
A Probable Anti-COVID Phytochemical (1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione) Screened Computationally from the Rhizome of Curcuma longa †

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Formulation of the Ligand Library
2.2.2. Retrieval of Structure of SARS-CoV-2 Spike Protein
2.2.3. Preparation of Retrieved Protein
2.2.4. Prediction of Binding Site and Generation of Grid for Docking
2.2.5. Protein Ligand Molecular Docking
Docking a Standard Repurposed Drug for Spike Protein
Prediction of the ADMET Properties
3. Results
3.1. Prepared Protein Structure of the RBD of SARS-CoV-2 (7EAM_1) for Molecular Docking
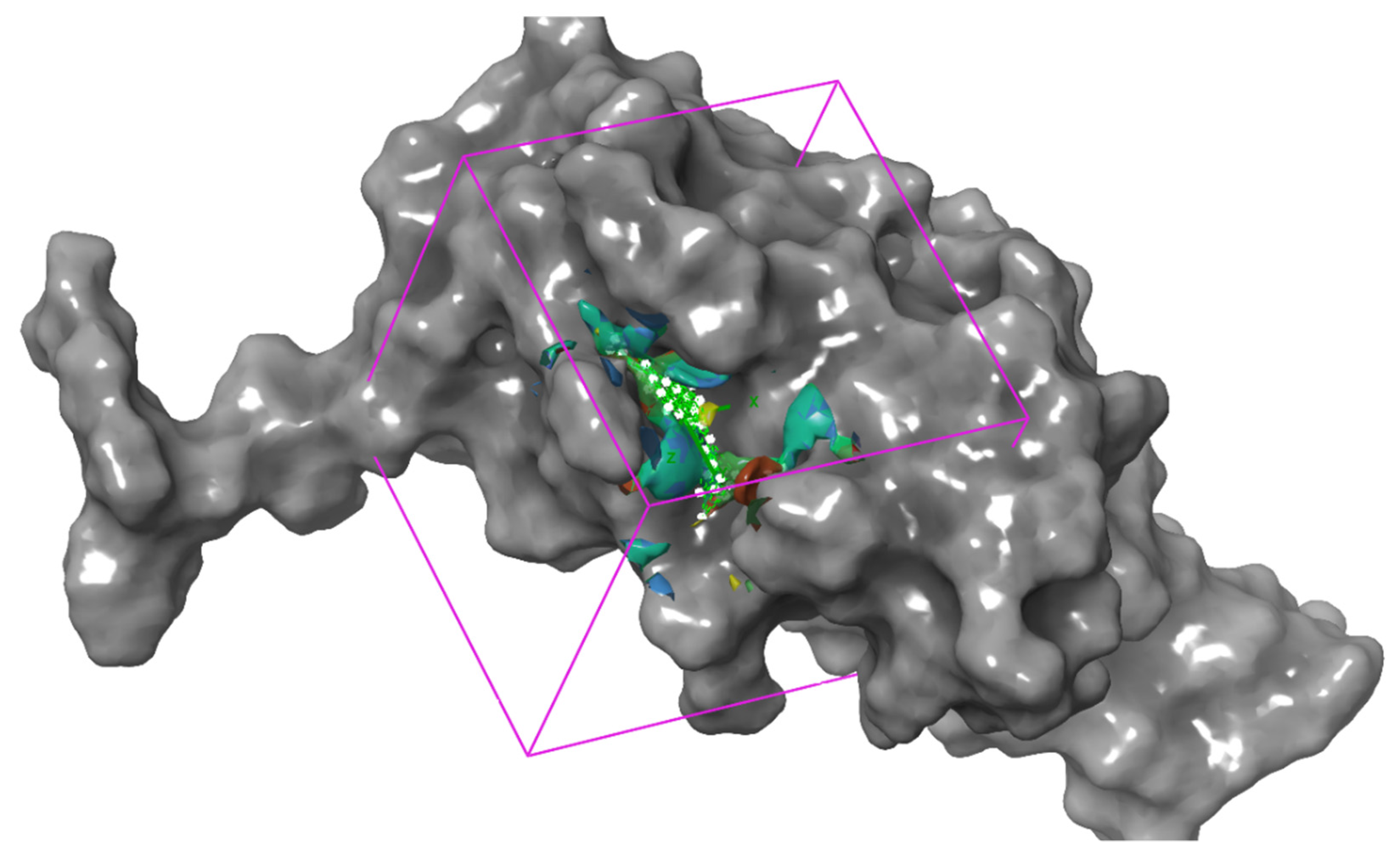
3.2. Predicting the Best Binding Site for Molecular Docking in the Protein Structure
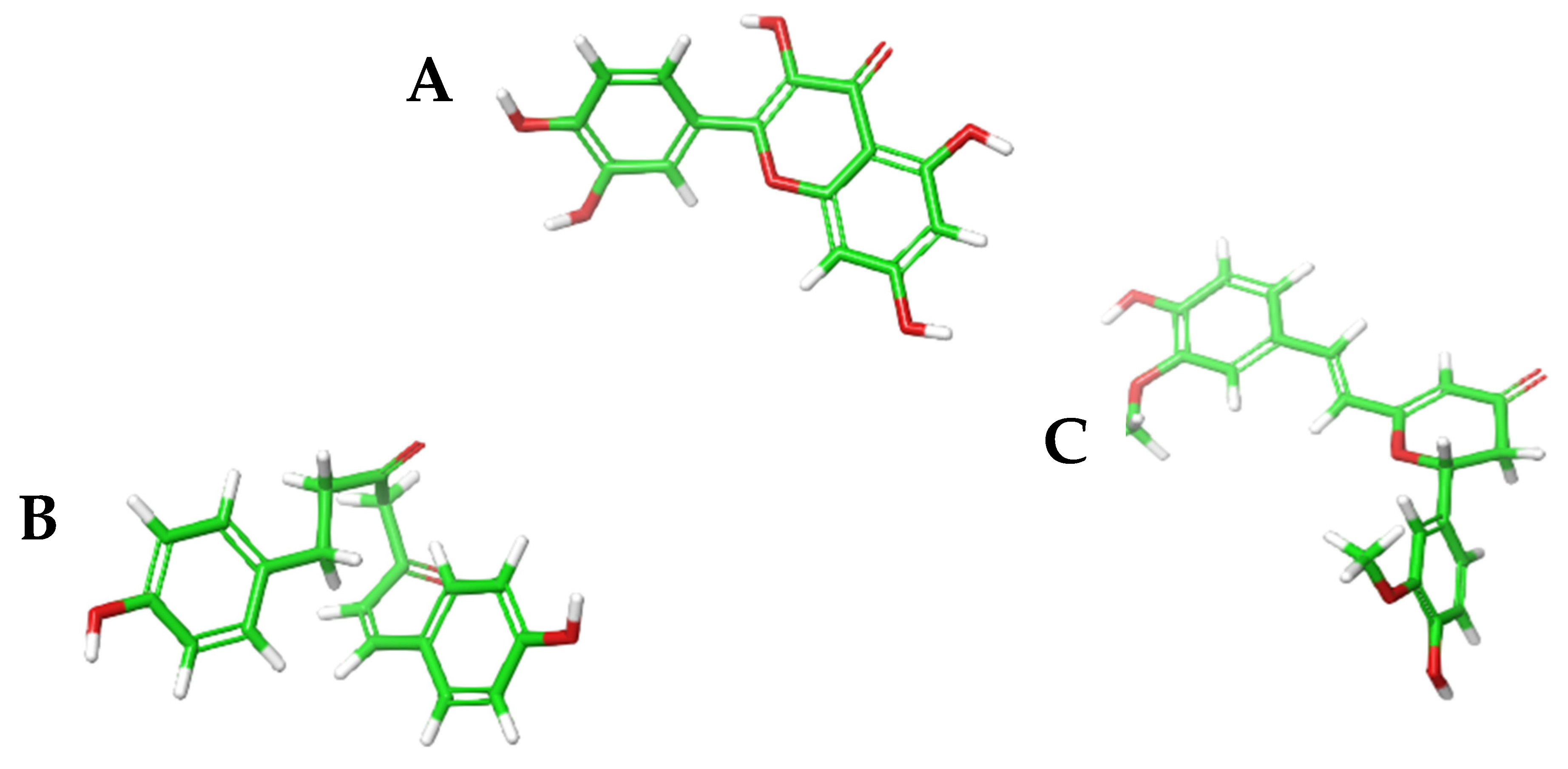
3.3. Molecular Docking of the Ligand Library of Curcuma longa to the Protein Binding Pockets
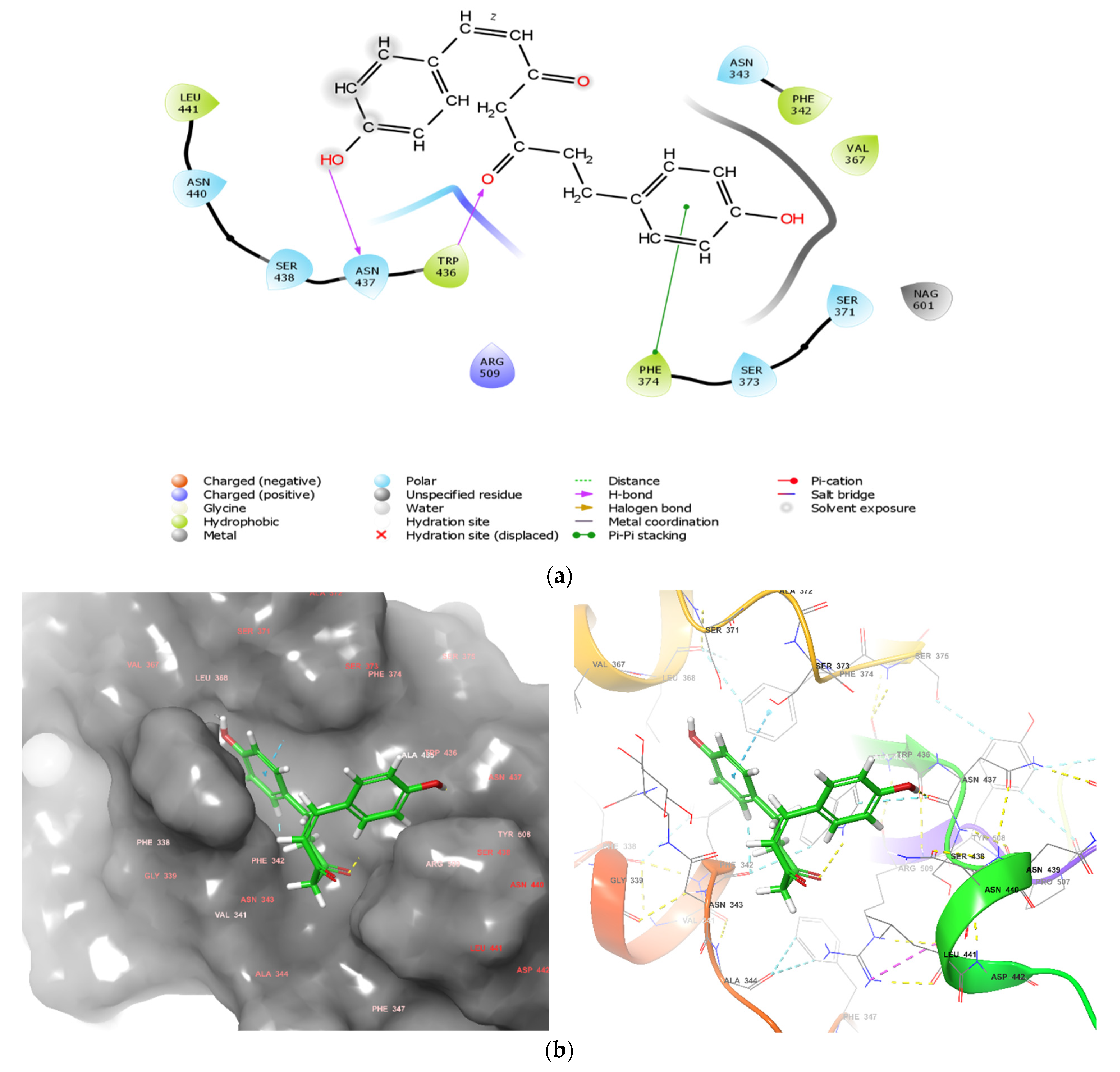
3.4. Examining the Interactions of 1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione with Amino-Acids Residues in the Binding Site of RBD of SARS-CoV-2
3.5. ADME/Tox Properties of 1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [Green Version]
