
Application of Drug Efficiency Index Metric for Analysis of Post-Traumatic Stress Disorder and Treatment Resistant Depression Gene Expression Profiles

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. PTSD Gene Expression Datasets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
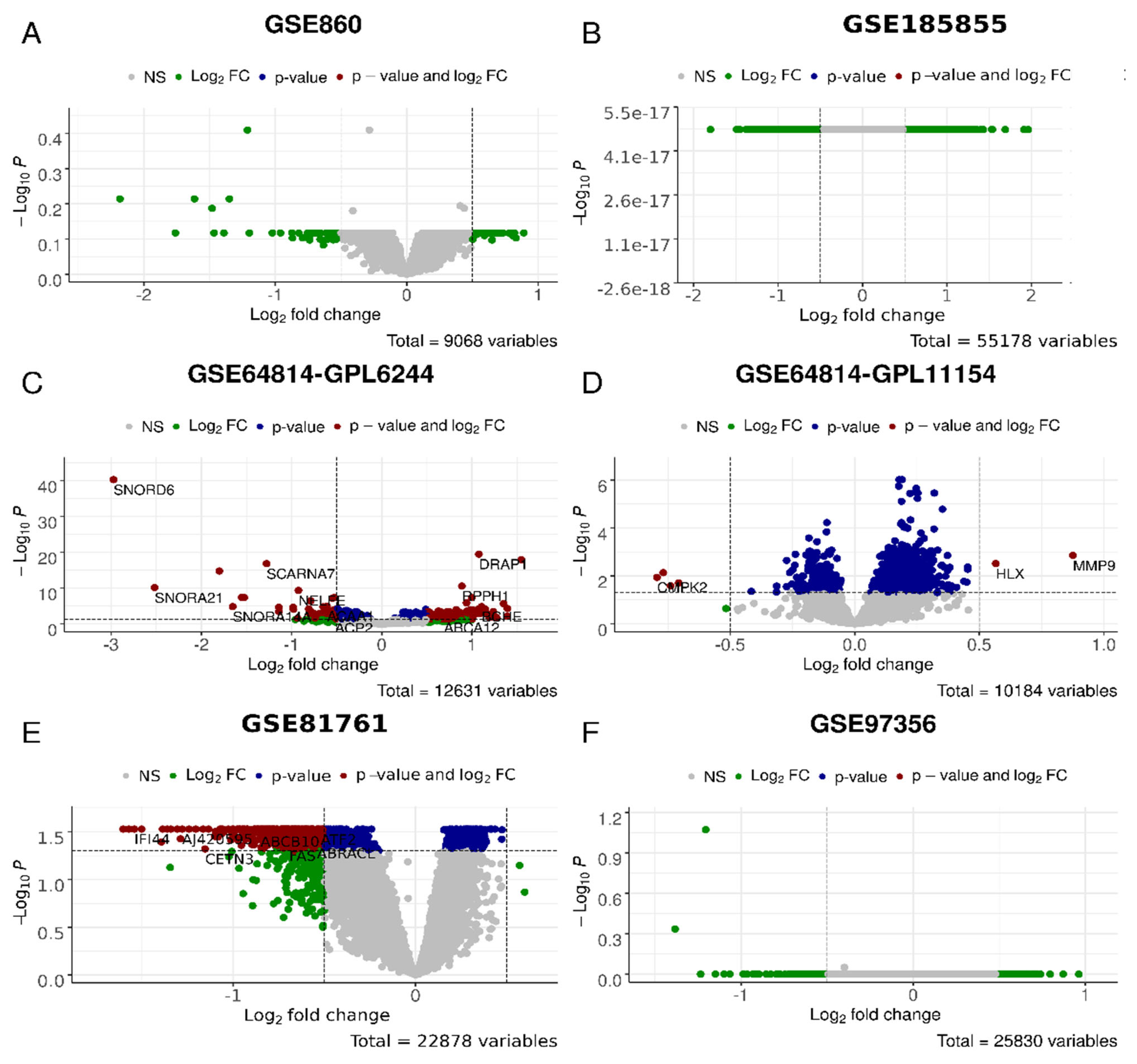{kind=link}
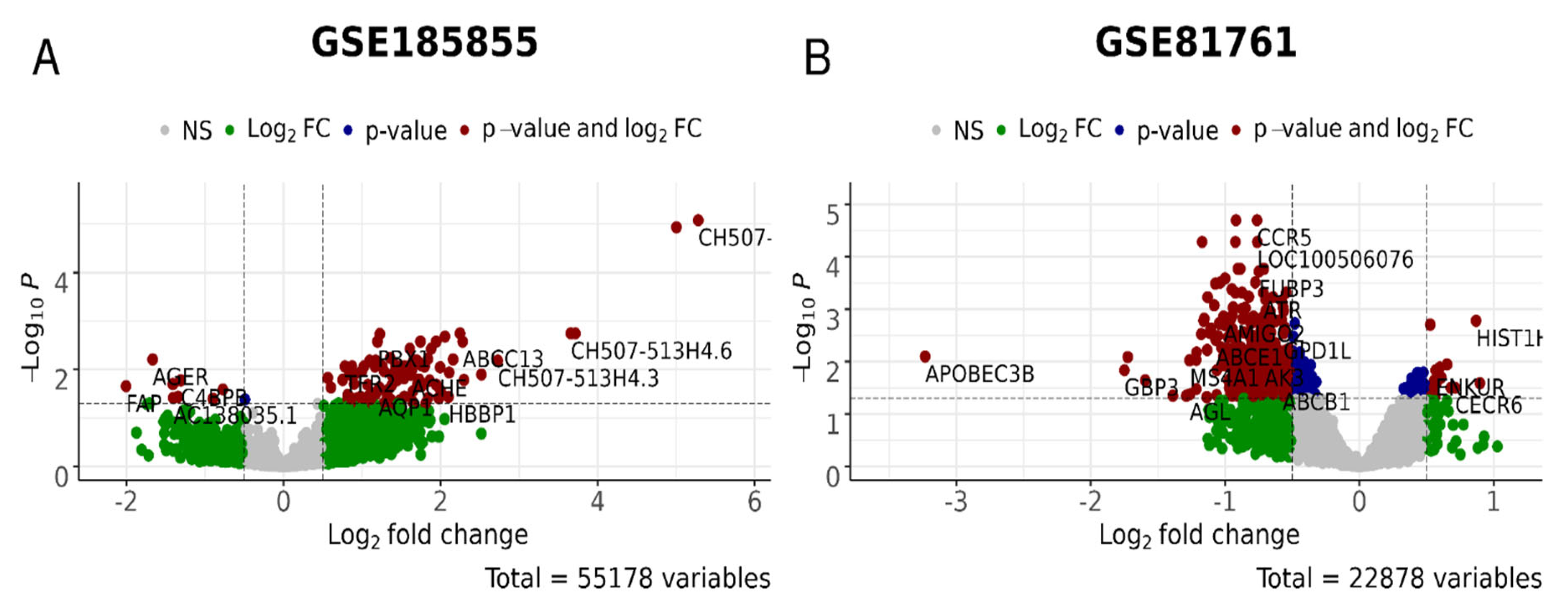{kind=link}
| Reference | [80] | [81] | [81] | [82] | [82] | [30,83] | [84] | [84] |
|---|---|---|---|---|---|---|---|---|
| GSE ID; clinical description | GSE860 | GSE64814-GPL6244 | GSE64814-GPL11154 | GSE81761 no improvement; non-responders | GSE81761 improvement; responders | GSE97356 | GSE185855 TRD: non- responders | GSE185855 TRD: responders |
| Profiling platform | Affymetrix U95A Array | Affymetrix Gene 1.0 ST Array | Illumina HiSeq 2000 | Affymetrix U133 Plus 2.0 Array | Affymetrix U133 Plus 2.0 Array | Illumina HiSeq 2000 | Illumina HiSeq 2000 | Illumina HiSeq 2000 |
| Treatment | Not specified | Not specified | Not specified | * | * | Not specified | Ketamine | Ketamine |
