
Exploring Synergistic Effect on the Stability of Ni-, Pd-, Ir-Doped TiN (N = 1–15) Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
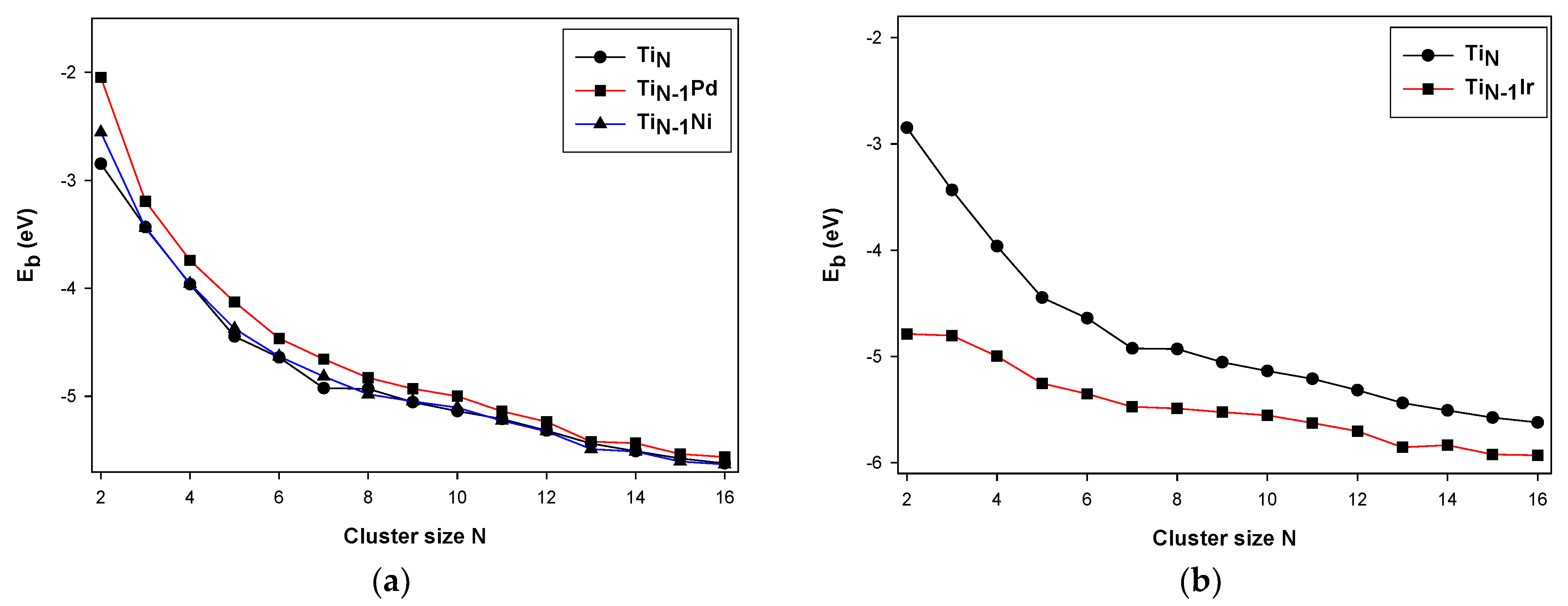
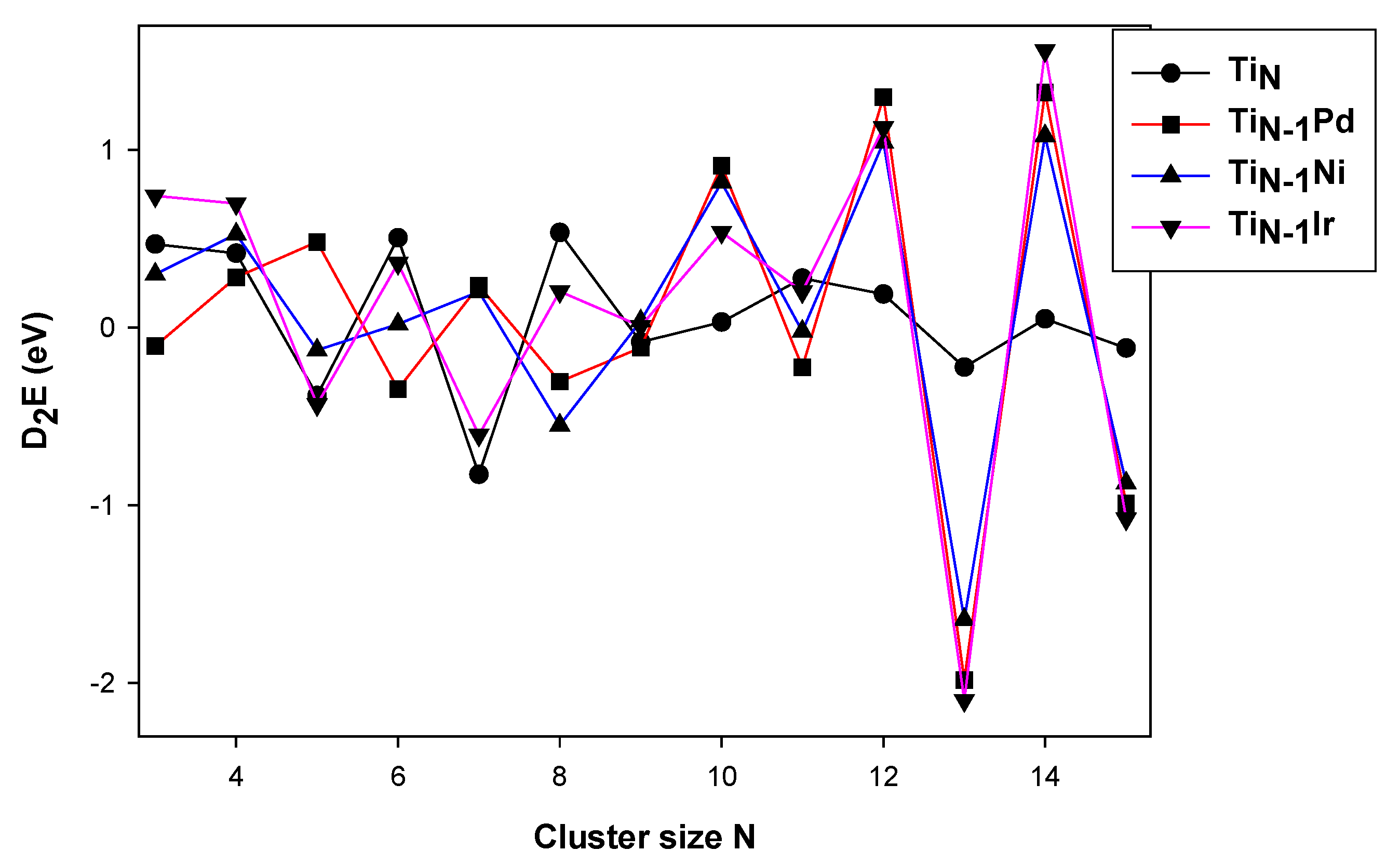
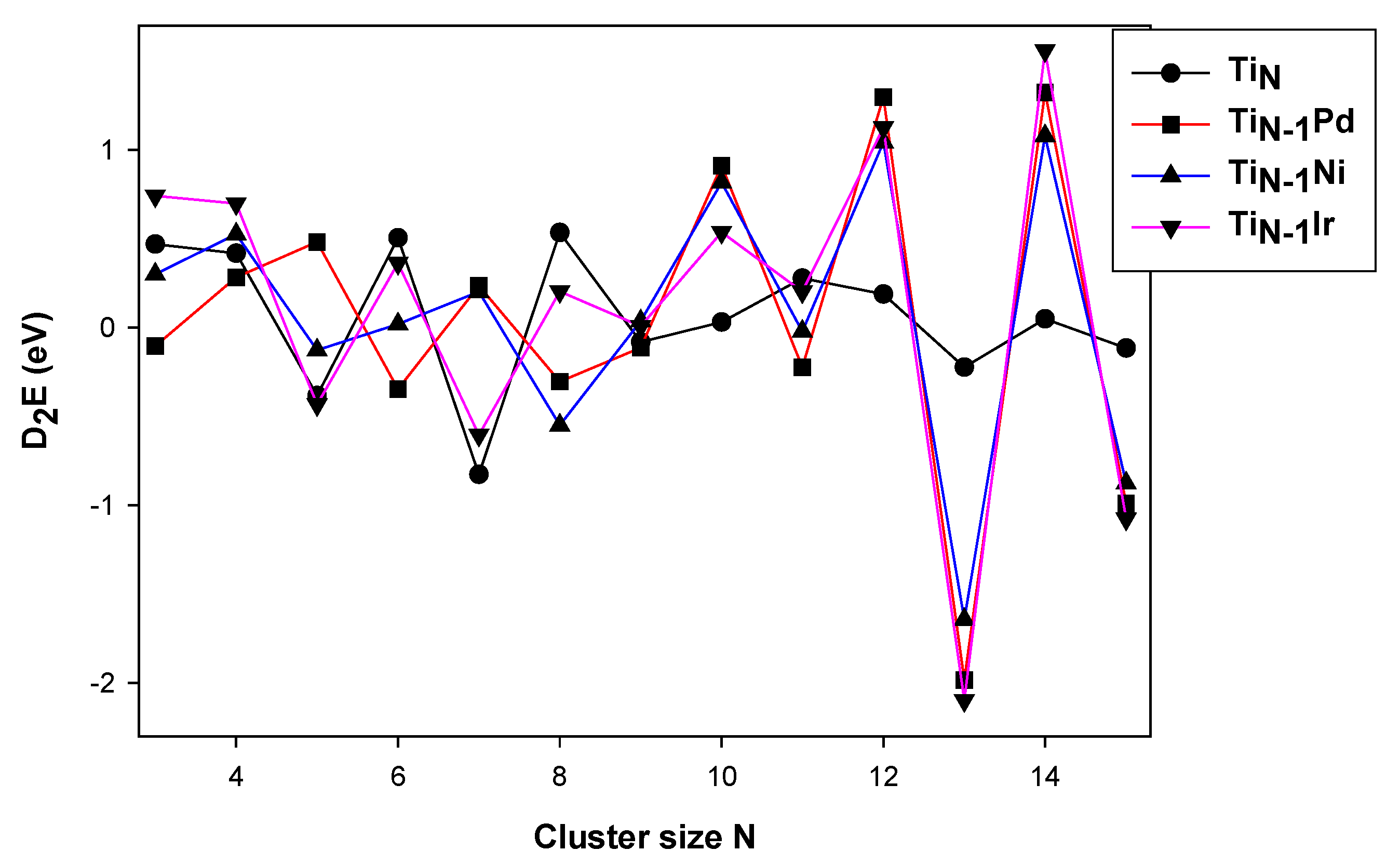
3.1. Stability of the TiNM (M = Pd, Ir, Ni)
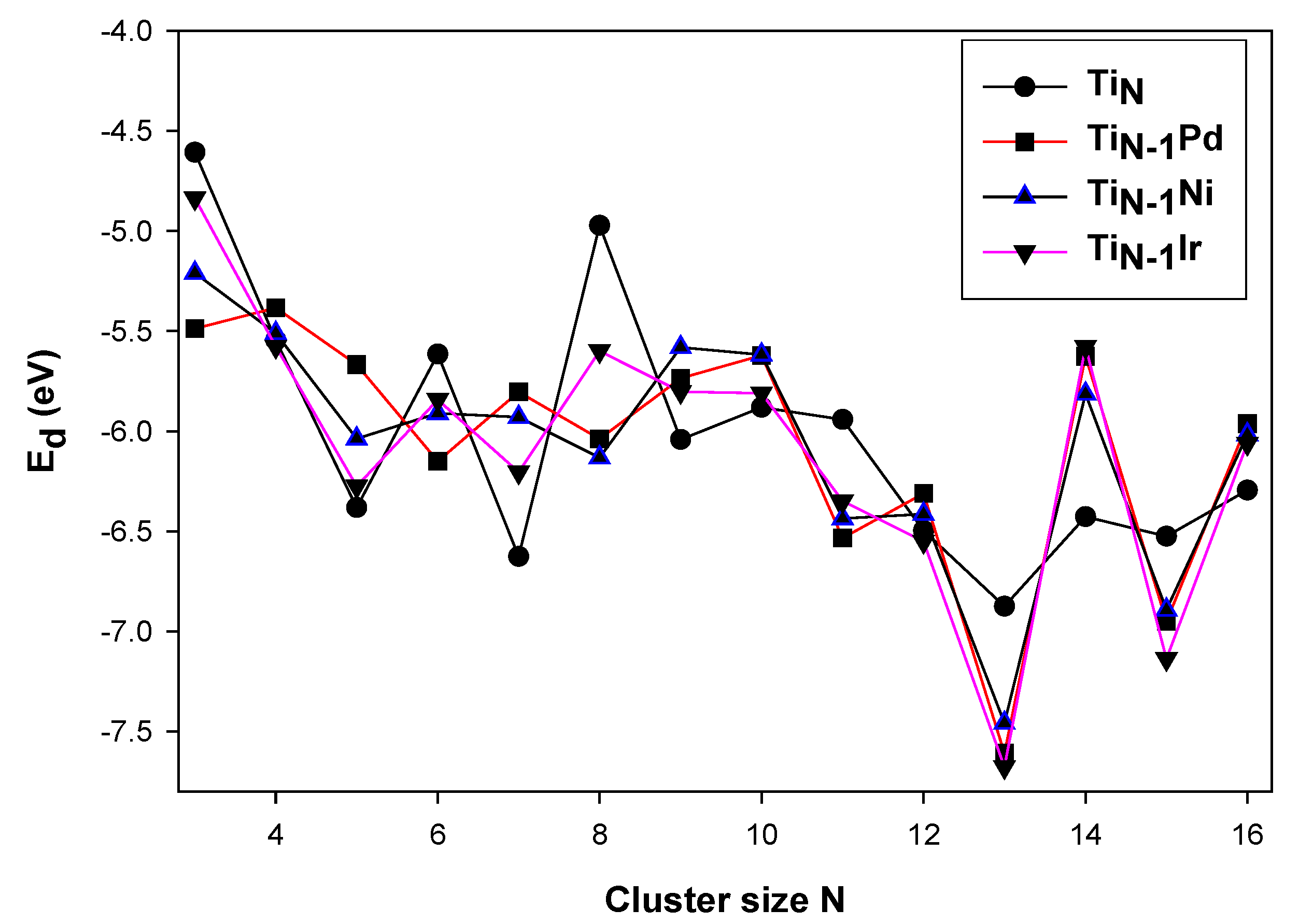
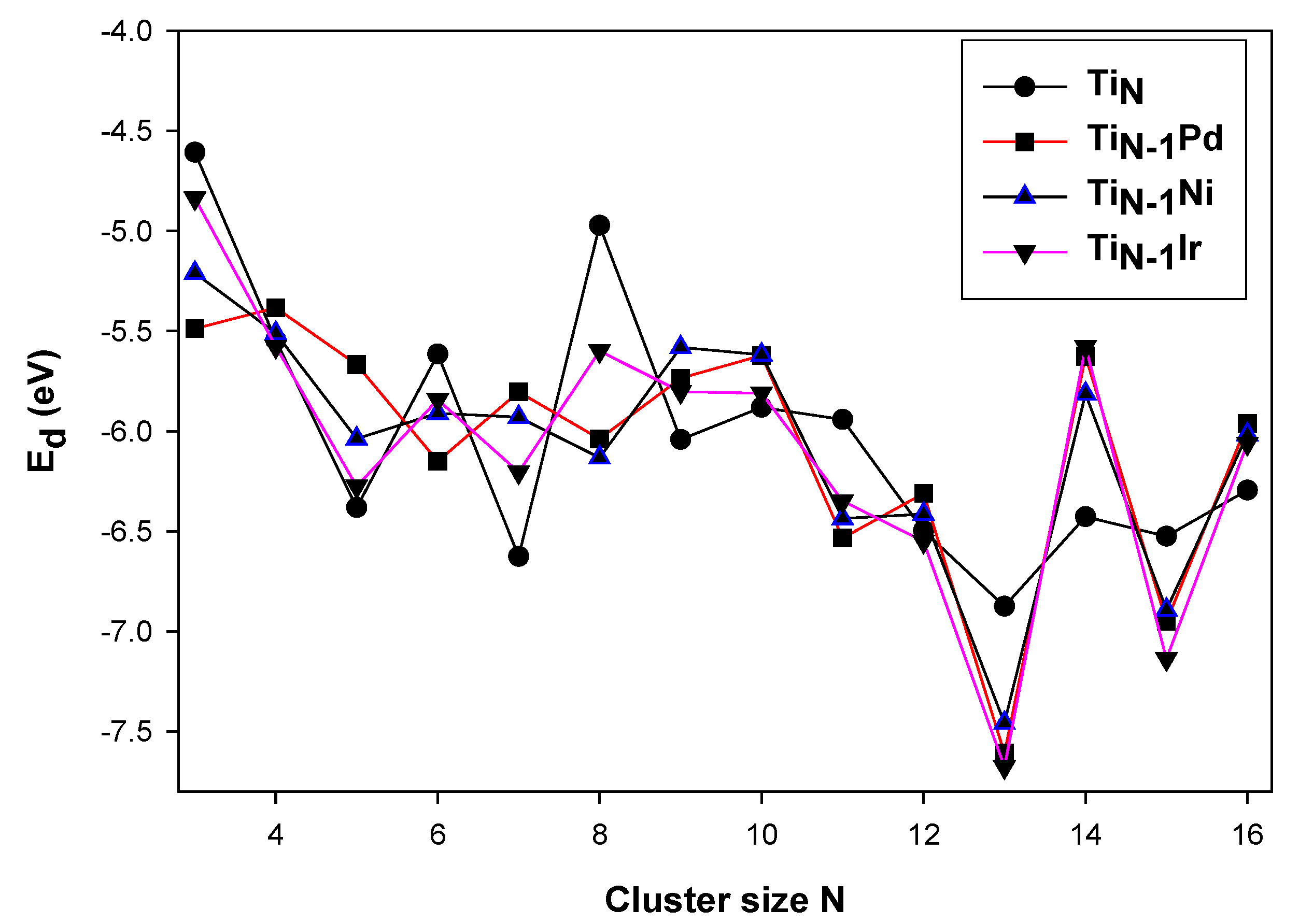
3.2. Electronic Properties
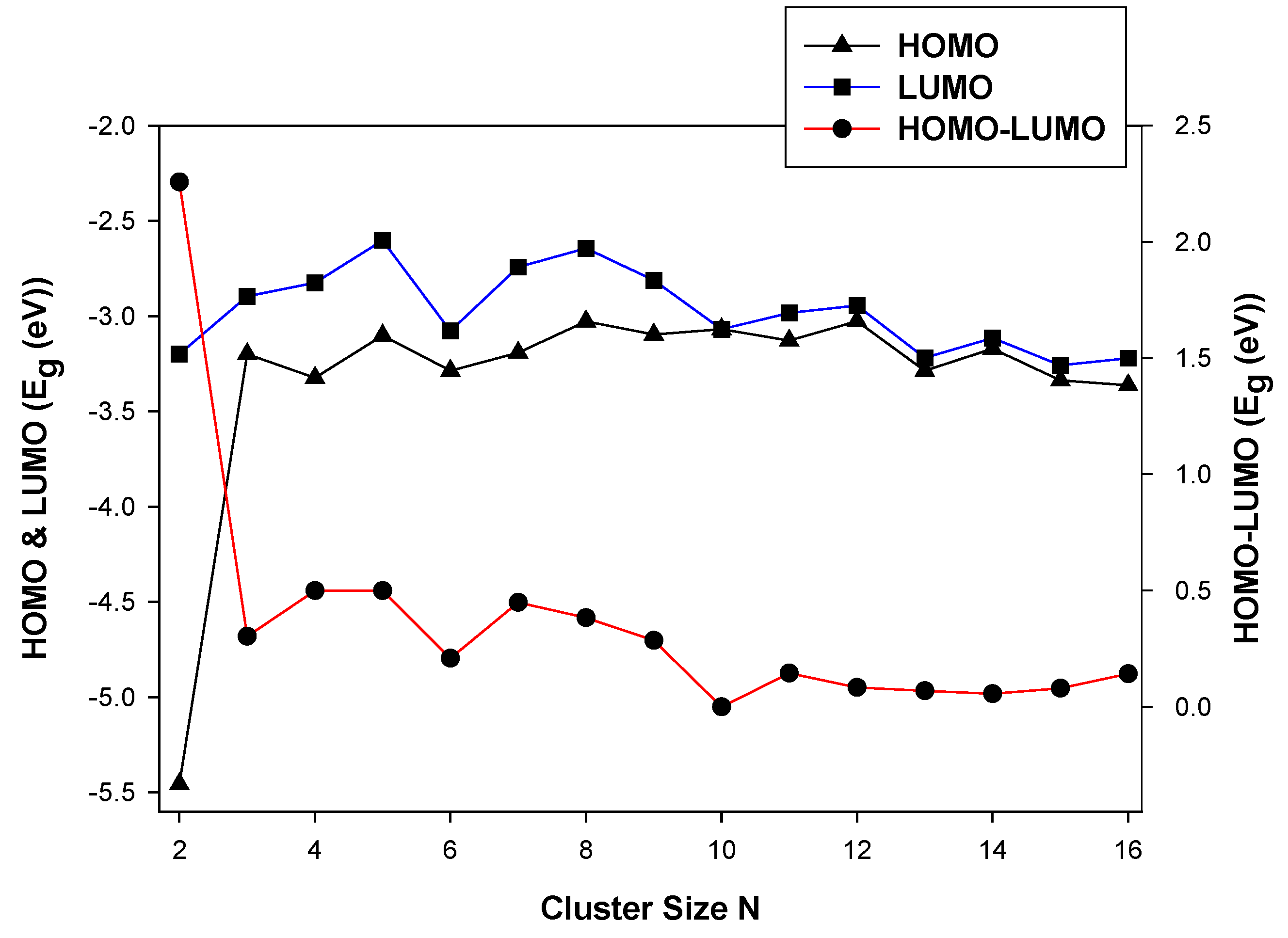
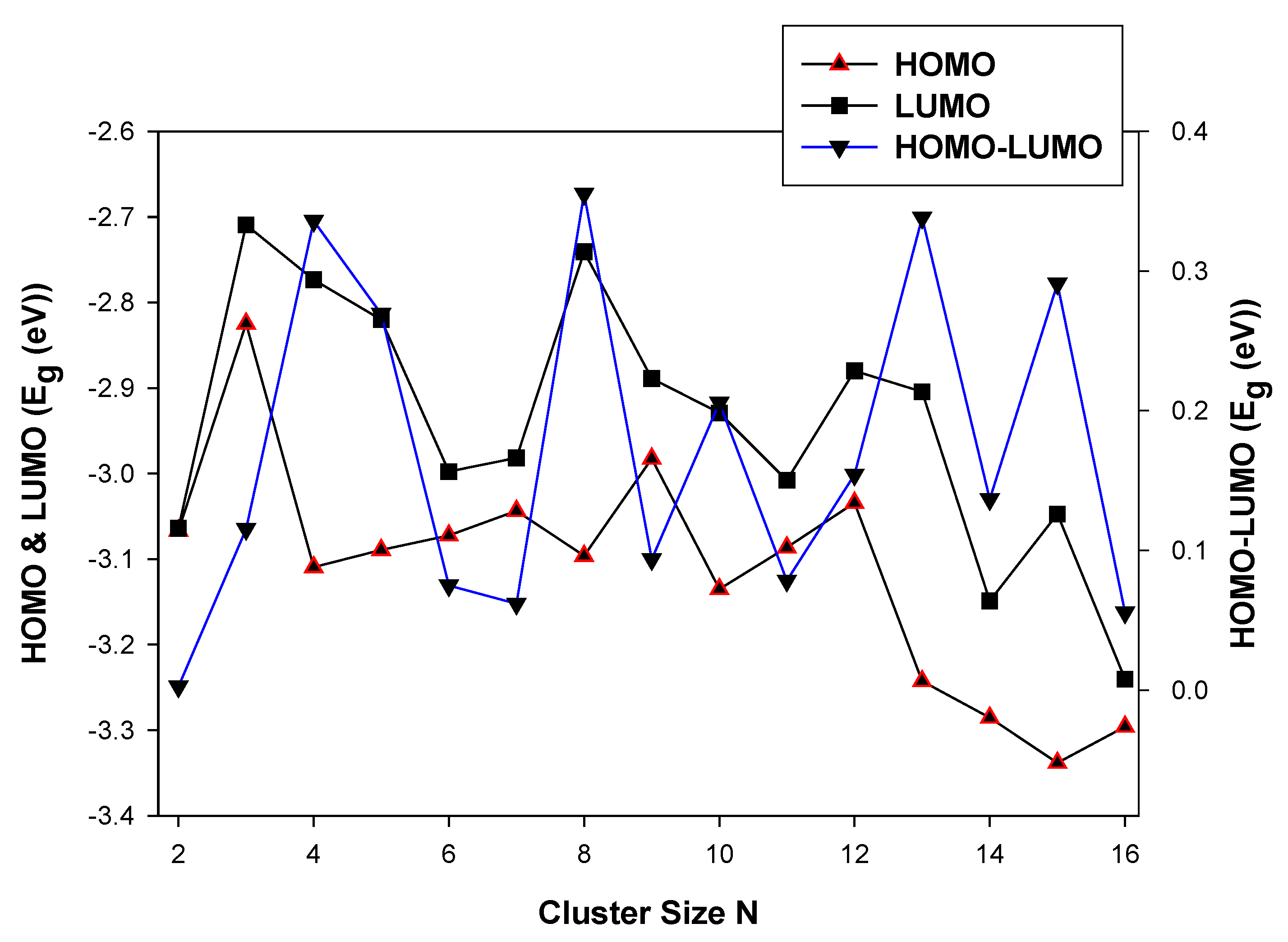
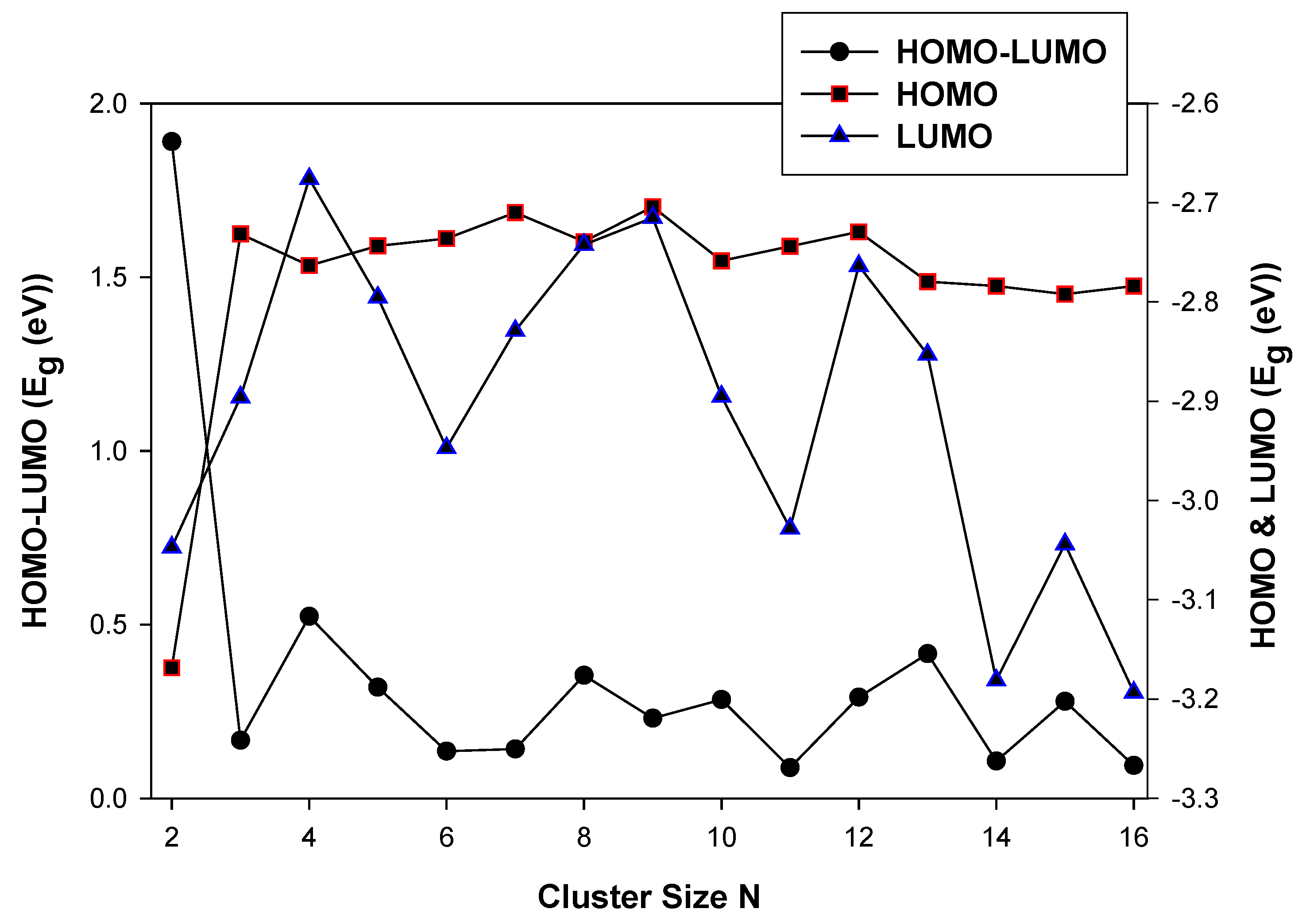
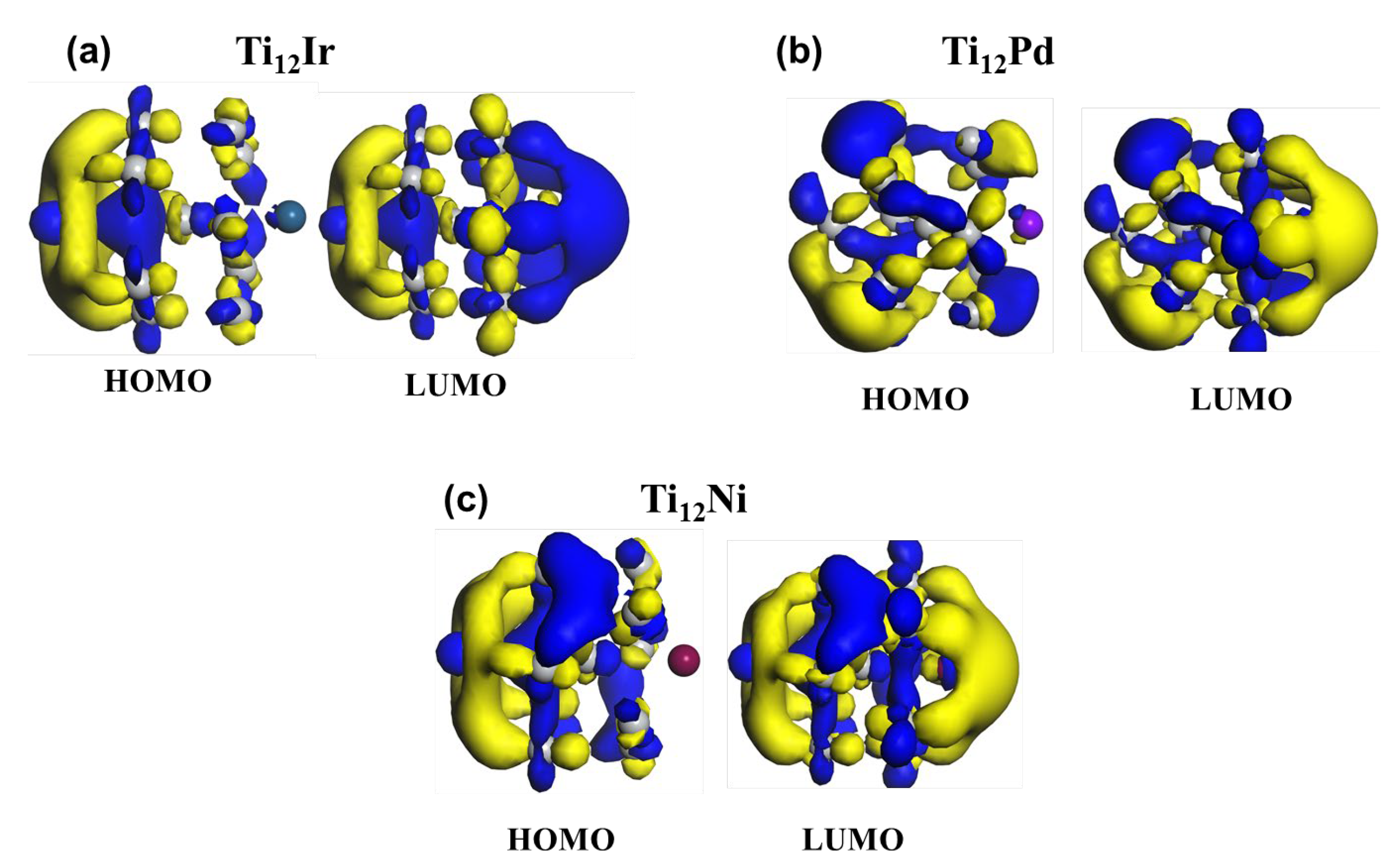
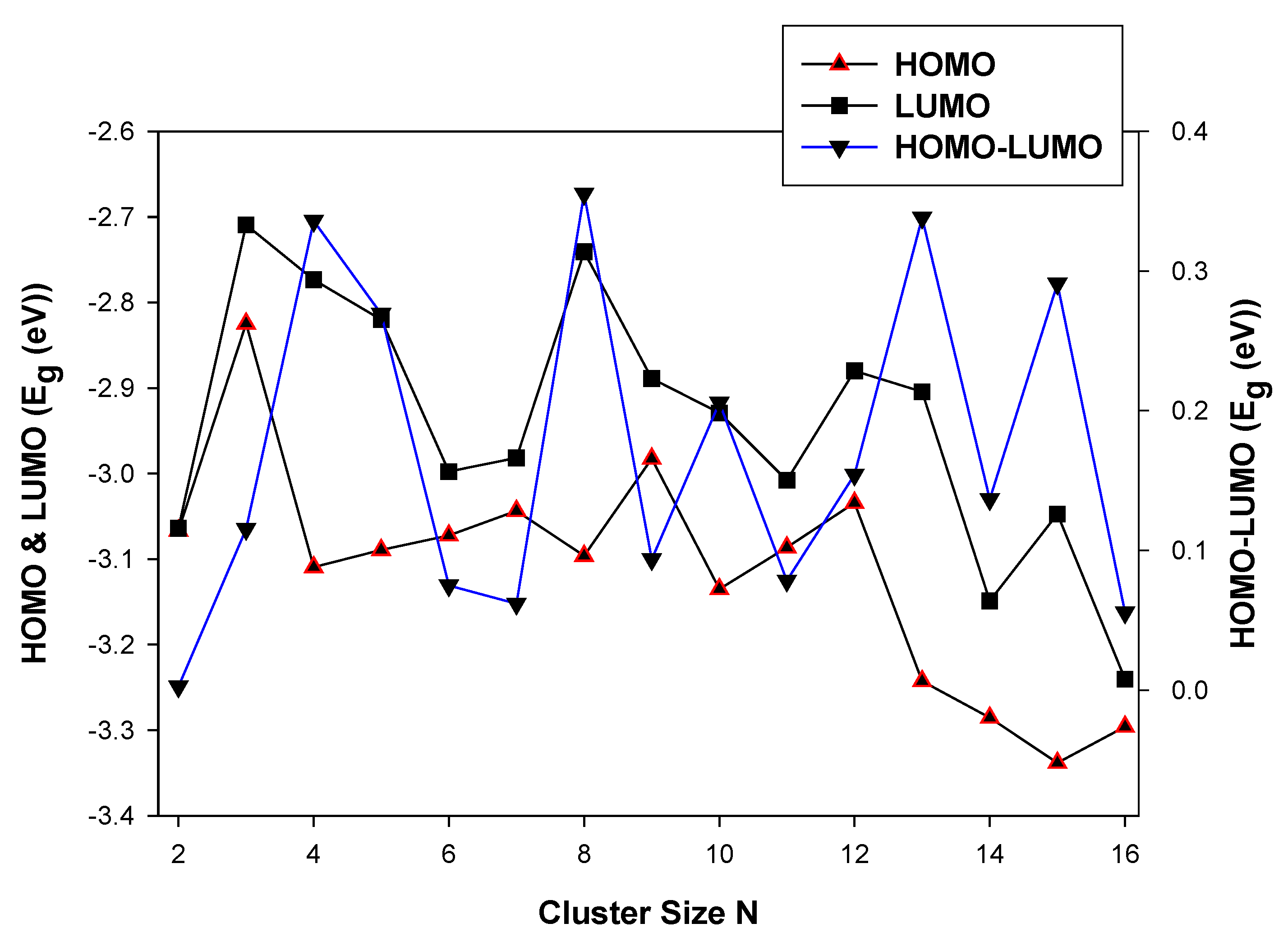
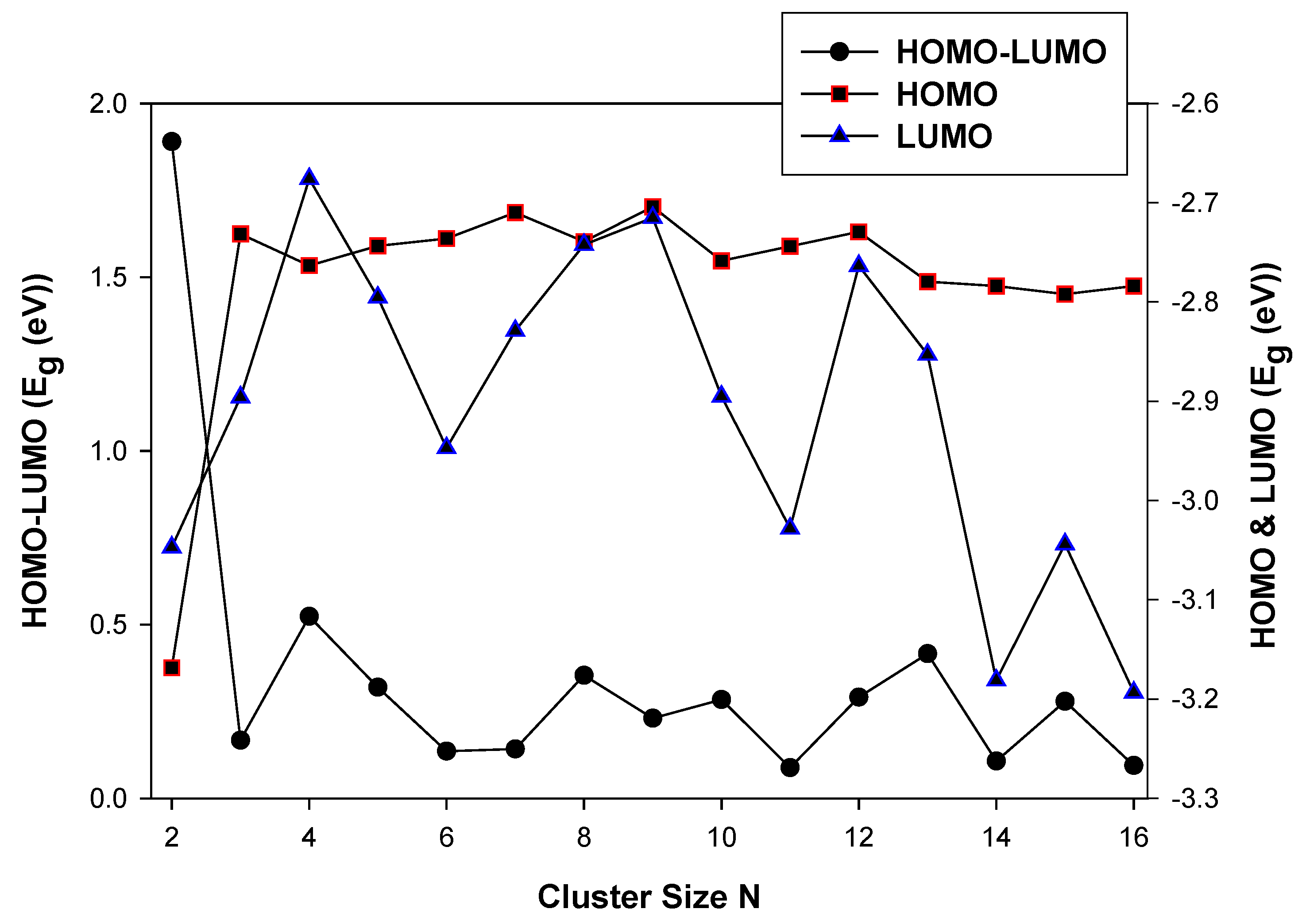
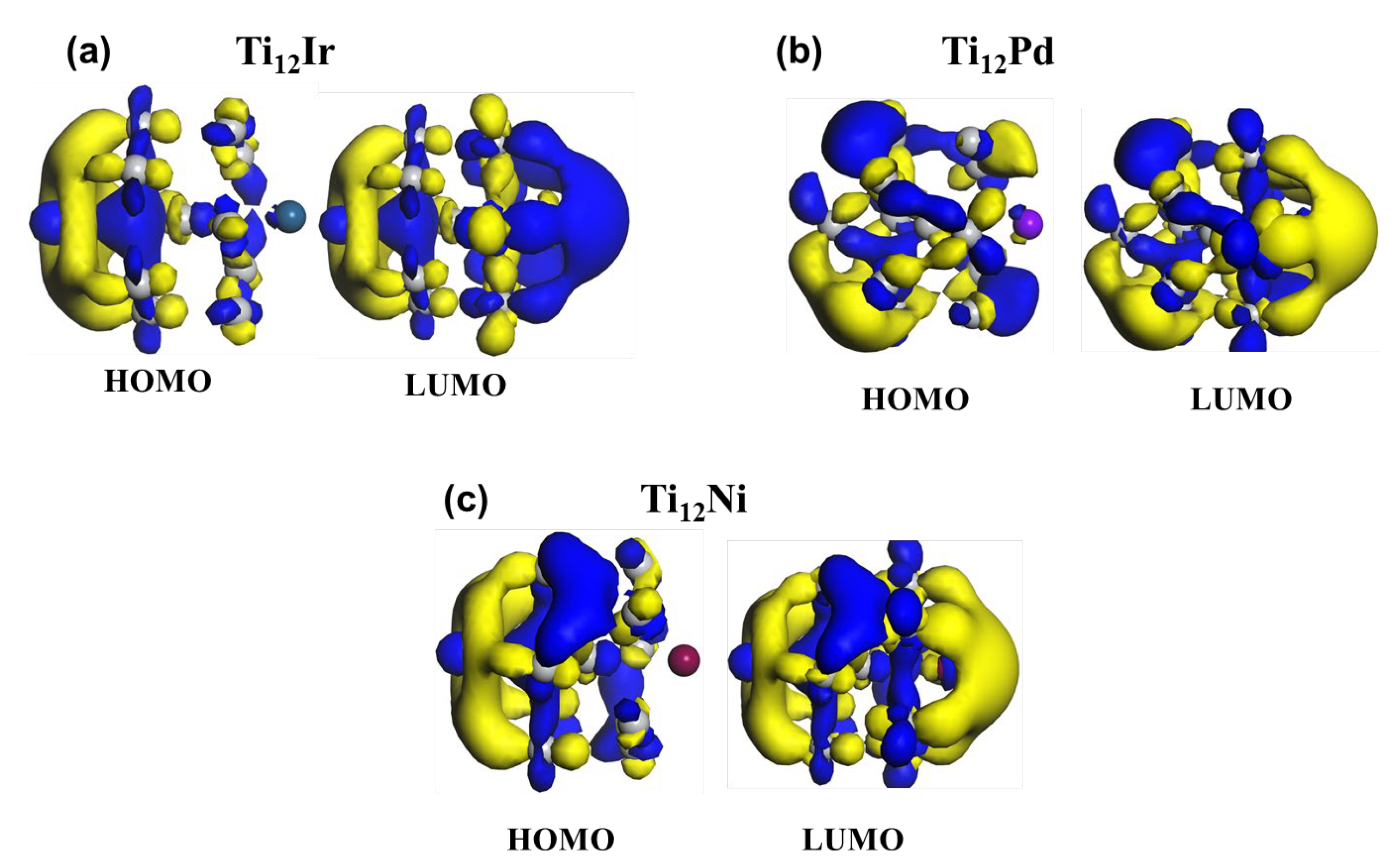
3.2.1. HOMO-LUMO Analysis for the Lowest-Energy Configurations
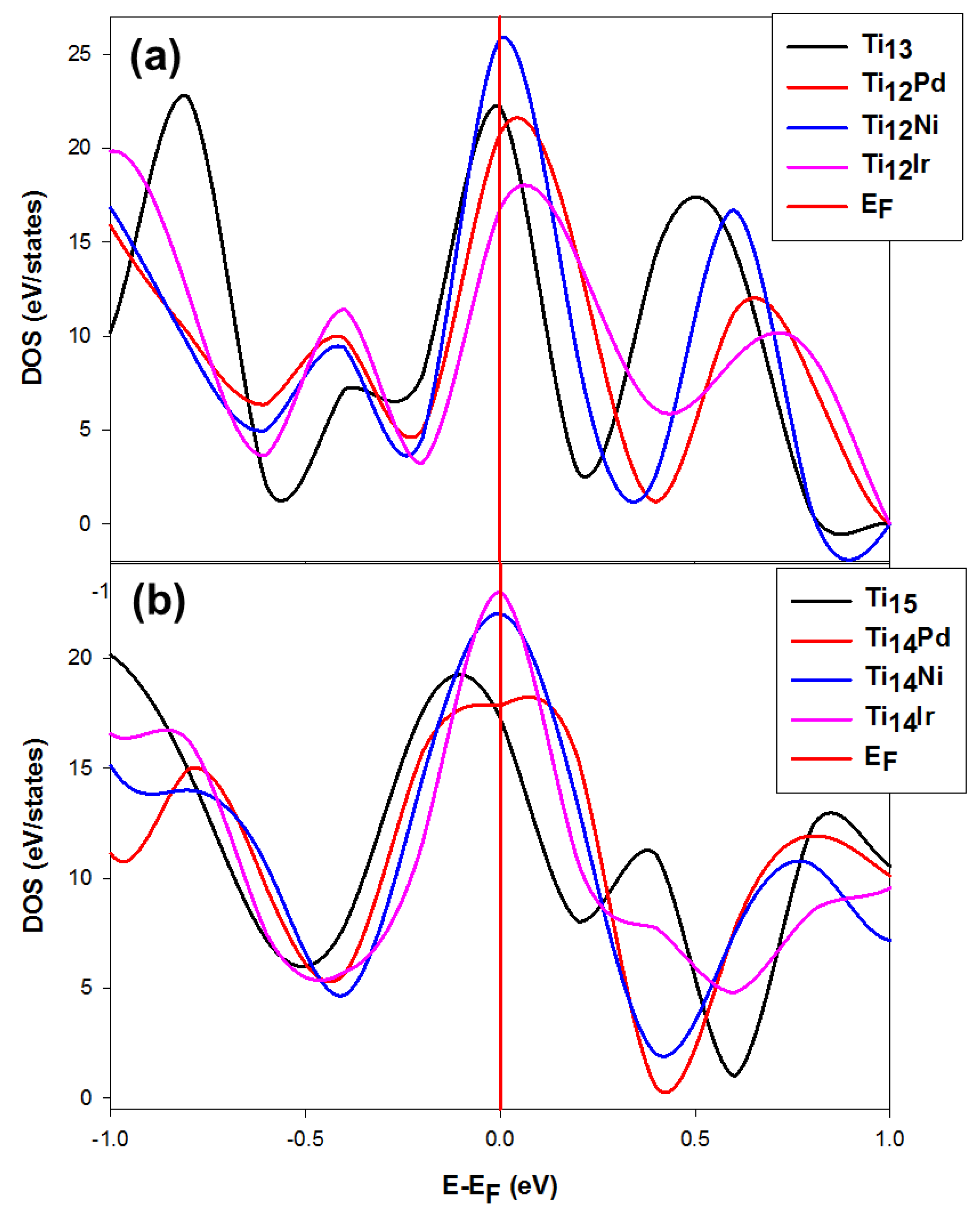
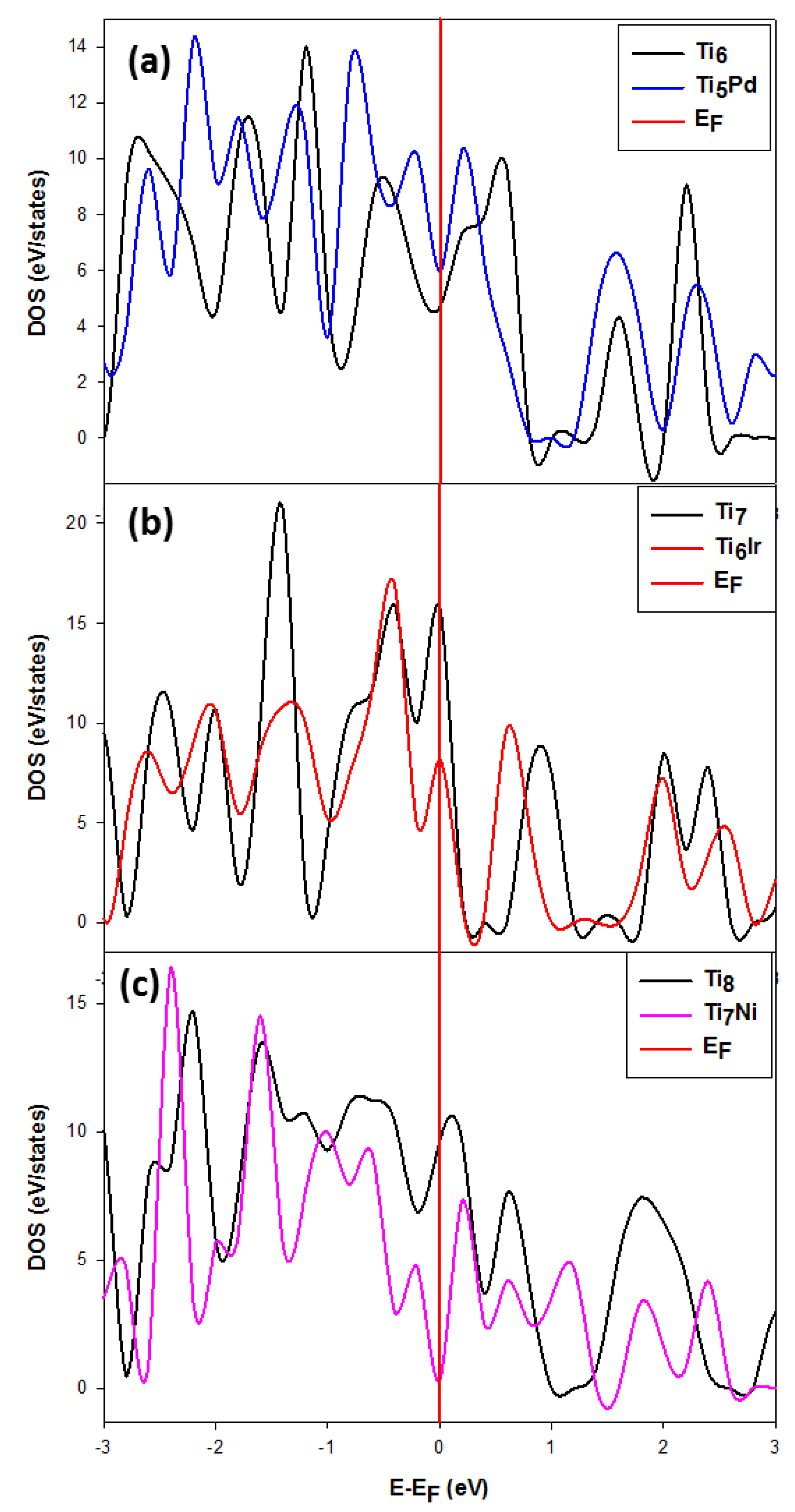
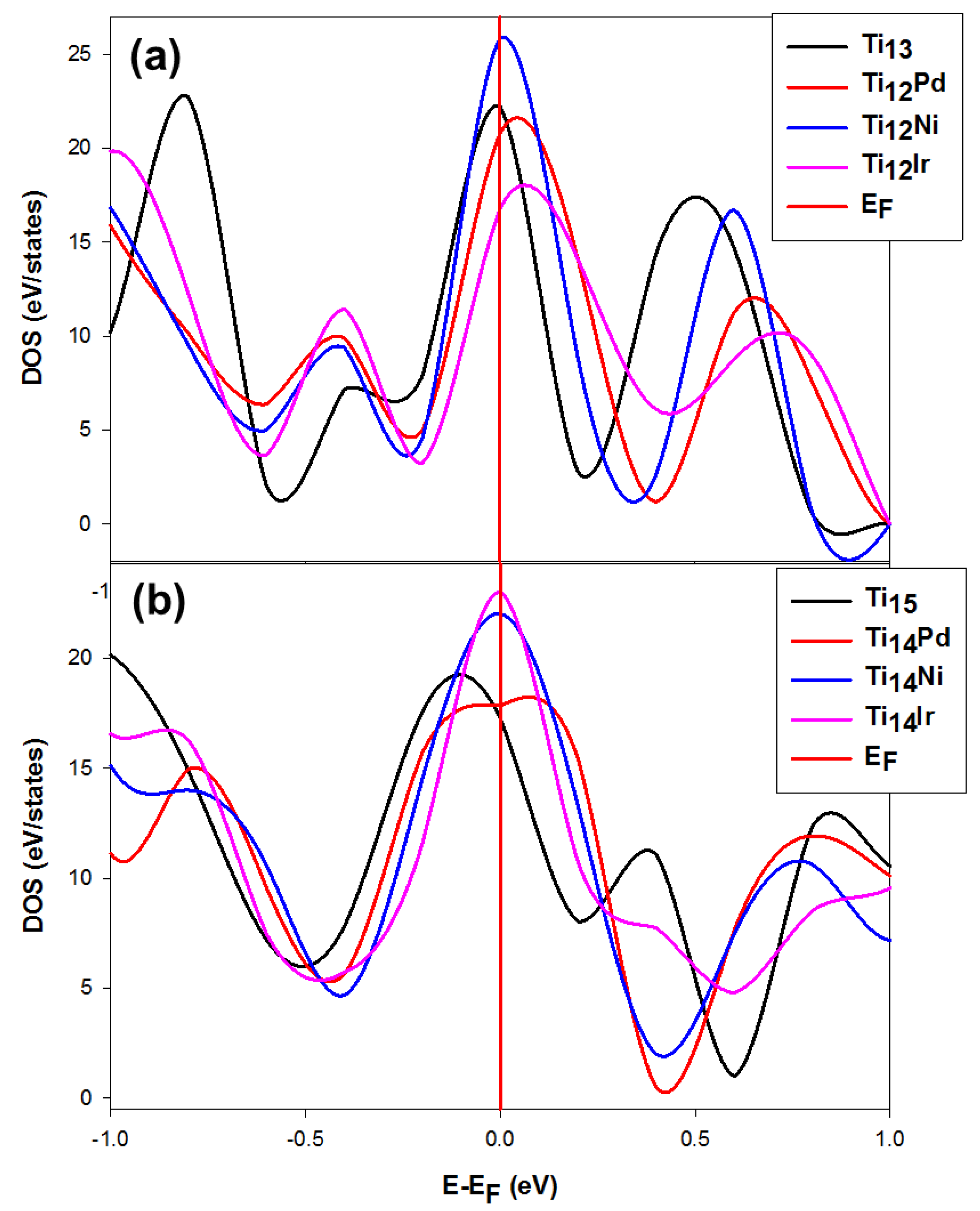
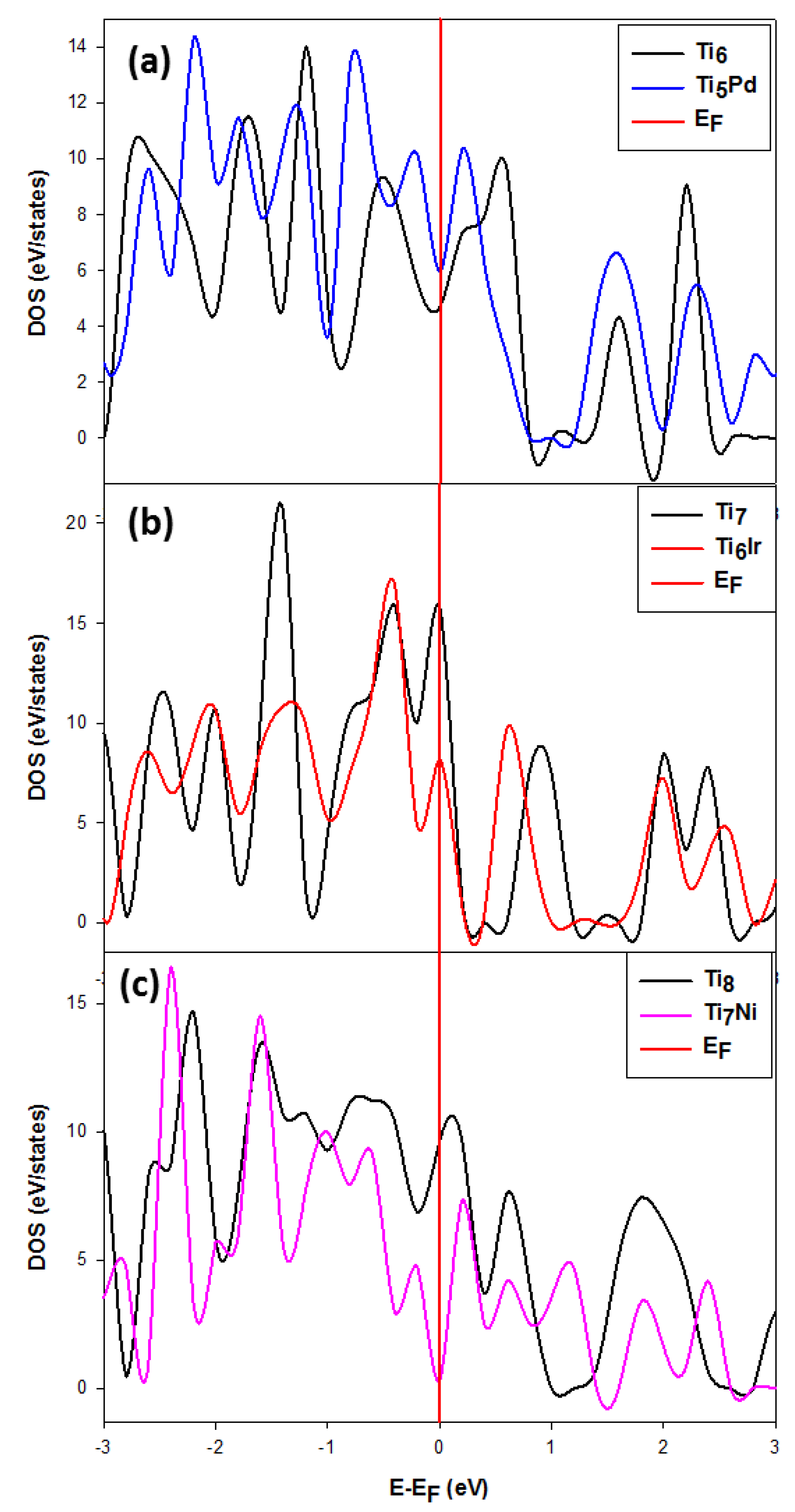
3.2.2. The Density of States (DOS) for the Stable and Metastable Doped Ti Nanoclusters
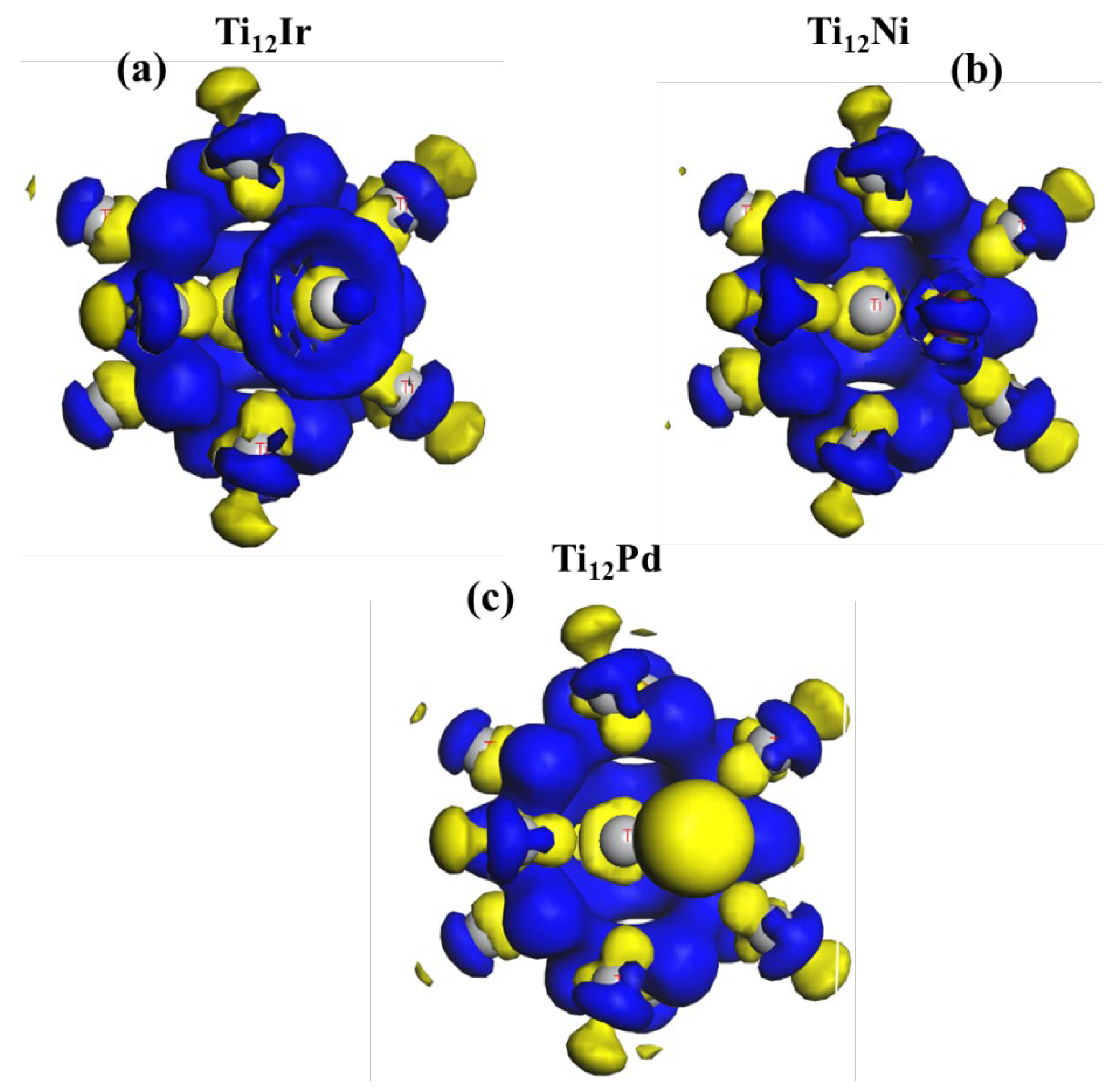
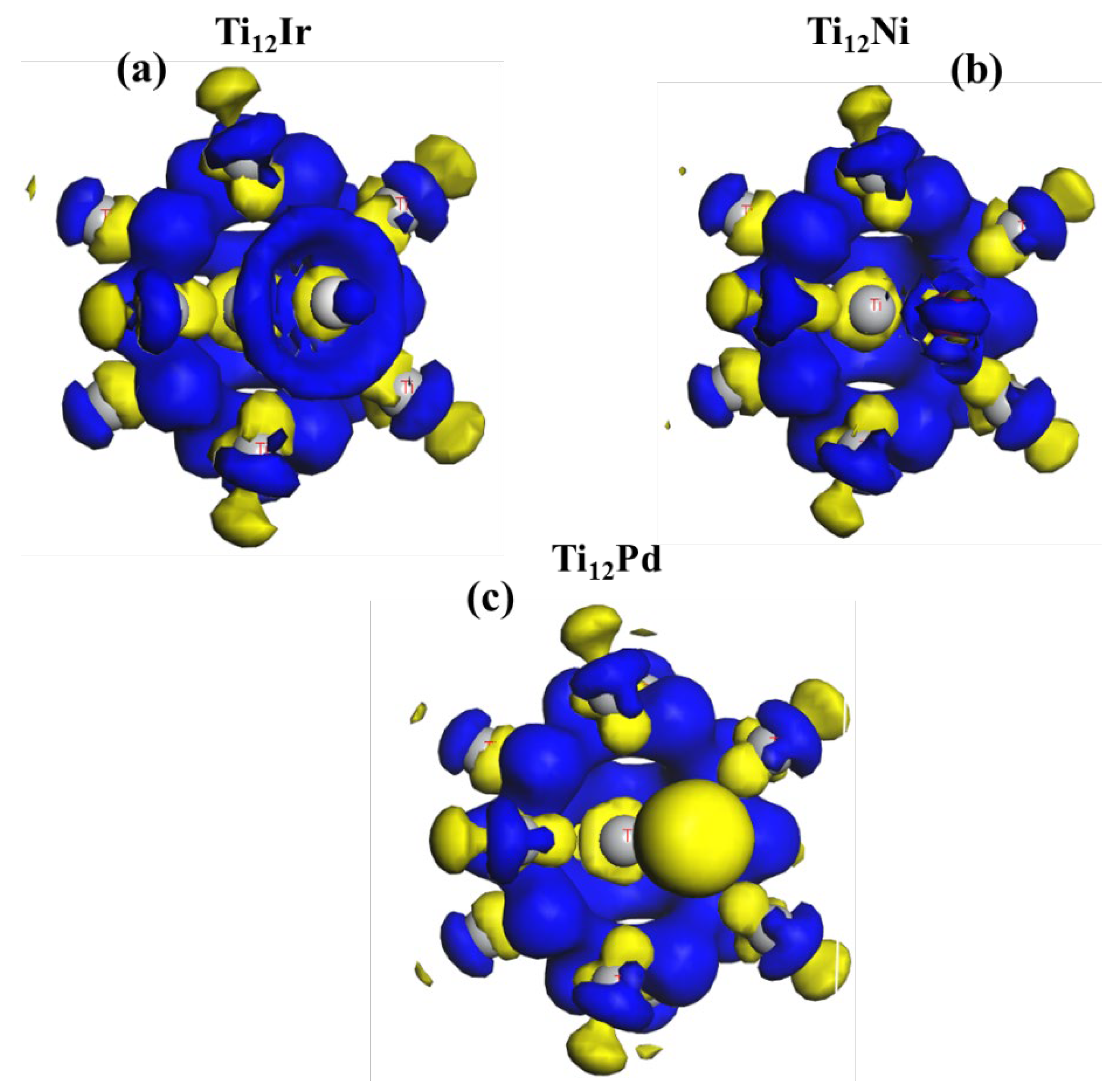
3.2.3. Charge Distribution Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Chen, O.; Angerhofer, A.; Cao, Y.C. On Doping CdS/ZnS Core/Shell Nanocrystals with Mn. J. Am. Chem. Soc. 2008, 130, 15649–15661. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Z.; Cao, L.; Chen, Y.; Zhu, E.; Lin, Z.; Li, M.; Yan, A.; Zettl, A.; Wang, Y.M.; et al. High-performance transition metal–doped Pt3Ni octahedra for oxygen reduction reaction. Science 2015, 348, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.J.; Efros, A.L.; Erwin, S.C. Doped Nanocrystals. Science 2008, 319, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Linic, S.; Christopher, P.; Xin, H.; Marimuthu, A. Catalytic and photocatalytic transformations on metal nanoparticles with targeted geometric and plasmonic properties. Acc. Chem. Res. 2013, 46, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Agasti, S.; Kim, C.; Li, X.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef]