- Idrees, I.; Khan, S.; Memon, N.H.; Zhang, Z. Effect of the Phytochemical Agents against the SARS-CoV and Some of them Selected for Application to COVID-19: A Mini-Review. Curr. Pharm. Biotechnol. 2021, 22, 444–450. [Google Scholar] [CrossRef]
- Attia, Y.A.; Alagawany, M.M.; Farag, M.R.; Alkhatib, F.M.; Khafaga, A.F.; Abdel-Moneim, A.-M.E.; Asiry, K.A.; Mesalam, N.M.; Shafi, M.E.; Al-Harthi, M.A.; et al. Phytogenic Products and Phytochemicals as a Candidate Strategy to Improve Tolerance to Coronavirus. Front. Vet. Sci. 2020, 783. [Google Scholar] [CrossRef]
- Attah, A.F.; Fagbemi, A.A.; Olubiyi, O.; Dada-Adegbola, H.; Oluwadotun, A.; Elujoba, A.; Babalola, C.P. Therapeutic Potentials of Antiviral Plants Used in Traditional African Medicine With COVID-19 in Focus: A Nigerian Perspective. Front. Pharmacol. 2021, 178. [Google Scholar] [CrossRef]
- Rajagopal, K.; Varakumar, P.; Baliwada, A.; Byran, G. Activity of phytochemical constituents of Curcuma longa (turmeric) and Andrographis paniculata against coronavirus (COVID-19): An in silico approach. Future J. Pharm. Sci. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Babaei, F.; Nassiri-Asl, M.; Hosseinzadeh, H. Curcumin (a constituent of turmeric): New treatment option against COVID-19. Food Sci. Nutr. 2020, 8, 5215–5227. