
First-Principles Study on Thermodynamic, Structural, Mechanical, Electronic, and Phonon Properties of tP16 Ru-Based Alloys

Abstract
1. Introduction
2. Methods
3. Results and Discussion
3.1. Crystal Structure
3.2. Mechanical Properties
3.2.1. Elastic Constants
3.2.2. Bulk, Shear, and Young’s Moduli
3.2.3. Elastic Anisotropy
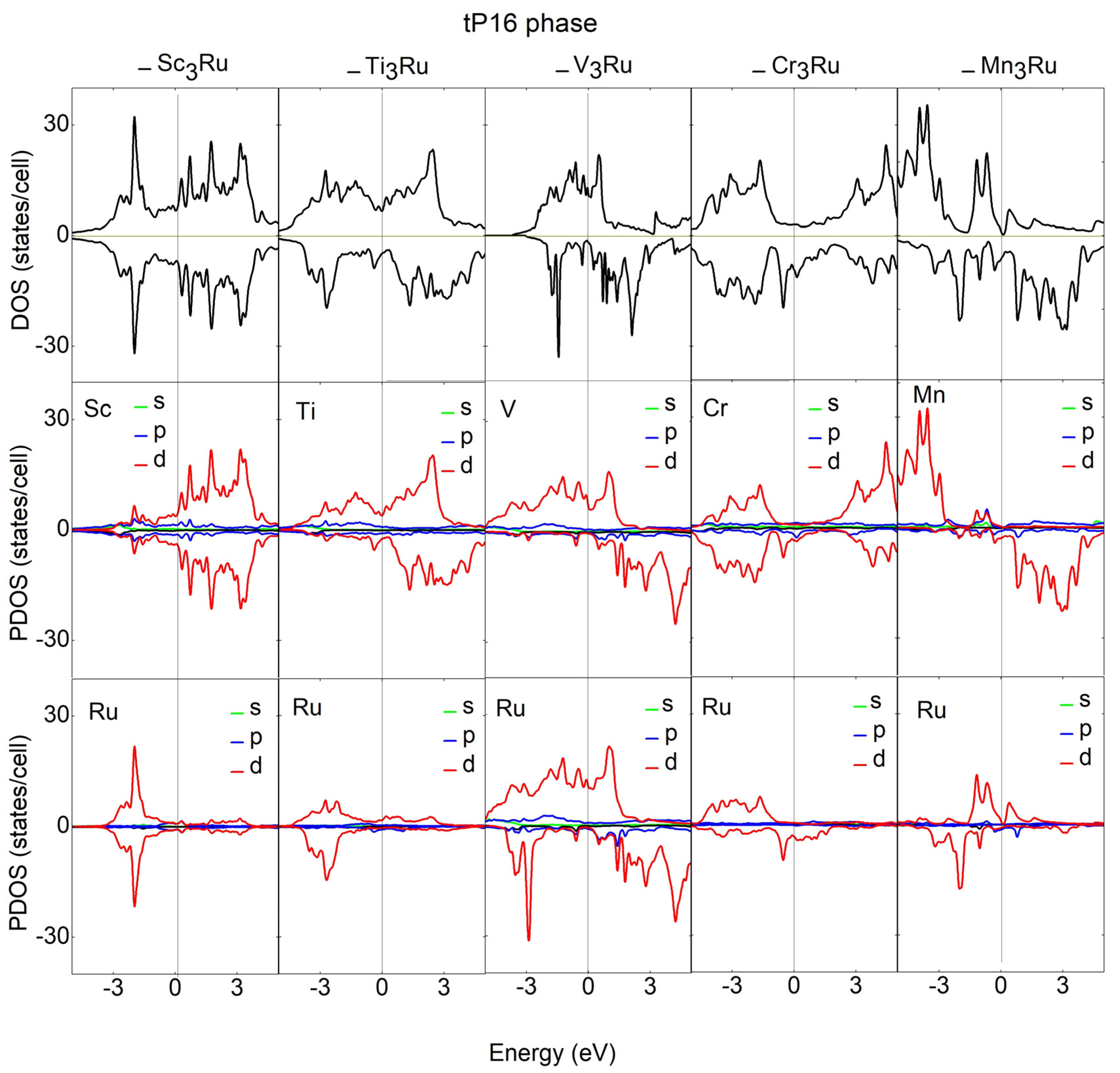
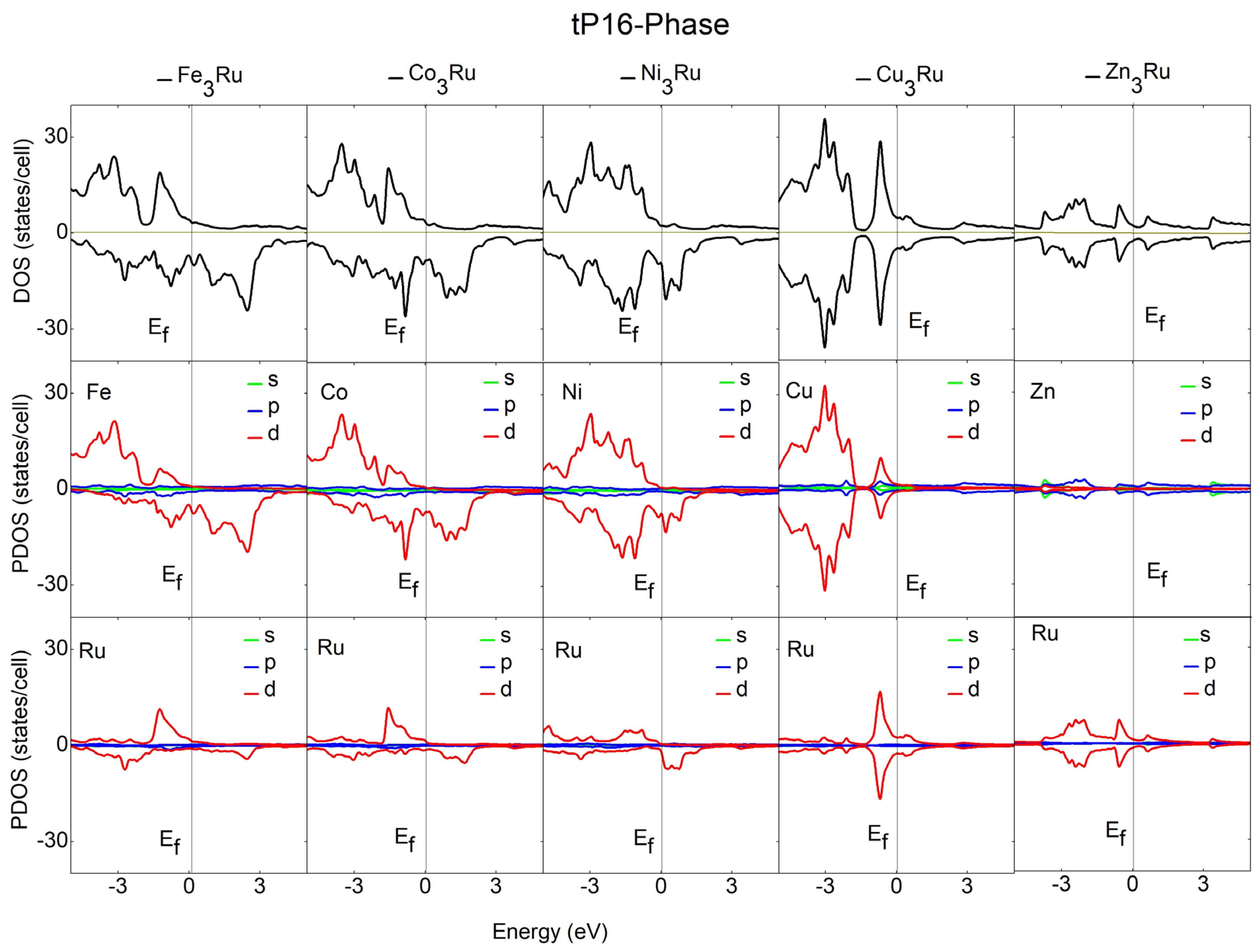
3.3. Electronic and Magnetic Properties
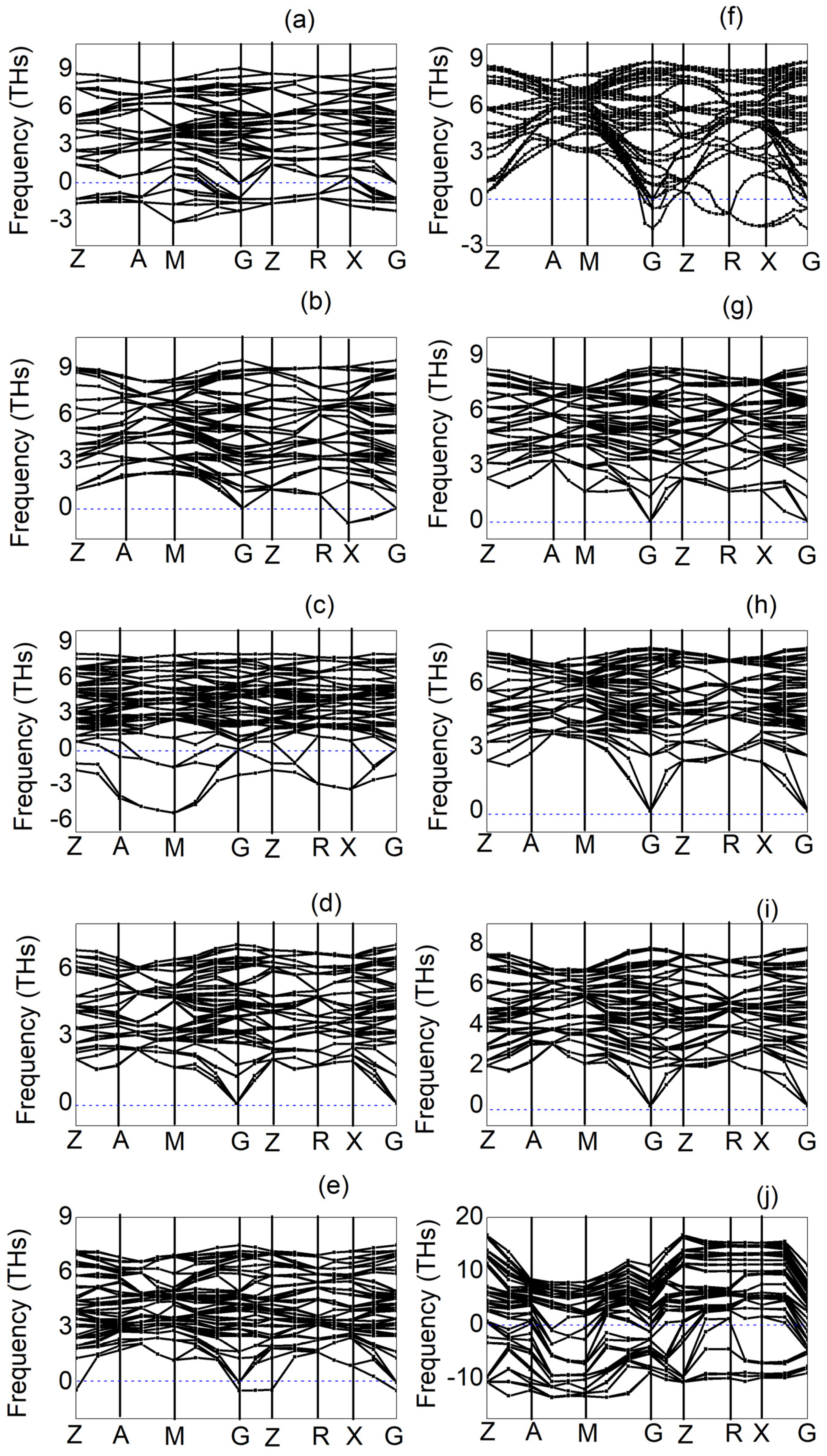
3.4. Phonon Dispersion Curves
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, P.J.; Yamabe-Mitarai, Y.; Wolff, I.M. High-temperature compression strengths of precipitation-strengthened ternary Pt-Al-X alloys. Scr. Mater. 2001, 44, 43–48. [Google Scholar] [CrossRef]
- Ravindran, P.; Subramoniam, G.; Asokamani, R. Ground-state properties and relative stability between the L12 and DOa phases of Ni3Al by Nb substitution. Phys. Rev. B 1996, 53, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ding, Q.; Wei, X.; Wang, J.; Zhang, Z.; Bei, H. The effects of key elements Re and Ru on the phase morphologies and microstructure in Ni-based single crystal superalloys. J. Alloys Compd. 2022, 926, 166835. [Google Scholar] [CrossRef]
- Lee, S.; Do, J.; Jang, K.; Jun, H.; Park, Y.; Choi, P.P. Promotion of topologically close-packed phases in a Ru-containing Ni-based superalloy. Scr. Mater. 2023, 222, 115041. [Google Scholar] [CrossRef]
- Wang, S.; Meng, F.; Wang, L.; Yu, H.; Sun, D. The Effect of Ru on the Evolution of the γ′ Phase in Ni-Al-Ru Alloys. Materials 2022, 15, 3344. [Google Scholar] [CrossRef] [PubMed]
- Sims, C.T.; Stoloff, N.S.; Hagel, W.C. Superalloys II; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Yamabe-Mitarai, Y.; Gu, Y.; Huang, C.; Völkl, R.; Harada, H. Platinum-group-metal-based intermetallics as high-temperature structural materials. JOM 2004, 56, 34–39. [Google Scholar] [CrossRef]
