
Synthesis and Antiproliferative Activity of Minor Hops Prenylflavonoids and New Insights on Prenyl Group Cyclization

 , and
, and
Abstract
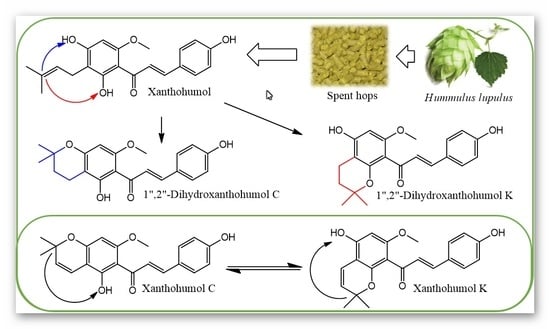
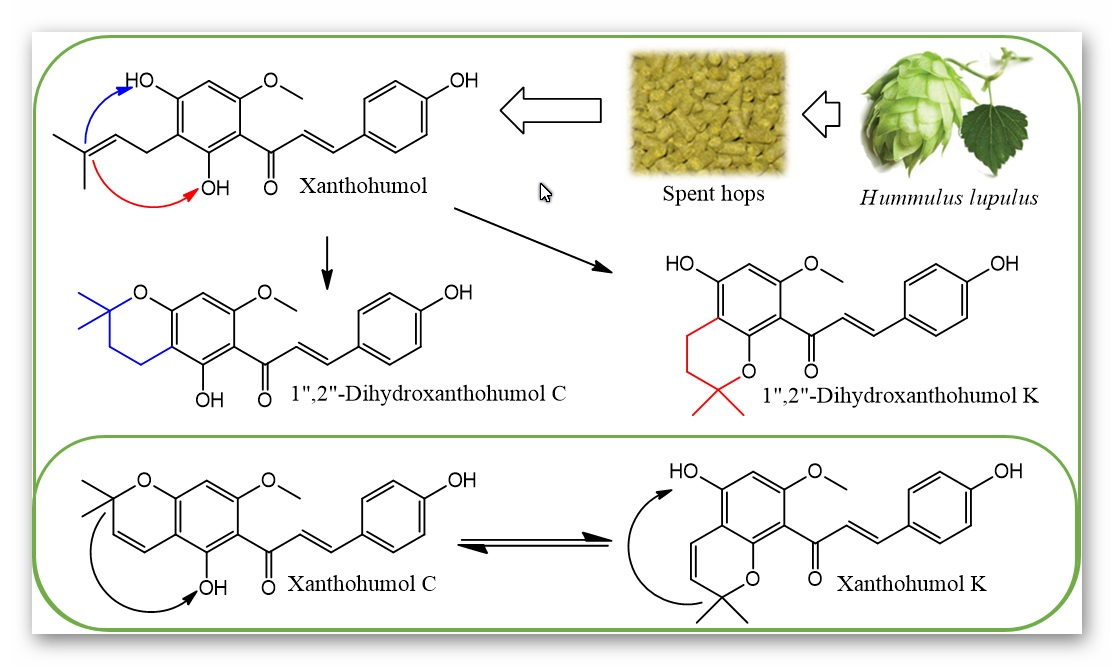
:
1. Introduction
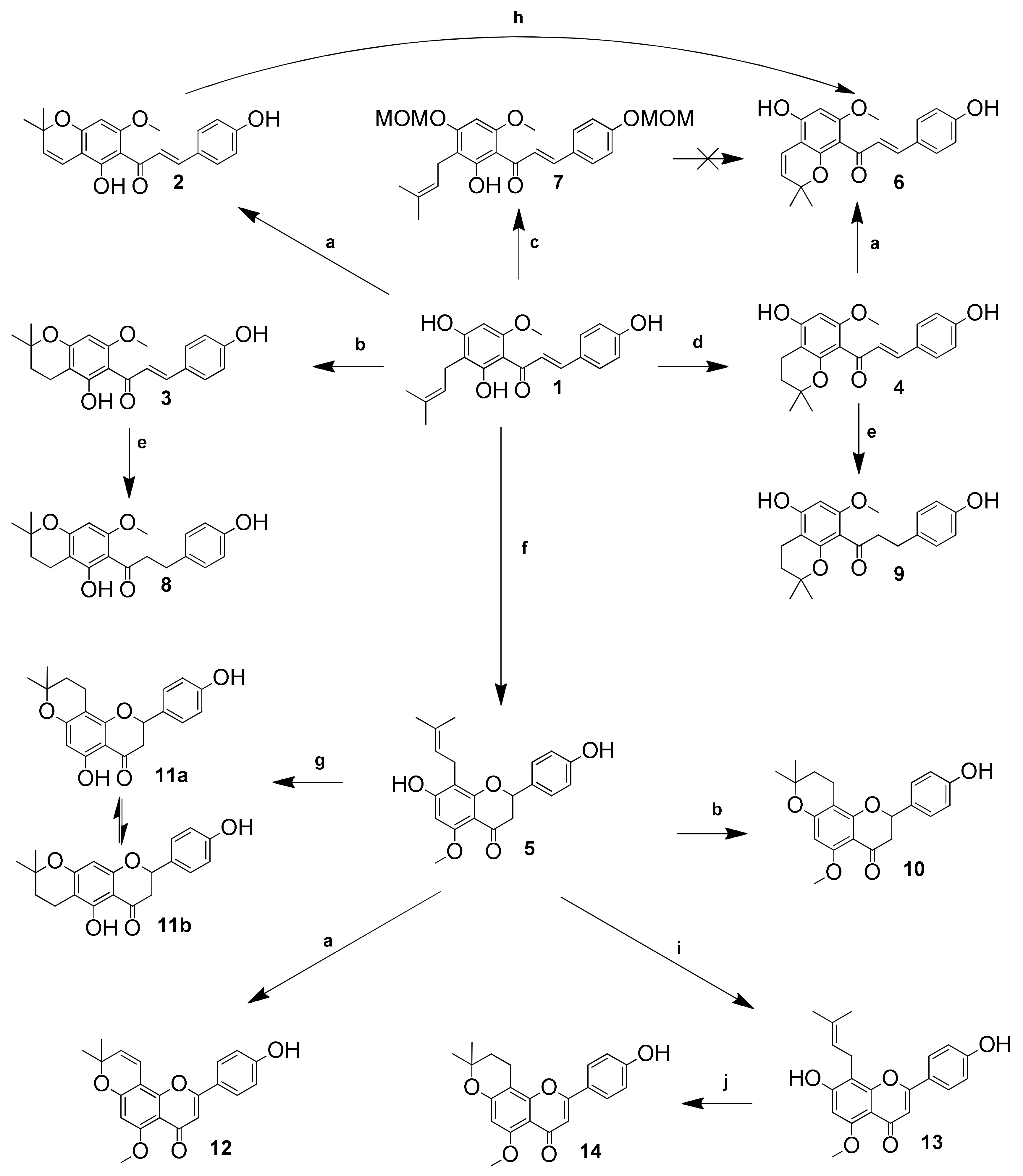
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Antiproliferative Assay in Vitro
3.2.1. Cells
3.2.2. SRB Assay
3.3. Chemical Synthesis
3.3.1. Xanthohumol (1)
3.3.2. Xanthohumol C (2)
3.3.3. 1″,2″-Dihydroxanthohumol C (3)
3.3.4. 1″,2″-Dihydroxanthohumol K (4)
3.3.5. Isoxanthohumol (5)
3.3.6. Xanthohumol K (6)
3.3.7. 4.4′-Dimethoxymethyl xanthohumol (7)
3.3.8. 1″,2″,α,β-Tetrahydroxanthohumol C (8)
3.3.9. 1″,2″,α,β-Tetrahydroxanthohumol K (9)
3.3.10. 1″,2″-Dihydroisoxanthohumol C (10)
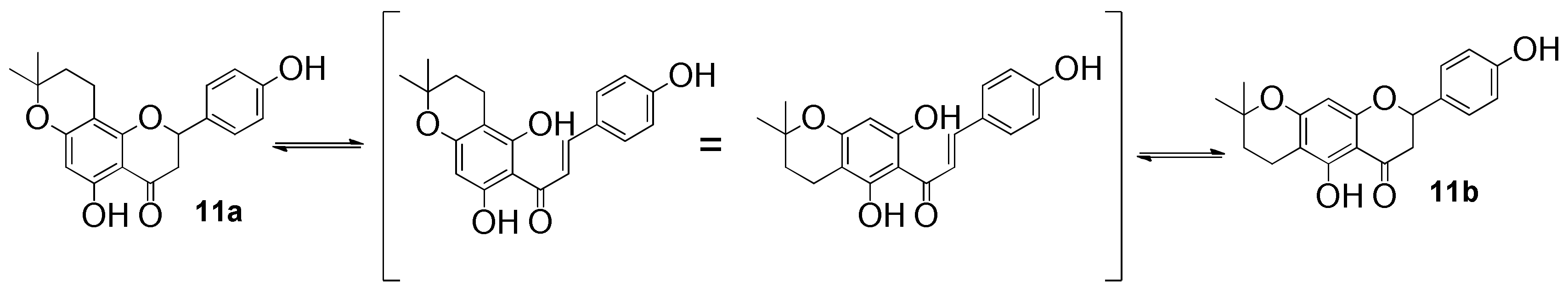
3.3.11. 5,4′-Dihydroxy-6″,6″-dimethyl-4″,5″-dihydropyrano-[2″,3″:7,8]flavanone (11a) and 5,4′-dihydroxy-6″,6″-dimethyl-4″,5″-dihydropyrano-[2″,3″:6,7]flavanone (11b)
3.3.12. 2,3-Dehydroisoxanthohumol C (12)
3.3.13. 2,3-Dehydroisoxanthohumol (13)
3.3.14. 1″,2″-Dihydro-2,3-dehydroisoxanthohumol C (14)
3.3.15. TFA Catalyzed Cyclisation of xanthohumol (1) with Various Catalyst Concentration and at Different Temperatures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hollman, P.C.; Katan, M.B. Dietary flavonoids: Intake, health effects and bioavailability. Food Chem. Toxicol. 1999, 37, 937–942. [Google Scholar] [CrossRef]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Burkard, M.; Biendl, M.; Lauer, U.M.; Frank, J.; Busch, C. Prenylated chalcones and flavonoids for the prevention and treatment of cancer. Nutrition 2016, 32, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Sharma, A.K. Anti-cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 2013, 3, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Kachadourian, R.; Leitner, H.M.; Day, B.J. Selected flavonoids potentiate the toxicity of cisplatin in human lung adenocarcinoma cells: A role for glutathione depletion. Int. J. Oncol. 2007, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.F.; Luthria, D.L.; Sasaki, T.; Heyerick, A. Flavonoids from Artemisia annua L. as antioxidants and their potential synergism with artemisinin against malaria and cancer. Molecules 2010, 15, 3135–3170. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Page, J.E. Xanthohumol and related prenylflavonoids from hops and beer: To your good health! Phytochemistry 2004, 65, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, L.R.; Pauli, G.F.; Farnsworth, N.R. The pharmacognosy of Humulus lupulus L. (hops) with an emphasis on estrogenic properties. Phytomedicine 2006, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, P.; Zavatti, M. Pharmacognostic and pharmacological profile of Humulus lupulus L. J. Etnopharmacol. 2008, 116, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hansen, P.E.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Ivanic, M.; Hsu, V.L.; Deinzer, M.L. Prenylflavonoids from Humulus lupulus. Phytochemistry 1997, 44, 1575–1585. [Google Scholar] [CrossRef]
- Wang, W.S.; Ye, Y.H.; Zhou, Y.W. New prenylchalcones from the hops of Humulus lupulus. J. Asian. Nat. Prod. Res. 2008, 10, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, F.; Hu, Z.; Ding, H.; Tang, H.; Ma, X.; Zhao, X. Novel prenylated bichalcone and chalcone from Humulus lupulus and their quinone reductase induction activities. Fitoterapia 2014, 93, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Frolich, S.; Schubert, C.; Bienzle, U.; Jenett-Siems, K. In vitro antiplasmodial activity of prenylated chalcone derivatives of hops (Humulus lupulus) and their interaction with haemin. J. Antimicrob. Chemother. 2005, 55, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Heilmann, J. Synthesis, cytotoxicity and antioxidative activity of minor prenylated chalcones from Humulus lupulus. J. Nat. Prod. 2008, 71, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Ohmayer, S.; Brunner, G.; Heilmann, J. Natural and non-natural prenylated chalcones: Synthesis, cytotoxicity and anti-oxidative activity. Bioorg. Med. Chem. 2008, 16, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Barbic, M.; Jurgenliemk, G.; Heilmann, J. Synthesis, cytotoxicity, anti-oxidative and anti-inflammatory activity of chalcones and influence of A-ring modifications on the pharmacological effect. Eur. J. Med. Chem. 2010, 45, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Popłoński, J.; Tronina, T.; Huszcza, E. Photochemical transformations of xanthohumol. Tetrahedron Lett. 2013, 54, 6035–6036. [Google Scholar] [CrossRef]