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Purohit, R. Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: An in-silico approach. Comput. Biol. Med. 2021, 139, 104965. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020. [Google Scholar] [CrossRef]
- Bormann, M.; Alt, M.; Schipper, L.; van de Sand, L.; Le-Trilling, V.T.K.; Rink, L.; Heinen, N.; Madel, R.J.; Otte, M.; Wuensch, K.; et al. Turmeric Root and Its Bioactive Ingredient Curcumin Effectively Neutralize SARS-CoV-2 In Vitro. Viruses 2021, 13, 1914. [Google Scholar] [CrossRef]
- Guijarro-Real, C.; Plazas, M.; Rodríguez-Burruezo, A.; Prohens, J.; Fita, A. Potential In Vitro Inhibition of Selected Plant Extracts against SARS-CoV-2 Chymotripsin-Like Protease (3CLPro) Activity. Foods 2021, 10, 1503. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Tetè, G.; Pregliasco, F.; Ronconi, G. The british variant of the new coronavirus-19 (Sars-cov-2) should not create a vaccine problem. J. Biol. Regul. Homeost. Agents 2021, 35, 1–4. [Google Scholar] [CrossRef]
- Ghildiyal, R.; Prakash, V.; Chaudhary, V.K.; Gupta, V.; Gabrani, R. Phytochemicals as Antiviral Agents: Recent Updates. In Plant-Derived Bioactives; Springer: Singapore, 2020; p. 279. [Google Scholar] [CrossRef]