- Shongwe, M.B.; Witcomb, M.J.; Cornish, L.A.; Papo, M.J. TEM studies of Pt-Al-Cr-Ru. Alloys 2012, 7, 12–14. [Google Scholar]
- Bewlay, B.P.; Jackson, M.R.; Zhao, J.-C.; Subramanian, P.R.; Mendiratta, M.G.; Lewandowski, J.J. Ultrahigh-temperature Nb-silicide-based composites. MRS Bull. 2003, 28, 646–653. [Google Scholar] [CrossRef]
- Harris, D.C.; Cabri, L.J. Nomenclature of platinum-group-element alloys: Review and revision. Can. Miner. 1991, 29, 231–237. [Google Scholar]
- Massalski, T.B.; Murray, J.L. Binary Phase Diagrams; ASM International: Almere, The Netherlands, 1990. [Google Scholar]
- Chattopadhyay, T.; Schubert, K. Kristallstruktur von Pt3Ga(r) und einigen phasen der mischung Pt–Al. J. Less Common Met. 1975, 41, 19–32. [Google Scholar] [CrossRef]
- Oya, Y.; Mishima, U.; Suzuki, T. L12 D0c martensitic transformation in Pt3Al and Pt3Ga. Z. Met. 1987, 78, 485–490. [Google Scholar]
- Kimmel, G. On the U3Si (Doc) crystallographic type. J. Less Common Met. 1978, 59, P83–P86. [Google Scholar] [CrossRef]
- Brandes, E.A.; Brook, G.B. Smithells Metals Reference Book; Butterworth: Butterworth, Malaysia, 1992. [Google Scholar]
- Tibane, M.M. Phase Stability Study of Pt-Cr and Ru-Cr Binary Alloys. Ph.D. Thesis, University of Limpopo, Polokwane, South Africa, 2011. [Google Scholar]
- Chauke, H.R.; Minisini, B.; Drautz, R.; Nguyen-Manh, D.; Ngoepe, P.E.; Pettifor, D.G. Theoretical investigation of the Pt3Al ground state. Intermetallics 2010, 18, 417–421. [Google Scholar] [CrossRef]
- Chen, M.; Liu, C.; Liu, M.; Kumar, U.P.; Li, Z.; Liu, L.; He, J.; Liang, T. Exploring the electronic, mechanical, and anisotropy properties of novel tetragonal B2CO phase. J. Mater. Res. 2019, 34, 3617–3626. [Google Scholar] [CrossRef]