| Untreated cases | 8 | 24 | 47 | 39 | 39 | 81 | 8 | 17 |
| Untreated controls | 7 | 48 | 94 | 27 | 27 | 201 | 21 | 21 |
| Treated cases | 9 | 24 | 47 | 5 | 6 | 42 | 8 | 13 |
| Treated controls | 9 | 48 | 94 | 6 | 6 | 201 | 21 | 21 |
| Number of genes | 9068 | 12,631 | 10,184 | 22,878 | 22,878 | 25,830 | 55,178 | 55,178 |
2.2. Pathway Activation Level Assessment
- Log-fold-change, LFCn, i.e., log2 ratio of gene n mRNA concentrations in the test sample and in the control pool (median value in the control group)
- Pathway p activation level, PALp = ∑n NIInp ∙ ARRnp ∙ LFCn/∑n |ARRn|. Here, the node involvement index, NIInp, is the Boolean flag for gene product n concerning the pathway p. The discrete value ARRnp (activator/repressor role) is determined for a gene n in the pathway p as follows:
- ARRnp = −1; gene product n is a repressor of pathway p
- −0.5; gene product n is more a repressor than an activator of pathway p
- 0; unclear repressor or activator role in pathway p
- 0.5; gene product n is more an activator than a repressor of pathway p
- 1; gene product n is an activator of pathway p.