- Jin, R.; Cao, Y.; Mirkin, C.A.; Kelly, K.L.; Schatz, G.C.; Zheng, J.G. Photoinduced Conversion of Silver Nanospheres to Nanoprisms. Science 2001, 294, 1901–1903. [Google Scholar] [CrossRef]
- Henglein, A. Small-particle research: Physicochemical properties of extremely small colloidal metal and semiconductor particles. Chem. Rev. 1989, 89, 1861–1873. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. Pd–Au bimetallic catalysts: Understanding alloy effects from planar models and (supported) nanoparticles. Chem. Soc. Rev. 2012, 41, 8009–8020. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Wang, Z.L.; El-Sayed, M.A.J. Alloy formation of gold–silver nanoparticles and the dependence of the plasmon absorption on their composition. Phys. Chem. B 1999, 103, 3529–3533. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Yeh, C.-S. A new approach for the formation of alloy nanoparticles: Laser synthesis of gold–silver alloy from gold–silver colloidal mixtures. Electronic supplementary information (ESI) available: Experimental details, UV–VIS spectra, TEM images and EDX analysis for molar ratios (Au:Ag) of 1:2 and 2:1. Chem. Commun. 2001, 4, 371–372. [Google Scholar] [CrossRef]
- Xiang, J.; Wei, S.H.; Yan, X.H.; You, J.Q.; Mao, Y.L. A density-functional study of Al-doped Ti clusters: TiNAl (N = 1–13). J. Chem. Phys. 2004, 120, 4251–4257. [Google Scholar] [CrossRef]
- Wang, B.L.; Zhao, J.J.; Shi, D.N.; Chen, X.S.; Wang, G.H. Geometric Transition and Electronic Properties of Titanium-Doped Aluminum Clusters. Phys. Rev. A 2005, 72, 023204. [Google Scholar] [CrossRef]
- Bergeron, D.E.; Castleman, A.W., Jr.; Morisato, T.; Khanna, S.N. Formation of Al13I-: Evidence for the Superhalogen Character of Al13. Science 2004, 304, 84–87. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Yang, C.J.; Li, S.T.J. Structures and stability of B-doped Al clusters: AlNB and AlNB2 (N = 1–7). Chem. Phys. 2005, 123, 204315. [Google Scholar] [CrossRef]