- Mnisi, B.O.; Benecha, E.; Tibane, M. Investigation of the thermodynamic, structural, electronic, mechanical and phonon properties of D0c Ru-based intermetallic alloys: An ab-initio Study. Mater. Res. Express 2024. [Google Scholar] [CrossRef]
- Mnisi, B.O.; Benecha, E.M.; Tibane, M.M. Computational Study of A15 Ru-Based Alloys for High-Temperature Structural Applications. In Ruthenium: An Element Loved by Researchers; IntechOpen: London, UK, 2021. [Google Scholar]
- Mnisi, B.O.; Benecha, E.M.; Chauke, H.R.; Ngoepe, P.E.; Tibane, M.M. Effect of transition metal doping on Cr–Ru alloys using first principles approach. Bull. Mater. Sci. 2020, 43, 120. [Google Scholar] [CrossRef]
- Miwa, K.; Fukumoto, A. First-principles study on (formula presented) transition-metal dihydrides. Phys. Rev. B 2002, 65, 155114. [Google Scholar] [CrossRef]
- La, P.; Wei, Y.; Lv, R.; Zhao, Y.; Yang, Y. Effect of Mn element on microstructure and mechanical properties of bulk nanocrystalline Fe3Al based materials prepared by aluminothermic reaction. Mater. Sci. Eng. A 2010, 527, 2313–2319. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Chen, S.; Pan, Y. Noble metal interlayer-doping enhances the catalytic activity of 2H–MoS2 from first-principles investigations. Int. J. Hydrogen Energy 2021, 46, 21040–21049. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, E. First-principles investigation of electronic and optical properties of H-doped FeS2. Int. J. Energy Res. 2021, 45, 11284–11293. [Google Scholar] [CrossRef]
- Chen, S.; Pan, Y. Influence of Group III and IV Elements on the Hydrogen Evolution Reaction of MoS2 Disulfide. J. Phys. Chem. C 2021, 125, 11848–11856. [Google Scholar] [CrossRef]