2.3. Evaluation of the Individual Drug Action
- Obtain the SPIA (PAL) values for each dataset and biological pathway.
- Calculate the values of the pathway weight (wp) factor as follows. For pathways with a positive mean SPIA (PAL) score of the case samples, wp = ((number of case samples with positive SPIA (PAL) score)/(total number of case samples)). For pathways with a negative mean SPIA (PAL) score of the case samples, wp = ((number of case samples with negative SPIA (PAL) score)/(total number of case samples)).
- Adjust the mean SPIA (PAL) score of each pathway by the weight factor,SPIAμ (PALμ) = mean (SPIA(PAL))·wp.
- Perform a Student’s t-test if the values of SPIAμ (PALμ) for the pool of case samples are different from 0 (for the pool of control samples, the values of SPIAμ (PALμ) are clearly equal to 0). During the Student’s t-test, the following case classes are taken into account: the untreated case (U), i.e., the pathological state before drug application, should be far from the control (C).
- 4.1
- The treated case (T), i.e., the pathological state after drug application, should be close to the control;
- 4.2
- The following values are the results for such calculations: |tU| = absolute t-value for the Student’s t-test for U-vs-C profiles;
- 4.3
- |tT| = absolute t-value for the Student’s t-test for T-vs-C profiles.
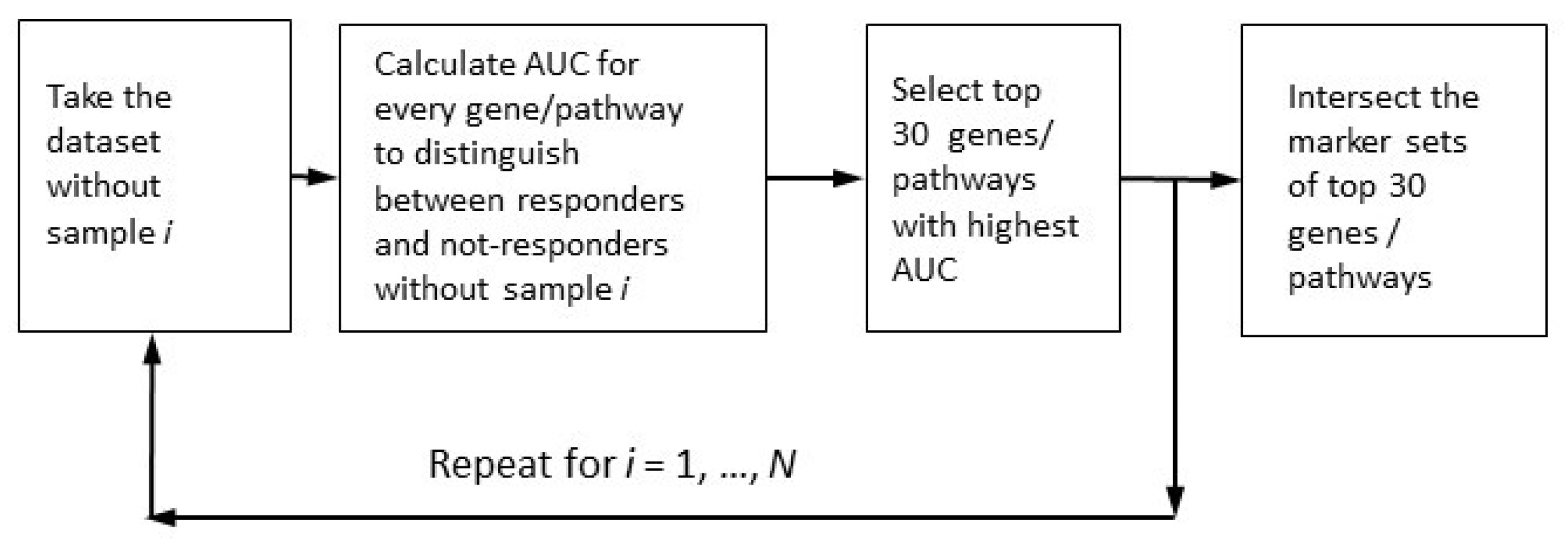
2.4. Robust Marker Gene and Pathway Analysis
- (1)
- Case samples from control samples (PTSD or TRD), the disease-vs-healthy, D_vs_H test;
- (2)
- Case samples after treatment or/and observation from those before treatment (untreated), the post-vs-ante, P_vs_A test;
- (3)
- Patients positively responding to the treatment from negatively or non-responding patients, the responders-vs-non-responders, R_vs_N test.
