1. Introduction
Most of the prickly pear’s genetic materials originate from Mexico. This fruit shows great variability in its anatomy (different colors and sizes), which makes it attractive for commercialization. The most important physiological characteristic of these fruits is that their varieties can present different ripening behaviors; they can be classified into early ripening, intermediate-ripening and late-ripening varieties [
1]. Prickly pears have two disadvantages for their commercialization: the number and size of their seeds and the presence of thorns. It would be of great interest to identify those factors that generate these characteristics. To date, the reports related to these characteristics are scarce or null, in addition to considering that there is no genomic sequence reported for the nopal, which will help a possible improvement scheme [
2].
MicroRNAs (miRNAs) are a class of small, non-coding RNAs that regulate gene expression in eukaryotes. They are involved in different processes of plant development, including fruit development, and very recently, they have been reported as part of the response mechanisms to diseases and other types of stress [
3]. miRNAs play an essential role in the post-transcriptional regulation of genes and target a large number of transcription factors and other regulatory proteins, suggesting a role in the control of regulatory pathways and development [
3]. In order to identify the function of microRNAs, it is necessary to analyze their expression and that of the targets, with which they interact negatively. A better proof of the function of microRNAs is their expression in transformed plants, where their effect on the processes of interest can be evaluated [
4].
With the scope of the objectives of this work, important information would be generated to understand the development of the fruit and its different characteristics in this interesting model, which could be applicable to other models and for possible genetic improvement systems.
2. Materials and Methods
2.1. Plant Material
For the different molecular analyzes, fruits of the nopal morphospecie with an intermediate ripening behavior “Reina” (
Opuntia robusta) were collected. Four stages of development were collected: flower bud, fertilized bud, green fruit and mature fruit. The fruits were processed as previously described [
4]. Total RNA was extracted and used for miRNA in microarrays and transcriptome analysis.
2.2. Transcriptomic Analysis of miRNAs during Fruit Development
In order to have a more complete picture of the behavior of the miRNAs, the expression of the miRNAs in prickly pear fruit was analyzed with a hybridization of microarrays and by analysis of the transcriptome of the miRNAs of the different stages (flower bud, fertilized bud, green and ripe). For the conserved miRNAs, the GeneChip
® miRNA 4.0 Array (Affymetrix) was used. Total RNA of prickly pear in different stages were used and hybridized in the Genotyping and Expression Analysis Unit (National Institute of Genomic Medicine-INMEGEN, Mexico). For the specific miRNAs, the transcriptomes were sequenced in the Sequencing Unit of the Institute of Biotechnology of UNAM (National University of Mexico). The data were analyzed with the R software of the Bioconductor Project (
http://www.bioconductor.org (accessed on 20 January 2020)) for normalization of the data before comparing and evaluating the differential expression. The differential expression was selected with at least twofold change using the oligo packages [
5] lime [
6].
2.3. Amplification of miRNA Precursors
In order to validate the differential expression of the identified miRNAs, total RNA was extracted from 100 mg of tissue. The precursors of the identified miRNAs were amplified according to the stem loop amplification method described by Li et al. (2009) [
7].
2.4. Establishment of Expression Networks during Fruit Development
From the data of the transcriptomes and the arrangement, the possible expression networks generated by the expression of the miRNAs related to the development of the fruit were constructed. Different packages were used for massive data analysis (Blast to go, KEGG). The results were related to the phenotypic analysis to find the association of the different molecules.
3. Results
3.1. RNA Isolation
The extractions were made with three biological replicates. After extraction, the quantity and quality (based on absorbance at 260 and 280 nm) of the RNA was determined with the nanodrop 2000 equipment (Thermo Scientific, Waltham, MA, USA). The main concentration was 12.41 ng/µL, and the A260/A280 ratio was 1.85.
3.2. Microarray Hybridization
To establish a point of comparison within fruit development as part of sexual development, it was necessary to establish a reference point in the array hybridization, the FF (Flower) transcripts, and the contrasting conditions, FV (green fruit) and FM (ripe fruit) (
Table 1). To obtain a complete picture within the fruit’s development history, a third confirmatory hybridization was performed, using FV transcripts as reference and FM as contrasting condition (FF-FM) (
Table 1). The expression level ×2 (greater than or equal to 2 and less than or equal to -2, 2× foldchange) was used to determine if the expression was differential. From the total miRNAs analyzed, 20.8% are differentially expressed in FF-FV, 60.7% in FF-FM and 55% in FV-FM. This result suggests a possible role in the expression of miRNAs, especially in the most advanced stages of fruit development (FV and FM).