- Zorofchian Moghadamtousi, S.; Abdul Kadir, H.; Hassandarvish, P.; Tajik, H.; Abubakar, S.; Zandi, K. A review on antibacterial, antiviral, and antifungal activity of curcumin. Biomed Res. Int. 2014, 2014, 186864. [Google Scholar] [CrossRef]
- Parham, S.; Kharazi, A.Z.; Bakhsheshi-Rad, H.R.; Nur, H.; Ismail, A.F.; Sharif, S.; RamaKrishna, S.; Berto, F. Antioxidant, Antimicrobial and Antiviral Properties of Herbal Materials. Antioxidants 2020, 9, 1309. [Google Scholar] [CrossRef]
- Silveira, D.; Prieto-Garcia, J.M.; Boylan, F.; Estrada, O.; Fonseca-Bazzo, Y.M.; Jamal, C.M.; Magalhães, P.O.; Pereira, E.O.; Tomczyk, M.; Heinrich, M. COVID-19: Is There Evidence for the Use of Herbal Medicines as Adjuvant Symptomatic Therapy? Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Sahni, H.; Sharma, H. Role of social media during the COVID-19 pandemic: Beneficial, destructive, or reconstructive? Int. J. Acad. Med. 2020, 6, 70. [Google Scholar] [CrossRef]
- Padhi, A.K.; Seal, A.; Khan, J.M.; Ahamed, M.; Tripathi, T. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: Insights from atomistic simulations. Eur. J. Pharmacol. 2021, 894, 173836. [Google Scholar] [CrossRef]