2.5. Alternative Methods for Differential Gene Expression and Enrichment Analysis
3. Results
3.1. Cohorts Used in the Analysis
3.2. Comparison of DEI Options for Different Approaches to the Assessment of Pathway Activation Levels
3.3. Robust Marker Gene/Pathway Analysis, Differentially Expressed Gene (DEG) Analysis, and Gene Ontology (GO) Enrichment Analysis
4. Future Perspectives
- The Big Data full expression profiles, obtained via ether microarray hybridization (MH) or next-generation sequencing (NGS) of mRNA are used as input data for PAL calculations;
- For each gene, we calculate the case-to-control log-fold changes (LFCs) using full expression profiles;
- Case samples (PTSD or TRD) from control samples;
- Case samples after treatment or/and observation from before treatment (untreated);
- Samples from patients positively responding to the treatment from responding negatively or non-responding.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| APAP | Automatic positive airway pressure |
| ARR | Activator/repressor role |
| AUC | Area under curve |
| BEP | Brief eclectic psychotherapy |
| CBT-i | Cognitive behavioral therapy for insomnia |
| CDEI | Cannabis drug efficiency index |
| DEG | Differentially expressed genes |
| DEI | Drug efficiency index |
| DEIb | Balanced drug efficiency index |
| DEIm | Mirrored drug efficiency index |
| DES | Drug efficiency score |
| DESeq2 | Differential gene expression analysis in sequencing 2 |
| EMDR | Eye movement desensitization and reprocessing |
| FloWPS | Floating-window projective separator |
| GAD | Generalized anxiety disorder |
| GO | Gene ontology |
| iPANDA | In silico pathway activation network decomposition analysis |
| LFC | Log-fold change |
| MDD | Major depressive disorder |
| MH | Microarray hybridization |
| ML | Machine learning |
| NET | Narrative exposure therapy |
| NGS | Next-generation sequencing |
| NII | Node involvement index |
| PAL | Pathway activation level |
| PE | Pathway-express |
| PTSD | Post-traumatic stress disorder |
| QN | Quantile normalization |
| SNRI | Serotonin–norepinephrine reuptake inhibitor |
| SPIA | Signaling pathway impact analysis |
| SSRI | Selective serotonin reuptake inhibitors |
| TAPPA | Topology analysis of pathway phenotype association |
| TBScore | Topology-based score |
| TRD | Treatment-resistant depression |
References
- Tarhan, N.; Konuk, M.; Karahan, M.; Özcan, Ö.Ö.; Öztürk Ayvaz, S.; Hızlı Sayar, G.; Ülküer, N.; Ayas, H.; Zeynep Güder, F. New diagnosis and treatment approaches to post-traumatic stress disorder. In Stress-Related Disorders; Ovuga, E., Ed.; IntechOpen: London, UK, 2022; ISBN 978-1-80355-363-4. [Google Scholar]
- Ehlers, A.; Hackmann, A.; Michael, T. Intrusive Re-experiencing in Post-traumatic Stress Disorder: Phenomenology, Theory, and Therapy. Memory 2004, 12, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Bisson, J.I.; Cosgrove, S.; Lewis, C.; Robert, N.P. Post-Traumatic Stress Disorder. BMJ 2015, 351, h6161. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, M.; Pearce, J.; Bethell, A.; Dankova, L.; Barbui, C.; Tol, W.A.; van Ommeren, M.; de Jong, J.; Seedat, S.; Chen, H.; et al. Pharmacotherapy for Post-Traumatic Stress Disorder: Systematic Review and Meta-Analysis. Br. J. Psychiatry 2015, 206, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D. An Enduring Somatic Threat Model of Posttraumatic Stress Disorder Due to Acute Life-Threatening Medical Events: Enduring Somatic Threat Model. Soc. Personal. Psychol. Compass 2014, 8, 118–134. [Google Scholar] [CrossRef]