- Bergeron, D.E.; Roach, P.J.; Castleman, A.W., Jr.; Jones, N.O.; Khanna, S.N. Al Cluster Superatoms as Halogens in Polyhalides and as Alkaline Earths in Iodide Salts. Science 2005, 307, 231–235. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold Catalysts Prepared by Coprecipitation for Low-Temperature Oxidation of Hydrogen and of Carbon Monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Sanchez, A.; Abbet, S.; Heiz, U.; Schneider, W.D.; Håkkinen, H.; Barnett, R.N.; Landman, U. When Gold Is Not Noble: Nanoscale Gold Catalysts. J. Phys. Chem. A 1999, 103, 9573–9578. [Google Scholar] [CrossRef]
- Neumaier, M.; Weigend, F.; Hampe, O.; Kappes, M.M. Binding energies of CO on gold cluster cations Aun+ (n = 1−65): A radiative association kinetics study. J. Chem. Phys. 2005, 122, 104702. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Bernhardt, T.M.; Chernyy, V.; Bakker, J.M.; Barnett, R.N.; Landman, U. Selective C-H bond cleavage in methane by small gold clusters. Angew. Chem. Int. Ed. 2017, 56, 13406–13410. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Vanbuel, J.; Janssens, E.; Lievens, P. Tuning the reactivity of small metal clusters by heteroatom doping. Acc. Chem. Res. 2018, 51, 3174–3182. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the over potential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Reitz, W. Handbook of fuel cells: Fundamentals, technology, and applications, (volume 2), in: W. Vielstich, A. Lamm, H.A. Gasteiger (editors). Mater. Manuf. 2007, 22, 789. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Blizanac, B.B.; Arenz, M.; Stamenkovic, V.R.; Ross, P.N.; Markovic, N.M. The impact of geometric and surface electronic properties of Pt-catalysts on the particle size effect in electrocatalysis. J. Phys. Chem. B 2005, 109, 14433–14440. [Google Scholar] [CrossRef]