3.3. RNA Seq Analysis
For the RNA-seq analysis, the same tissues were used (FF, FV and FM), and the homology search was performed only against Arabidopsis thaliana with blastn-short under an identity criterion greater than 90%, a maximum 1 mismatch and an alignment length of 19–24 nt.
With a probability greater than 80%, 67 differential miRNAs were detected between FF and FV, 89 between FV and FM and 99 between FF and FM. The participation of the families miR156, miR162, miR164, miR165 and miR169 from
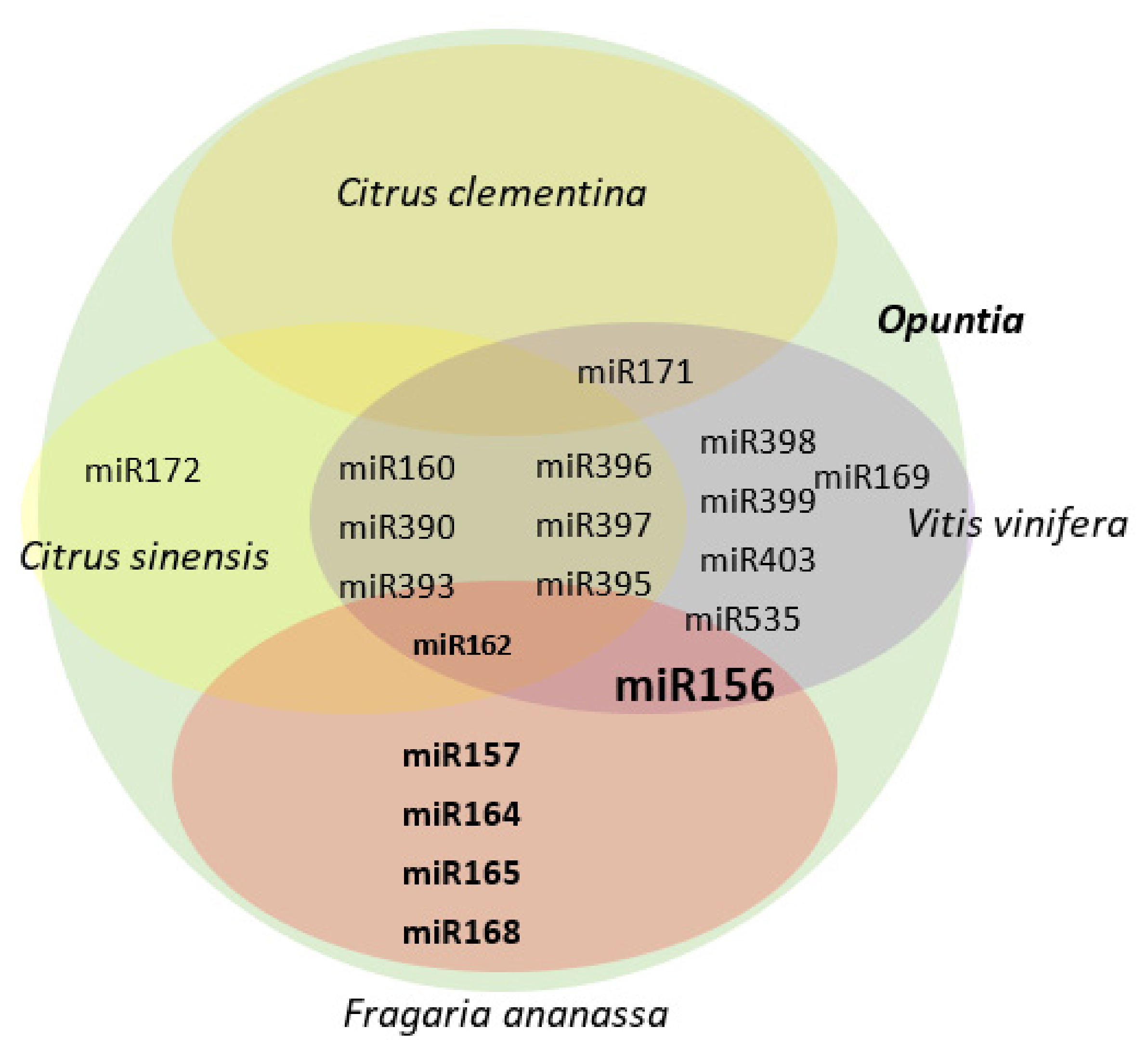
Arabidopsis thaliana was found. The criteria for the establishment of candidates of miRNAs with participation in the development and ripening of prickly pear were as follows: (a) families of miRNAs with the highest prevalence among the different expression groups according to the dendrogram and categorization and (b) miRNAs with a higher level of expression or repression in fruit development transitions. With these criteria, 30 families of candidate miRNAs were found, a strong conservation was observed within the plant kingdom, as well as a total of 14 miRNAs conserved in both the plant and animal kingdoms, and 7 are reported only in animals. From the list of species with homologous miRNAs, there are three non-climacteric fruits, such as prickly pear:
Citrus clementina,
Citrus sinensis and
Vitis vinifera (
Figure 1). Sixteen families of candidate miRNAs are expressed in at least one of these three species. A search for the expression of miRNAs was carried out in other non-climacteric fruits, and the expression of seven families of miRNAs was found: miR156, 157, 159, 162, 164, 165 and 168 [
7]. miR156 and 159 were are also expressed in
Vitis vinifera and miR162 in
Citrus clementina and
Vitis vinifera. A total of 43 miRNAs were identified with 26 different targets in relation to the fruit development from flower induction. It highlights the importance of miR172 and miR395 as key candidates in the fruit-ripening time. miR397 is also established as an opportunity for the generation of varieties for seed production (
Table 1).