- Zhu, N.; Guo, Y.; Zhang, X.; Wang, F. The elastic anisotropy, electronic and thermodynamic properties of TM5Si4 (TM = Sc, Y, Ti, Zr and Hf) silicides from first-principles calculations. Vacuum 2021, 194, 110586. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Y.J.; Li, Y.; Li, Q.; Duan, W.; Wen, H.H. Comparative studies on superconductivity in Cr3Ru compounds with bcc and A15 structures. J. Phys. Condens. Matter 2022, 34, 475602. [Google Scholar] [CrossRef] [PubMed]
- Mehl, M.J.; Papaconstantopoulos, D.A. Applications of a tight-binding total-energy method for transition and noble metals: Elastic constants, vacancies, and surfaces of monatomic metals. Phys. Rev. B 1996, 54, 4519. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc: Hoboken, USA, 2005. [Google Scholar]
- Hong, S.; Fu, C.L. Phase stability and elastic moduli of Cr2Nb by first-principles calculations. Intermetallics 1999, 7, 5–9. [Google Scholar] [CrossRef]
- Li, Z.; Xiong, K.; Sun, Y.; Jin, C.; Zhang, S.; He, J.; Mao, Y. First-principles study of mechanical and thermodynamic properties of intermetallic Pt3M (M = Al, Hf, Zr, Co, Y, Sc). Comput. Condens. Matter 2020, 23, e00462. [Google Scholar] [CrossRef]
- Heid, R.; Bohnen, K.-P.; Renker, B.; Wolf, T.; Schober, H. Ab initio lattice dynamics and electron-phonon coupling in the refractory compounds ZrN and HfN. Phys. Rev. B 2005, 71, 92302. [Google Scholar] [CrossRef]
- Ding, W.-J.; Yi, J.-X.; Chen, P.; Li, D.-L.; Peng, L.-M.; Tang, B.-Y. Elastic properties and electronic structures of typical Al–Ce structures from first-principles calculations. Solid State Sci. 2012, 14, 555–561. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Q.; Tang, B.; Zheng, J.; Zheng, Z.; Zhou, W.; Tian, J.; Wang, J. First-principles calculations of mechanical and thermodynamic properties of tetragonal Be12Ti. RSC Adv. 2019, 9, 5302–5312. [Google Scholar] [CrossRef]