- Han, B.C.; Miranda, C.R.; Ceder, G. Effect of particle size and surface structure on adsorption of O and OH on platinum nanoparticles: A first-principles study. Phys. Rev. B 2008, 77, 075410. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Toyoda, E.; Hatanaka, T.; Morimoto, Y. First principles calculations on site-dependent dissolution potentials of supported and unsupported Pt particles. J. Phys. Chem. C 2010, 114, 17557–17568. [Google Scholar] [CrossRef]
- Antolini, E.; Salgado, J.; Gonzalez, E. The stability of Pt-M (M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells: A literature review and tests on a Pt Co catalyst. J. Power Sources 2006, 160, 957–968. [Google Scholar] [CrossRef]
- Piotrowski, M.; Piquini, P.; Da Silva, J. Platinum-based nanoalloys PtnTM55-n (TM = Co, Rh, Au): A density functional theory investigation. J. Phys. Chem. C 2021, 116, 18432–18439. [Google Scholar] [CrossRef]
- Guedes-Sobrinho, D.; Nomiyama, R.K.; Chaves, A.S.; Piotrowski, M.J.; Da Silva, J.L.F. Structure, electronic, and magnetic properties of binary PtnTM55-n (TM = Fe, Co, Ni, Cu, Zn) nanoclusters: A density functional theory investigation. J. Phys. Chem. C 2015, 119, 15669–15679. [Google Scholar] [CrossRef]
- Du, J.G.; Sun, X.Y.; Jiang, G. A DFT study on small M-doped titanium (M = V, Fe, Ni) clusters: Structures, chemical bonds and magnetic properties. J. Eur. Phys. D 2009, 55, 111–120. [Google Scholar] [CrossRef]
- Du, J.; Sun, X.; Chen, J.; Jiang, G. The changes in the geometrical, electronic and magnetic properties of titanium clusters as one titanium atom is substituted by boron. J. Phys. B At. Mol. Opt. Phys. 2010, 43, 205103. [Google Scholar] [CrossRef]