- Gu, Y.-Y.; Zhang, M.; Cen, H.; Wu, Y.-F.; Lu, Z.; Lu, F.; Liu, X.-S.; Lan, H.-Y. Quercetin as a potential treatment for COVID-19-induced acute kidney injury: Based on network pharmacology and molecular docking study. PLoS ONE 2021, 16, e0245209. [Google Scholar] [CrossRef]
- Di Pierro, F.; Iqtadar, S.; Khan, A.; Mumtaz, S.U.; Chaudhry, M.M.; Bertuccioli, A.; Derosa, G.; Maffioli, P.; Togni, S.; Riva, A.; et al. Potential Clinical Benefits of Quercetin in the Early Stage of COVID-19: Results of a Second, Pilot, Randomized, Controlled and Open-Label Clinical Trial. Int. J. Gen. Med. 2021, 14, 2807–2816. [Google Scholar] [CrossRef]
- Di Pierro, F.; Derosa, G.; Maffioli, P.; Bertuccioli, A.; Togni, S.; Riva, A.; Allegrini, P.; Khan, A.; Khan, S.; Khan, B.A.; et al. Possible Therapeutic Effects of Adjuvant Quercetin Supplementation Against Early-Stage COVID-19 Infection: A Prospective, Randomized, Controlled, and Open-Label Study. Int. J. Gen. Med. 2021, 14, 2359–2366. [Google Scholar] [CrossRef]
- ClinicalTrials.gov, Quercetin in the Treatment of SARS-COV 2. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04853199 (accessed on 15 August 2021).
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482. [Google Scholar] [CrossRef]
- Price, G.; Patel, D.A. Drug Bioavailability. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557852/ (accessed on 15 August 2021).