- Popoola, A.I. Computational Study of Noble Metal Alloys; University of Witwatersand: Johannesburg, South Africa, 2014. [Google Scholar]
- Skinner, D.J.; Zedalis, M. Elastic modulus versus melting temperature in aluminum based intermetallics. Scr. Metall. 1988, 22, 1783–1785. [Google Scholar] [CrossRef]
- Blackman, M. On the calculation of characteristic temperatures from the Elastic constants. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1951, 42, 1441–1442. [Google Scholar] [CrossRef]
- Fine, M.E.; Brown, L.D.; Marcus, H.L. Elastic constants versus melting temperature in metals. Scr. Metall. 1984, 18, 951–956. [Google Scholar] [CrossRef]
- Popoola, A.I.; Lowther, J.E. Computational Study of Platinum Group Superalloys. Int. J. Mod. Phys. B 2014, 28, 1450066. [Google Scholar] [CrossRef]
- Reuss, A. Calculation of the flow limits of mixed crystals on the basis of the plasticity of monocrystals. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Liu, K.; Dong, B.; Zhou, X.L.; Wang, S.M.; Zhao, Y.S.; Chang, J. Structural, elastic, and thermodynamic properties of hexagonal molybdenum nitrides under high pressure from first principles. J. Alloys Compd. 2015, 632, 830–836. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.-X.; Duan, L.; Fan, J.-B.; Ni, L.; Ji, V. A comparison study of the structural and mechanical properties of cubic, tetragonal, monoclinic, and three orthorhombic phases of ZrO2. J. Alloys Compd. 2018, 749, 283–292. [Google Scholar] [CrossRef]
- Salje, E. Phase transitions in ferroelastic and co-elastic crystals. Ferroelectrics 1990, 104, 111–120. [Google Scholar] [CrossRef]
- Falk, F. Model free energy, mechanics, and thermodynamics of shape memory alloys. Acta Metall. 1980, 28, 1773–1780. [Google Scholar] [CrossRef]