- Rodríguez-Kessler, P.L.; Rodríguez-Domínguez, A.R. Structures and Electronic Properties of TiNV (N = 1−16) Clusters: First-Principles Calculations. J. Phys. Chem. A 2016, 120, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Verkhovtsev, A.V.; Hanauske, M.; Yakubovich, A.V.; Solov’yov, A.V. Characterization of small pure and Ni-doped titanium clusters: Ab initio versus classical approaches. Comput. Mater. Sci. 2013, 76, 80–88. [Google Scholar] [CrossRef]
- Phaahla, T.M.; Ngoepe, P.E.; Catlow, C.R.A.; Chauke, H.R. The effect of doping with pt impurity on ti clusters: A density functional theory study. Suid-Afrik. Tydskr. Natwet. Tegnol. 2021, 40, 75–78. [Google Scholar] [CrossRef]
- Woodley, S.M. Knowledge Led Master Code Search for Atomic and Electronic Structures of LaF3 Nanoclusters on Hybrid Rigid Ion–Shell Model–DFT Landscapes. J. Phys. Chem. C 2013, 117, 24003–24014. [Google Scholar] [CrossRef]
- Farrow, M.R.; Chow, Y.; Woodley, S.M. Structure prediction of nanoclusters; a direct or a pre-screened search on the DFT energy landscape? Phys. Chem. Chem. Phys. 2014, 16, 21119–21134. [Google Scholar] [CrossRef] [PubMed]
- Lazauskas, T.; Sokol, A.A.; Buckeridge, J.; Catlow, C.R.A.; Escher, S.G.E.T.; Farrow, M.R.; Mora-Fonz, D.; Blum, V.W.; Phaahla, T.M.; Chauke, H.R.; et al. Thermodynamically accessible titanium clusters TiN, N = 2–32. Phys. Chem. Chem. Phys. 2018, 20, 13962–13973. [Google Scholar] [CrossRef]
- Mattson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing meaningful density functional theory calculations in materials science. J. Mod. Simul. Mater. Sci. Eng. 2005, 13, 1–32. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Lazauskas, T.; Sokol, A.A.; Woodley, S.M. An efficient genetic algorithm for structure prediction at the nanoscale. Nanoscale 2017, 9, 3850–3864. [Google Scholar] [CrossRef]
- Escher, S.G.; Lazauskas, T.; Zwijnenburg, M.A.; Woodley, S.M. Structure prediction of (BaO) n nanoclusters for n ≤ 24 using an evolutionary algorithm. Comput. Theor. Chem. 2017, 1107, 74–81. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Mora-Fonz, D.; Lazauskas, T.; Farrow, M.R.; Catlow, C.R.A.; Woodley, S.M.; Sokol, A.A. Why are polar surfaces of ZnO stable? Chem. Mater. 2017, 29, 5306–5320. [Google Scholar] [CrossRef]