- Nuti, E.; Bassani, B.; Camodeca, C.; Rosalia, L.; Cantelmo, A.; Gallo, C.; Baci, D.; Bruno, A.; Orlandini, E.; Nencetti, S.; et al. Synthesis and antiangiogenic activity study of new hop chalcone Xanthohumol analogues. Eur. J. Med. Chem. 2017, 138, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Li, X.; Yang, K.; Li, X.; Zhang, Z.; Wang, L.; Deng, Z.; Song, B.; Yan, Z.; Zhang, Y.; et al. Synthesis and antioxidant evaluation of desmethylxanthohumol analogs and their dimers. Eur. J. Med. Chem. 2017, 125, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Herath, W.; Ferreira, D.; Khan, S.I.; Khan, I.A. Identification and biological activity of microbial metabolites of xanthohumol. Chem. Pharm. Bull. 2003, 11, 1237–1240. [Google Scholar] [CrossRef]
- Tronina, T.; Bartmańska, A.; Popłoński, J.; Huszcza, E. Transformation of xanthohumol by Aspergillus ochraceus. J. Basic Microbiol. 2014, 54, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Tronina, T.; Bartmańska, A.; Filip-Psurska, B.; Wietrzyk, J.; Popłoński, J.; Huszcza, E. Fungal metabolites of xanthohumol with potent antiproliferative activity on human cancer cell lines in vitro. Bioorg. Med. Chem. 2013, 21, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; Guangcheng, W.; Xiuxia, L.; Dong, C.; Zhuang, Y.; Liang, M.; Haoyu, Y.; Xiaolin, L.; Yan, R.; Jinying, C.; et al. Rational design, synthesis and pharmacological properties of pyranochalcone derivatives as potent anti-inflammatory agents. Eur. J. Med. Chem. 2012, 54, 278–280. [Google Scholar]
- Guangcheng, W.; Wenshuang, W.; Fei, P.; Dong, C.; Zhuang, Y.; Liang, M.; Neng, Q.; Haoyu, Y.; Xiaolei, H.; Jinying, C.; et al. Design, synthesis and structure reactivity relationship studies of novel millepachine derivatives as potent antiproliferative agents. Eur. J. Med. Chem. 2012, 54, 793–803. [Google Scholar]
- Dong, C.; Xiaolei, H.; Guangcheng, W.; Zhuang, Y.; Fei, P.; Liang, M.; Ronghong, Z.; Haoyu, Y.; Minghai, T.; Wenshuang, W.; et al. Synthesis and biological evaluation of novel pyranochalcone derivatives as a new class of microtubule stabilizing agents. Eur. J. Med. Chem. 2013, 62, 579–589. [Google Scholar]
- Bartmańska, A.; Huszcza, E.; Tronina, T. Transformations of isoxanthohumol by fungi. J. Mol. Catal. B: Enzym. 2009, 61, 221–244. [Google Scholar] [CrossRef]