- Poirier, J.-P. Introduction to the Physics of the Earth’s Interior; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Frantsevich, I.N.; Voronov, F.F.; Bokuta, S.A. Elastic Constants and Elastic Moduli of Metals and Insulators Handbook; Frantsevich, I.N., Ed.; Naukova Dumka: Kiev, Ukraine, 1983; pp. 60–180. [Google Scholar]
- Chen, X.-Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Bai, J.; Raulot, J.M.; Zhang, Y.D.; Esling, C.; Zhao, X.; Zuo, L. Crystallographic, magnetic, and electronic structures of ferromagnetic shape memory alloys Ni2XGa (X = Mn, Fe, Co) from first-principles calculations. J. Appl. Phys. 2011, 109, 014908. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Liu, X.; Ge, X.; Si, R.; Zhang, M.; Xu, X. A survey investigation on the stability, electronic and elastic properties of hexagonal ternary transition-metal borides by first-principles calculations. Vacuum 2020, 177, 109432. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alloy | a (Å) | c (Å) | V0 (Å3) | ΔHf (eV/Atom) | Magnetic Moments (µB/Atom) | Density (g/cm3) | c/a |
|---|---|---|---|---|---|---|---|
| Sc3Ru | 6.23 | 8.97 | 348.07 | −0.18 | 0.02 | 4.50 | 1.44 |
| Ti3Ru | 5.40 | 9.28 | 270.39 | −0.41 | 0.89 | 6.01 | 1.72 |
| V3Ru | 5.73 | 8.11 | 266.05 | −0.27 | 2.19 | 6.34 | 1.42 |
| Cr3Ru Cr3Ru [16] | 6.04 5.20 | 7.28 | 265.89 | 0.11 0.24 | 0.68 | 6.42 | 1.21 |
| Mn3Ru | 5.75 | 8.14 | 269.02 | −1.61 | 3.25 | 6.56 | 1.42 |
| Fe3Ru | 5.07 | 8.02 | 206.15 | 0.07 | 0.95 | 8.65 | 1.58 |
| Co3Ru | 5.20 | 7.34 | 198.30 | 0.10 | 1.82 | 9.31 | 1.41 |
| Ni3Ru | 5.15 | 7.28 | 192.89 | 0.92 | 1.08 | 9.55 | 1.41 |
| Cu3Ru | 5.21 | 7.37 | 200.43 | 0.31 | 0.00 | 9.67 | 1.41 |
| Zn3Ru | 5.36 | 7.57 | 217.32 | −0.23 | 0.00 | 9.08 | 1.41 |