- Mora-Fonz, D.; Buckeridge, J.; Logsdail, A.J.; Scanlon, D.O.; Sokol, A.A.; Woodley, S.M.; Catlow, C.R.A. Morphological features and band bending at nonpolar surfaces of ZnO. J. Phys. Chem. C 2015, 119, 11598–11611. [Google Scholar] [CrossRef]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Vanlenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Burke, K.; Perdew, J.P.; Ernzerhof, M. Why the generalized gradient approximation works and how to go beyond it. Int. J. Quantum Chem. 1997, 61, 287–293. [Google Scholar] [CrossRef]
- Milman, V.; Winkler, B.; White, J.A.; Pickard, C.J.; Payne, M.C.; Akhmatskaya, E.V.; Nobes, R.H. Electronic structure of materials. Int. J. Quantum Chem. 2000, 77, 799–926. [Google Scholar]
- Segall, M.D.; Lindan, P.L.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalised gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1999, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Kumar, V.; Kawazoe, Y. Metal-encapsulated fullerenelike and cubic caged clusters of silicon. Phys. Rev. Lett. 2001, 87, 045503. [Google Scholar] [CrossRef]
- Kumar, V.; Kawazoe, Y. Metal-encapsulated caged clusters of germanium with large gaps and different growth behavior than silicon. Phys. Rev. Lett. 2002, 88, 235504. [Google Scholar] [CrossRef]
- Sakurai, M.; Watanabe, K.; Sumiyama, K.; Suzuki, K. Magic numbers in transition metal (Fe, Ti, Zr, Nb, and Ta) clusters observed by Time-of-flight mass spectroscopy. J. Chem. Phys. 1999, 111, 235–238. [Google Scholar] [CrossRef]
- Wu, H.; Desai, S.R.; Wang, L.-S. Electronic structure of small titanium clusters: Emergence and evolution of the 3d band. Phys. Rev. Lett. 1996, 76, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qiu, Q.; Wang, B.; Wang, J.; Wang, G. Geometric and electronic properties of titanium clusters studied by ultrasoft pseudopotential. Solid State Commun. 2001, 118, 157–161. [Google Scholar] [CrossRef]