- Andrade-Carrera, B.; Clares, B.; Noé, V.; Mallandrich, M.; Calpena, A.C.; García, M.L.; Garduño-Ramírez, M.L. Cytotoxic Evaluation of (2S)-5,7-Dihydroxy-6-prenylflavanone Derivatives Loaded PLGA Nanoparticles against MiaPaCa-2 Cells. Molecules 2017, 22, 1553. [Google Scholar] [CrossRef] [PubMed]
- Nerander, T.; Reddy, K.P. BF3-Et2O mediated biogenetic type synthesis of chromanochalcones from prenylated chalcones via regioselective cyclisation reaction. Tetrahedron Lett. 2007, 48, 7628–7632. [Google Scholar] [CrossRef]
- Mizobuchi, S. Study on the Structure of 5,7,4′-Trihydroxyflavanones. Agric. Biol. Chem. 1985, 49, 2195–2196. [Google Scholar]
- Popłoński, J.; Sordon, S.; Tronina, T.; Huszcza, E. Selective hydrogenation of xanthohumol to α,β-dihydroxanthohumol. Przem. Chem. 2014, 93, 1916–1918. [Google Scholar]
- Simmler, C.; Hajirahimkhan, A.; Lankin, D.C.; Bolton, J.L.; Jones, T.; Soejarto, D.D.; Chen, S.-N.; Pauli, G.F. Dynamic Residual Complexity of the Isoliquiritigenin-Liquiritigenine Interconversion During Bioassay. J. Agric. Food. Chem. 2013, 61, 2145–2157. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Y.; Liu, T.; Wu, P.; Gao, J.; Xu, J.; Yang, B.; Hu, Y. Flavonoids as Vasorelaxant Agents: Synthesis, Biological Evaluation and Quantitative Structure Activities Relationship (QSAR) Studies. Molecules 2011, 16, 8257–8272. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sugino, T. Efficient conversion of 2′-hydroxychalcones into flavanones and flavanols in a water suspension medium. Green Chem. 2001, 3, 133–134. [Google Scholar] [CrossRef]
- Aitmambetov, A.; Kubzheterova, A. An Improved Method for the Synthesis of Flavanones. Russ. J. Bioorg. Chem. 2002, 28, 165–166. [Google Scholar] [CrossRef]
- Touli, Y.S.; Fellous, A.; Scherman, D.; Chabot, G.G. Flavonoid-induced morphological modifications of endothelial cells through microtubule stabilization. Nutr. Cancer. 2009, 61, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddam, G.; Ebrahimi, S.A.; Rahbar-Roshandel, N.; Foroumadi, A. Antiproliferative Activity of Flavonoids: Influence of the Sequential Methoxylation State of the Flavonoid Structure. Phytother. Res. 2012, 26, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Antiproliferative Activity of Flavonoids on Several Cancer Cell Lines. Biosci. Biotechnol. Biochem. 