| tI16-Pt3Al [17] | 5.4 | −0.79 | 1.45 | ||||
| tP16-Pt3Ga [12] | 5.5 | 7.80 | 6.14 | ||||
| L12-Pt3Al [37] | 3.9 |
| Phase | C11 | C12 | C13 | C33 | C44 | C66 | MT ± 300 K |
|---|---|---|---|---|---|---|---|
| Sc3Ru | 120.46 | 64.81 | 62.42 | 95.48 | 38.42 | 8.42 | 858.60 |
| Ti3Ru | 172.26 | 77.43 | 91.89 | 162.47 | 154.62 | 3.50 | 1114.49 |
| V3Ru | 192.82 | 24.35 | 73.52 | 154.00 | 58.86 | 47.65 | 1163.46 |
| Cr3Ru | 51.53 | 36.48 | 53.03 | 5.39 | −3.65 | 15.73 | 516.68 |
| Mn3Ru | 190.79 | 29.40 | 83.55 | 130.89 | 77.64 | 25.21 | 1122.71 |
| Fe3Ru | 361.31 | 77.92 | 141.14 | 237.96 | 39.08 | −42.41 | 1794.87 |
| Co3Ru | 330.98 | 68.68 | 170.26 | 270.15 | 130.85 | 36.12 | 1602.33 |
| Ni3Ru | 337.15 | 115.37 | 166.10 | 286.49 | 117.29 | 63.19 | 1795.19 |
| Cu3Ru | 262.26 | 87.26 | 153.90 | 192.07 | 94.72 | 28.84 | 1428.89 |
| Zn3Ru | 247.59 | 68.25 | 122.39 | 177.61 | 89.16 | 53.27 | 1363.19 |
| L12-Ni3Al [45] L12-Pt3Al [37] | 235 317 | 157 177 | 128 111 | 1691.00 |
| tP16 Phase | BV | GV | BR | GR | BH | GH | EH | BH/GH | HV | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sc3Ru | 79.52 | 26.84 | 78.21 | 19.61 | 78.86 | 23.22 | 63.43 | 3.40 | 0.366 | 0.01 |
| Ti3Ru | 114.38 | 78.93 | 114.35 | 14.34 | 114.37 | 46.64 | 83.28 | 2.45 | 0.321 | 3.63 |
| V3Ru | 98.05 | 57.62 | 97.94 | 53.86 | 97.99 | 55.74 | 140.57 | 1.76 | 0.261 | 8.35 |
| Cr3Ru | 43.73 | −0.58 | 45.44 | 12.43 | 44.58 | −6.51 | −20.53 | −6.85 | 0.577 | UND |
| Mn3Ru | 100.61 | 57.16 | 100.56 | 42.22 | 100.58 | 49.69 | 127.99 | 2.02 | 0.288 | 5.61 |
| Fe3Ru | 186.78 | 47.18 | 184.48 | 96.20 | 185.63 | 71.69 | 190.54 | 2.59 | 0.329 | 5.00 |
| Co3Ru | 194.50 | 94.42 | 193.07 | 67.62 | 193.79 | 81.02 | 213.33 | 2.39 | 0.317 | 6.43 |