- Salazar-Villanueva, M.; Hernandez Tejeda, P.H.; Pal, U.; Rivas-Silva, J.F.; Rodriquez Mora, J.L.; Ascencio, J.A. Stable TiN (n = 2–15) clusters and their geometries: DFT calculations. J. Phys. Chem. A 2006, 110, 10274–10278. [Google Scholar] [CrossRef]
- Hurtado, R.B.; Cortez-Valadez, M.; Gámez-Corrales, R.; Flores-Acosta, M. Structural and vibrational properties of gold-doped titanium clusters: A first-principles study. Comput. Theor. Chem. 2018, 1124, 32–38. [Google Scholar] [CrossRef]
- Hoffmann, R.; Howell, J.M.; Rossi, A.R. Bicapped tetrahedral, trigonal prismatic, and octahedral alternatives in main and transition group six-coordination. J. Am. Chem. Soc. 1976, 98, 2484–2492. [Google Scholar] [CrossRef]
- Hejazi, S.H.; Trevelyan, P.M.J.; Azaiez, J.; De Wit, A. Viscous fingering of a miscible reactive A + B → C interface: A linear stability analysis. J. Fluid Mech. 2010, 652, 501–528. [Google Scholar] [CrossRef]
- Smith, A.M.; Nie, S. Semiconductor Nanocrystals: Structure, Properties, and Band Gap Engineering. Acc. Chem. Res. 2010, 43, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P. Perspectives on the physical chemistry of semiconductor nanocrystals. J. Phys. Chem. 1996, 100, 13226–13239. [Google Scholar] [CrossRef]
- Mahlangu, R.; Phasha, M.J.; Chauke, H.R.; Ngoepe, P.E. Structural, elastic and electronic properties of equiatomic PtTi as potential high-temperature shape memory alloy. Intermetallics 2013, 33, 27–32. [Google Scholar] [CrossRef]
- Chauke, H.R.; Minisini, B.; Drautz, R.; Nguyen-Manh, D.; Ngoepe, P.E.; Pettifor, D.G. Theoretical investigation of the Pt3Al ground state. Intermetallics 2010, 18, 417–421. [Google Scholar] [CrossRef]