1999, 63, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, P.; Maruthavanan, T. Synthesis of substituted flavone derivatives as potent antimicrobial agents. Bull. Chem. Soc. Ethiop. 2011, 25, 419–425. [Google Scholar] [CrossRef]
- Tronina, T.; Bartmańska, A.; Milczarek, M.; Wietrzyk, J.; Popłoński, J.; Rój, E.; Huszcza, E. Antioxidant and antiproliferative activity of glycosides obtained by biotransformation of xanthohumol. Bioorg. Med. Chem. Lett. 2013, 23, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Nat. Cancer. Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.L.; Stevens, J.F.; Helmrich, A.; Henderson, M.C.; Rodriguez, R.J.; Yang, Y.-H.; Deinzer, M.L.; Barnes, D.W.; Buhler, D.R. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem. Toxicol. 1999, 37, 271–285. [Google Scholar] [CrossRef]
- Kim, J.-A.; Kang, Y.-R.; Thapa, D.; Lee, J.-S.; Park, M.-A.; Lee, K.-H.; Lyoo, W.-S.; Lee, Y.-R. Anti-invasive and anti-angiogenic effects of xanthohumol and its synthetic derivatives. Biomol. Ther. 2009, 17, 422–429. [Google Scholar] [CrossRef]
- Oberbauer, E.; Urmannc, C.; Steffenhagen, C.; Bieler, L.; Brunner, D.; Furtner, T.; Humpeld, C.; Bäumere, B.; Bandtlow, C.; Couillard-Despres, S.; et al. Chroman-like cyclic prenylflavonoids promote neuronal differentiation and neurite outgrowth and are neuroprotective. J. Nutr. Biochem. 2013, 24, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Nookandeh, A.; Frank, N.; Steiner, F.; Ellinger, R.; Schneider, B.; Gerhäuser, C.; Becker, H. Xanthohumol metabolites in faeces of rats. Phytochemistry 2004, 65, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Helle, J.; Kräker, K.; Bader, M.I.; Keiler, A.M.; Zierau, O.; Vollmer, G.; Welsh, J.; Kretzschmar, G. Assessment of the proliferative capacity of the flavanones 8-prenylnaringenin, 6-(1,1-dimethylallyl)naringenin and naringenin in MCF-7 cells and the rat mammary gland. Mol. Cell Endocrinol. 2014, 392, 125–135. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors for possible research projects in cooperation. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature 2 | TFA Amount 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| °C | % 4 of 1 | % 4 of 3 | % 4 of 4 | 4/3 | % (v/v) | % 4 of 1 | % 4 of 3 | % 4 of 4 | 4/3 |