4. Discussion
In the present work, the expression of miRNAs in different stages of fruit ripening was analyzed, from bud flower to ripe fruit, with greater emphasis on the transition from VF to FM, using massive data management methodologies. In total, 1047 miRNAs were differentially expressed, and 30 miRNA families were selected based on prevalence in the expression of miRNA families among the different expression groups, as well as intensity of expression. A search was carried out in databases and other sources, as well as tools for the prediction of miRNA–mRNA interactions, and we found 43 miRNAs and 26 targets with a role within the ripening process. The importance of miR172 and miR395 is highlighted, as key candidates in the time of fruit ripening, as well as that of miR397as an opportunity for the generation of varieties for seed production.
5. Conclusions
A total of 43 miRNAs with 26 different targets were identified in relation to fruit development from flower induction. It highlights the importance of miR172 and miR395, as key candidates in the fruit ripening time. miR397 is also established, as an opportunity for the generation of varieties for control of seed production.
Author Contributions
Conceptualization, A.C.-H. and P.L.L.d.A.; methodology, S.G.-G. and D.A.-M.; software and validation, C.A.C.-J. and O.-M.F.; writing—original draft preparation, A.C.-H.; writing—review and editing, P.L.L.d.A.; project administration, A.C.-H.; funding acquisition, A.C.-H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Universidad De La Salle Bajio grant number 2021-01. Project: Estudio de la expresión de miR156, miR160 y miR397 en el desarrollo de la tuna (Opuntia sp.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Thanks to CENGUA-INIFAP, for providing the materials used for the experiments. The individuals (CENGUA-INIFAP) consented the acknowledgement.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pimienta-Barrios, E. Prickly pear (Opuntia spp.): A valuable fruit crop for the semiarid lands of México. J. Arid Environ. 1994, 28, 1–11. [Google Scholar] [CrossRef]
- Cedillo-Jiménez, C.A.; Cruz-Ramirez, L.A.; Mondragón-Jacobo, C.; Aguilar-Ruiz, C.A.; González-Márquez, M.A.; Perusquia, A.A.; Cruz-Hernández, A. miRNAs involvement on fruit development in prickly pear (Opuntia sp.). Acta Hortic. 2020, 1297, 635–638. [Google Scholar] [CrossRef]
- Cedillo-Jiménez, C.A.; Hernández–Salazar, M.; Escobar-Feregrino, T.; Caballero-Pérez, J.; Arteaga-Vázquez, M.; Cruz-Ramírez, A.; Torres-Pacheco, I.; Guevara-González, R.; Cruz-Hernandez, A. MicroRNAs Sequencing for Understanding the Genetic Regulation of Plant Genomes. In Plant Genomes; Intech Publishing: London, UK, 2016; ISBN 978-953-51-4622-3. [Google Scholar]
- Rosas-Cárdenas, F.; Escobar-Guzmán, R.; Cruz-Hernández, A.; Marsch-Martínez, N.; DeFolter, S. An efficient method for miRNA detection and localization in crop plants. Front. Plant Sci. 2015, 6, 99. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Huang, F.; Chang, L.; Ma, Y. MicroRNA expression profiles in conventional and micropropagated strawberry (Fragaria × ananassa Duch.) plants. Plant Cell Rep. 2009, 28, 891–902. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).





{kind=link}