- Tshwane, D.M.; Modiba, R.; Govender, G.; Ngoepe, P.E.; Chauke, H.R. The adsorption of halogen molecules on Ti (110) surface. J. Mater. Res. 2020, 36, 592–601. [Google Scholar] [CrossRef]
- Corneliu, I.O.; Mihai, A.G. Structure and electronic properties of TiO2 nanoclusters and dye-nanocluster systems appropriate to model hybrid photovoltaic or photocatalytic applications. J. Nanomater. 2019, 357, 7–10. [Google Scholar]
- Bader, R.F.W.; Henneker, W.H. Molecular charge distributions and chemical binding. J. Chem. Phys. 1967, 46, 3361–3363. [Google Scholar] [CrossRef]
- Dutta, A.; Mondal, P. Density functional theory study of structure, electronic and magnetic properties of non-metal (Group 13) doped stable Rhn (n = 2–8) clusters and catalytic activities towards methanol activities. J. Chem. Sci. 2018, 130, 1–22. [Google Scholar] [CrossRef]
- Bautista Hernandez, A.; Cortes-Arriagada, D.; Camacho Garcia, H.; Chigo Anota, E.; Salaza Villanueva, M. Quantum molecular study on doping effect in titanium and vanadium clusters: Their application to remove some chemical species. Appl. Nanosci. 2020, 10, 37–49. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phaahla, T.; Ngoepe, P.; Chauke, H. Exploring Synergistic Effect on the Stability of Ni-, Pd-, Ir-Doped TiN (N = 1–15) Nanoparticles. Alloys 2024, 3, 15-30. https://doi.org/10.3390/alloys3010002
Phaahla T, Ngoepe P, Chauke H. Exploring Synergistic Effect on the Stability of Ni-, Pd-, Ir-Doped TiN (N = 1–15) Nanoparticles. Alloys. 2024; 3(1):15-30. https://doi.org/10.3390/alloys3010002
Chicago/Turabian StylePhaahla, Tshegofatso, Phuti Ngoepe, and Hasani Chauke. 2024. "Exploring Synergistic Effect on the Stability of Ni-, Pd-, Ir-Doped TiN (N = 1–15) Nanoparticles" Alloys 3, no. 1: 15-30. https://doi.org/10.3390/alloys3010002
APA StylePhaahla, T., Ngoepe, P., & Chauke, H. (2024). Exploring Synergistic Effect on the Stability of Ni-, Pd-, Ir-Doped TiN (N = 1–15) Nanoparticles. Alloys, 3(1), 15-30. https://doi.org/10.3390/alloys3010002