- Ditzinger, F.; Price, D.J.; Ilie, A.-R.; Köhl, N.J.; Jankovic, S.; Tsakiridou, G.; Aleandri, S.; Kalantzi, L.; Holm, R.; Nair, A.; et al. Lipophilicity and hydrophobicity considerations in bio-enabling oral formulations approaches—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 464–482. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Number | Site Score | D-Score | Volume |
|---|---|---|---|
| Site 1 | 0.682529 | 0.628868 | 94.325 |
| Site 2 | 0.708399 | 0.585154 | 131.0260 |
| Site 3 | 0.665813 | 0.647446 | 80.26200 |
| Phytochemical Compounds | Entry ID | Canonical SMILES | Docking Score | |
|---|---|---|---|---|
| 1. | Quercetin | CID 5280343 | C1=CC(=C(C=C1C2=C(C(=O)C3=C(C=C(C=C3O2)O)O)O)O)O | −6.903 |
| 2. | 1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione (BHHD) | CID 9796708 | C1=CC(=CC=C1CCC(=O)CC(=O)C=CC2=CC=C(C=C2)O)O | −4.349 |
| 3. | Cyclocurcumin | CID 69879809 | COC1=C(C=CC(=C1)C=CC2=CC(=O)CC(O2)C3=CC(=C(C=C3)O)OC)O | −4.195 |
| 4. | *Umifenovir (Arbidol)*—Standard | CID 131411 | CCOC(=O)C1=C(N(C2=CC(=C(C(=C21)CN(C)C)O)Br)C)CSC3=CC=CC=C3 | −4.176 |
| 5. | 1,7-Bis-(4-hydroxy-3-methoxyphenyl)-1,4,6-heptatrien-3-one | CID 10904292 | COC1=C(C=CC(=C1)C=CC=CC(=O)C=CC2=CC(=C(C=C2) | −3.719 |
| 6. | Epi-procurcumenol | CID 10263440 | CC1=CC(=O)C(=C(C)C)CC2C1CCC2(C)O | −3.719 |
| ADME/Tox | Parameter | Value |
|---|---|---|
| Physicochemical Properties | Formula | C19H18O4 |
| Molecular weight | 310.34 g/mol | |
| Num. heavy atoms | 23 | |
| Num. arom. heavy atoms | 12 | |
| Num. rotatable bonds | 7 | |
| Num. H-bond acceptors | 4 | |
| Num. H-bond donors | 2 | |
| Topological PSA | 74.60 Å2 | |
| Lipophilicity and water solubility | Log Po/w (iLOGP) | 1.94 |
| Consensus Log Po/w | 2.88 | |
| H2O solubility class | Moderately soluble | |
| Pharmacokinetics | GI absorption | High |
| BBB permeant | Yes | |
| P-gp substrate | No | |
| Drug likeness | Lipinski | Yes: 0 violation |
| Bioavailability score | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezeorba, T.P.C.; Uchendu, N.O.; Nweze, E.J.; Okoroafor, C.K.; Ogbu, P.O.; Okpara, M.C.; Asomadu, R.O.; Joshua, P.E. A Probable Anti-COVID Phytochemical (1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione) Screened Computationally from the Rhizome of Curcuma longa . Med. Sci. Forum 2021, 7, 6. https://doi.org/10.3390/ECMS2021-10845
Ezeorba TPC, Uchendu NO, Nweze EJ, Okoroafor CK, Ogbu PO, Okpara MC, Asomadu RO, Joshua PE. A Probable Anti-COVID Phytochemical (1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione) Screened Computationally from the Rhizome of Curcuma longa . Medical Sciences Forum. 2021; 7(1):6. https://doi.org/10.3390/ECMS2021-10845
Chicago/Turabian StyleEzeorba, Timothy P. C., Nene O. Uchendu, Ekene J. Nweze, Chibuzo K. Okoroafor, Pascal O. Ogbu, Miracle C. Okpara, Rita O. Asomadu, and Parker E. Joshua. 2021. "A Probable Anti-COVID Phytochemical (1,7-Bis-(4-hydroxyphenyl)-1-heptene-3,5-dione) Screened Computationally from the Rhizome of Curcuma longa " Medical Sciences Forum 7, no. 1: 6. https://doi.org/10.3390/ECMS2021-10845