| Ni3Ru | 206.21 | 93.77 | 206.21 | 85.46 | 206.21 | 89.61 | 234.82 | 2.30 | 0.310 | 7.46 |
| Cu3Ru | 167.41 | 65.09 | 167.39 | 42.36 | 167.40 | 53.72 | 145.59 | 3.12 | 0.355 | 2.44 |
| Zn3Ru | 144.32 | 70.30 | 144.01 | 58.49 | 144.17 | 64.39 | 168.14 | 2.24 | 0.306 | 5.90 |
| Li2-Ni3Al [45] | 183.00 | 92.00 | 237.00 | 1.98 | 0.280 | |||||
| Li2-Pt3Al [37] | 2.43 |
| Phase | A1 | A2 | AU | AB |
|---|---|---|---|---|
| Sc3Ru | 0.30 | 1.69 | 1.86 | 0.83 |
| Ti3Ru | 0.07 | 4.10 | 22.53 | 0.01 |
| V3Ru | 0.57 | 1.18 | 0.35 | 0.06 |
| Cr3Ru | 2.09 | 0.30 | −4.80 | −1.92 |
| Mn3Ru | 0.31 | 2.01 | 1.77 | 1.77 |
| Fe3Ru | −0.30 | 0.49 | −2.53 | 3.45 |
| Co3Ru | 0.28 | 2.01 | 1.99 | 0.37 |
| Ni3Ru | 0.57 | 1.61 | 0.49 | 0.00 |
| Cu3Ru | 0.33 | 2.59 | 2.68 | 0.01 |
| Zn3Ru | 0.59 | 1.98 | 1.01 | 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mnisi, B.O.; Benecha, M.E.; Tibane, M.M. First-Principles Study on Thermodynamic, Structural, Mechanical, Electronic, and Phonon Properties of tP16 Ru-Based Alloys. Alloys 2024, 3, 126-139. https://doi.org/10.3390/alloys3020007
Mnisi BO, Benecha ME, Tibane MM. First-Principles Study on Thermodynamic, Structural, Mechanical, Electronic, and Phonon Properties of tP16 Ru-Based Alloys. Alloys. 2024; 3(2):126-139. https://doi.org/10.3390/alloys3020007
Chicago/Turabian StyleMnisi, Bhila Oliver, Moseti Evans Benecha, and Malebo Meriam Tibane. 2024. "First-Principles Study on Thermodynamic, Structural, Mechanical, Electronic, and Phonon Properties of tP16 Ru-Based Alloys" Alloys 3, no. 2: 126-139. https://doi.org/10.3390/alloys3020007
APA StyleMnisi, B. O., Benecha, M. E., & Tibane, M. M. (2024). First-Principles Study on Thermodynamic, Structural, Mechanical, Electronic, and Phonon Properties of tP16 Ru-Based Alloys. Alloys, 3(2), 126-139. https://doi.org/10.3390/alloys3020007