| 0 | 54.6 | 9.5 | 35.9 | 3.8 | 0.1 | 97.5 | 0.4 | 2.1 | 5.3 |
| 22 | 16.7 | 16.7 | 66.6 | 4.0 | 0.5 | 46.5 | 9.8 | 43.7 | 4.5 |
| 39 | 0 | 18.7 | 81.3 | 4.3 | 1 | 10.3 | 16.8 | 72.9 | 4.3 |
| Compound | Cancer Cell Line, IC50 (µM) | |||
|---|---|---|---|---|
| PC-3 | HT-29 | MCF-7 | ||
| 1 |  | 7.0 ± 1.5 | 10.1 ± 1.1 | 8.1 ± 0.8 |
| 2 |  | 10.1 ± 3.0 | 10.6 ± 0.6 | 15.0 ± 1.8 |
| 3 |  | 49.6 ± 6.6 | 16.7 ± 6.9 | 15.9 ± 3.6 |
| 4 |  | 10.7 ± 5.9 | 12.5 ± 1.5 | 9.0 ± 6.4 |
| 6 |  | 74.9 ± 25.6 | 87.5 ± 3.2 | 83.8 ± 14.6 |
| 8 |  | 59.9 ± 5.4 | 62.7 ± 13.2 | 16.2 ± 2.1 |
| 9 |  | 68.0 ± 14.2 | 89.1 ± 3.2 | 36.0 ± 3.6 |
| 10 |  | 51.3 ± 21.2 | 85.4 ± 2.1 | 35.3 ± 6.6 |
| 11a/11b |  | 48.9 ± 7.8 | 25.8 ± 2.9 | 13.2 ± 3.7 |
| 12 |  | 202.0 ± 0.0 | 217.1 ± 40.9 | 120.1 ± 3.6 |
| 13 |  | 13.8 ± 1.4 | 20.1 ± 1.5 | 7.9 ± 0.5 |
| 14 |  | 64.6 ± 10.7 | 91.2 ± 17.2 | 19.3 ± 6.1 |
| cisplatin (reference) | 12.33 ± 2.77 | 9.10 ± 1.13 | 8.27 ± 2.10 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popłoński, J.; Turlej, E.; Sordon, S.; Tronina, T.; Bartmańska, A.; Wietrzyk, J.; Huszcza, E. Synthesis and Antiproliferative Activity of Minor Hops Prenylflavonoids and New Insights on Prenyl Group Cyclization. Molecules 2018, 23, 776. https://doi.org/10.3390/molecules23040776
Popłoński J, Turlej E, Sordon S, Tronina T, Bartmańska A, Wietrzyk J, Huszcza E. Synthesis and Antiproliferative Activity of Minor Hops Prenylflavonoids and New Insights on Prenyl Group Cyclization. Molecules. 2018; 23(4):776. https://doi.org/10.3390/molecules23040776
Chicago/Turabian StylePopłoński, Jarosław, Eliza Turlej, Sandra Sordon, Tomasz Tronina, Agnieszka Bartmańska, Joanna Wietrzyk, and Ewa Huszcza. 2018. "Synthesis and Antiproliferative Activity of Minor Hops Prenylflavonoids and New Insights on Prenyl Group Cyclization" Molecules 23, no. 4: 776. https://doi.org/10.3390/molecules23040776
APA StylePopłoński, J., Turlej, E., Sordon, S., Tronina, T., Bartmańska, A., Wietrzyk, J., & Huszcza, E. (2018). Synthesis and Antiproliferative Activity of Minor Hops Prenylflavonoids and New Insights on Prenyl Group Cyclization. Molecules, 23(4), 776. https://doi.org/10.3390/molecules23040776