 μg/day, the leachable is to be assessed for an oncogenicity risk evaluation. If the desired toxicity information exists for the identified leachable compound, a toxicological risk evaluation can be conducted by searching previously published information for appropriate data on oncogenicity. If inadequate oncology databases are accessible, an in silico toxicity safety evaluation can be executed as per issued guidance, such as ICH M7 (R1). If the observed leachable compound is not potentially genotoxic due to it not holding a chemically alert group or moiety for genotoxicity, the observed leachable compound is to be accepted concerning genotoxicity. If the observed leachable compound is identified as an oncogene or genotoxic due to it holding a structurally alarming functional chemical group for genotoxicity, it should be monitored based on the guidance provided and the available limits in ICH M7 [10,20,21,22].
4. Conclusions
This research article documents a guided overview of extractable and leachable (E&L) screening study design, methodologies, instrumentation, and toxicological risk assessment for the evaluation of the risk associated with patient exposure to leachables in parenteral drug products and orally inhaled nasal drug products that are highly concerned with toxicity and that may originate from the drug products’ manufacturing process and/or equipment during the manufacturing process and with primary or secondary container closure systems that are used to package the finished drug products. However, there are some key challenges that are still present in the field of extractable and leachables (E&Ls), such as if the formulation contains complex matrixes, like oil-based emulsions or non-aqueous solvents like polyethylene glycol, benzyl alcohol, etc., then, it is not practically feasible to separate these emulsions or non-aqueous phases from the organic solvent before it is injected into highly sensitive hyphenated instrumental techniques like GC/MS and LC/MS. Some newly invented sampling techniques are also coming into the market for such kinds of matrices to reduce interference, such as solid-phase extraction (SPE), micro twisters, and tax absorbents, which are used to treat samples first and then inject them into instruments, but there are certain limitations present with these novel techniques concerning the product. Another key challenge is that there is no harmonized or universally acceptable published guidance available to conduct E&L studies, as at present, agencies accept dossiers based on scientific study design and approaches with the support of some published recommendations and guidance documents like the product quality research institute (PQRI). This is the major future scope, i.e., recognizing sources/agencies for issuing harmonized guidance that will be universally acceptable and practically feasible.
Author Contributions
A.S.G., S.C., I.M.A. and H.D.M.C. contributed to conceptualizing and designing the manuscript. A.S.G., I.M.A., H.D.M.C., J.P.M.d.L. and S.C. contributed to writing the manuscript. A.S.G., H.D.M.C. and S.C. contributed to the nano adsorbent and mechanism section in the manuscript. A.S.G., H.D.M.C. and S.C. conceptualized and analyzed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- USP-NF. <1664> Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems; USP-NF: North Bethesda, MD, USA, 2015. [Google Scholar]
- The United States Pharmacopeial Convention. General Chapter <1663> Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems; USP 41–NF 36; USP-NF: Rockville, MD, USA, 2018; 7910. [Google Scholar]
- PQRI. Safety Thresholds and Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products; PQRI Drug Product Technical Committee: Washington, DC, USA, 2006. [Google Scholar]
- PQRI. PQRI (Product Quality Research Institute) Parenteral and Ophthalmic Drug Products Leachables and Extractables Working Group. Report; PQRI: Washington, DC, USA, 2009; pp. 1–32. [Google Scholar]
- FDA. ICH M7(R2) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk; Guidance for Industry; FDA: Silver Spring, MD, USA, 2023; 7.
- United State Pharmacopeia (USP). <661> Plastic Packaging Systems and Their Materials of Construction; USP: Rockville, MD, USA, 2017. [Google Scholar]
- U.S. Pharmacopeia. General Chapter <1665> Characterization and Qualification of Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products. Available online: https://qbd.co.kr/USP1665RAsection (accessed on 1 February 2025).
- U.S. Pharmacopeia. <665> Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products; U.S. Pharmacopeia: Rockville, MD, USA, 2021. [Google Scholar]
- United State Pharmacopeia (USP). <1661> Evaluation of Plastic Packaging Systems and Their Materials of Construction with Respect to Their User Safety Impact; U.S. Pharmacopeia: Rockville, MD, USA, 2014. [Google Scholar]
- ICH. ICH Harmonized Tripartite Guideline ICH Q12—Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management; ICH Expert Working Groups: Geneva, Switzerland, 2020. [Google Scholar]
- ISO 10993-12:2021; Biological Evaluation of Medical Devices—Part 12: Sample Preparation and Reference Materials. International Organization for Standardization: Geneva, Switzerland, 2021.
- ISO 10993-18:2020; Biological Evaluation of Medical Devices—Part 18: Chemical Characterization of Medical Device Materials Within a Risk Management Process. International Organization for Standardization: Geneva, Switzerland, 2020. Available online: https://www.iso.org/standard/64750.html (accessed on 1 February 2025).
- International Organization for Standardization Biological Evaluation of Medical Devices-Part 18: Chemical Characterization of Medical Device Materials within a Risk Management Process. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/detail.cfm?standard__identification_no=43774 (accessed on 1 February 2025).
- International Scientific Guidelines Adopted in Australia EMEA Guidance: Guidelines on Plastic Immediate Packaging Materials. Available online: https://www.tga.gov.au/sites/default/files/2024-07/finalplasticimmediatepackagingmaterialscurrent.pdf (accessed on 1 February 2025).
- Guidance for Industry Container Closure Systems for Packaging Human Drugs and Biologics. 1999. Available online: https://www.fda.gov/media/70788/download (accessed on 1 February 2025).
- United State Pharmacopeia (USP). <660> EP-3.2.1, Glass Containers for Pharmaceutical Use. Available online: https://www.biomed.co.th/english/Article-USP-Chapter660-EP321-Glass-Containers for PharmaceuticalUse.php (accessed on 1 February 2025).
- ICH Harmonized Tripartite Guideline Quality Risk Management (Q9). 2006. Available online: https://database.ich.org/sites/default/files/Q9_Guideline.pdf (accessed on 1 February 2025).
- Food and Drug Administration. US FDA Guidance for Industry and FDA Staff: Current Good Manufacturing Practice for Combination Products; Stability studies on rFVIIIFc drug substance; Food and Drug Administration: Silver Spring, MD, USA, 2015; pp. 1–14. [Google Scholar]
- Paskiet, D.; Jenke, D.; Ball, D.; Houston, C.; Norwood, D.L.; Markovic, I. The Product Quality Research Institute (PQRI) Leachables and Extractables Working Group Initiatives for Parenteral and Ophthalmic Drug Product (PODP). PDA J. Pharm. Sci. Technol. 2013, 67, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. US FDA Guidance for Industry Q8(R2) Pharmaceutical Development; Food and Drug Administration: Silver Spring, MD, USA, 2009; Volume 8, pp. 81–100.
- Markovic, I. Challenges Associated with Extractable and/or Leachable Substances in Therapeutic Biologic Protein Products. Am. Pharm. Rev. 2006, 9, 20–27. [Google Scholar]
- Markovic, I. Regulatory Perspective on Extractables & Leachables in Biologics: Quality Considerations. In Proceedings of the UPS/PQORI E&L Workshop, Rockville, MD, USA, 28 April 2014. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).