- McHugh, P.R.; Treisman, G. PTSD: A Problematic Diagnostic Category. J. Anxiety Disord. 2007, 21, 211–222. [Google Scholar] [CrossRef]
- Brunet, A.; Akerib, V.; Birmes, P. Don’t Throw out the Baby with the Bathwater (PTSD Is Not Overdiagnosed). Can. J. Psychiatry 2007, 52, 501–502; discussion 503. [Google Scholar] [CrossRef]
- Kilpatrick, D.G.; Resnick, H.S.; Milanak, M.E.; Miller, M.W.; Keyes, K.M.; Friedman, M.J. National Estimates of Exposure to Traumatic Events and PTSD Prevalence Using DSM-IV and DSM-5 Criteria: DSM-5 PTSD Prevalence. J. Trauma. Stress 2013, 26, 537–547. [Google Scholar] [CrossRef]
- Breslau, N. The Epidemiology of Trauma, PTSD, and Other Posttrauma Disorders. Trauma Violence Abus. 2009, 10, 198–210. [Google Scholar] [CrossRef]
- Ditlevsen, D.N.; Elklit, A. Gender, Trauma Type, and PTSD Prevalence: A Re-Analysis of 18 Nordic Convenience Samples. Ann. Gen. Psychiatry 2012, 11, 26. [Google Scholar] [CrossRef]
- Powers, M.B.; Halpern, J.M.; Ferenschak, M.P.; Gillihan, S.J.; Foa, E.B. A Meta-Analytic Review of Prolonged Exposure for Posttraumatic Stress Disorder. Clin. Psychol. Rev. 2010, 30, 635–641. [Google Scholar] [CrossRef]
- Lee, C.W.; Cuijpers, P. A Meta-Analysis of the Contribution of Eye Movements in Processing Emotional Memories. J. Behav. Ther. Exp. Psychiatry 2013, 44, 231–239. [Google Scholar] [CrossRef]
- Amos, T.; Stein, D.J.; Ipser, J.C. Pharmacological Interventions for Preventing Post-Traumatic Stress Disorder (PTSD). Cochrane Database Syst. Rev. 2014, 8, CD006239. [Google Scholar] [CrossRef]
- Sloan, D.M.; Marx, B.P. Written Exposure Therapy for PTSD: A Brief Treatment Approach for Mental Health Professionals; American Psychological Association: Washington, DC, USA, 2019; ISBN 978-1-4338-3013-6. [Google Scholar]
- Brewin, C.R.; Andrews, B.; Valentine, J.D. Meta-Analysis of Risk Factors for Posttraumatic Stress Disorder in Trauma-Exposed Adults. J. Consult. Clin. Psychol. 2000, 68, 748–766. [Google Scholar] [CrossRef]
- Ozer, E.J.; Best, S.R.; Lipsey, T.L.; Weiss, D.S. Predictors of Posttraumatic Stress Disorder and Symptoms in Adults: A Meta-Analysis. Psychol. Bull. 2003, 129, 52–73. [Google Scholar] [CrossRef]
- Bleiberg, K.L.; Markowitz, J.C. A Pilot Study of Interpersonal Psychotherapy for Posttraumatic Stress Disorder. Am. J. Psychiatry 2005, 162, 181–183. [Google Scholar] [CrossRef]
- Markowitz, J.C. IPT and PTSD. Depress. Anxiety 2010, 27, 879–881. [Google Scholar] [CrossRef]
- Markowitz, J.C.; Milrod, B.; Bleiberg, K.; Marshall, R.D. Interpersonal Factors in Understanding and Treating Posttraumatic Stress Disorder. J. Psychiatr. Pract. 2009, 15, 133–140. [Google Scholar] [CrossRef]
- Lawrence, S.; De Silva, M.; Henley, R. Sports and Games for Post-Traumatic Stress Disorder (PTSD). Cochrane Database Syst. Rev. 2010, 2010, CD007171. [Google Scholar] [CrossRef]
- Roberts, N.P.; Kitchiner, N.J.; Kenardy, J.; Robertson, L.; Lewis, C.; Bisson, J.I. Multiple Session Early Psychological Interventions for the Prevention of Post-Traumatic Stress Disorder. Cochrane Database Syst. Rev. 2019, 8, CD006869. [Google Scholar] [CrossRef]
- World Health Organization; UNHCR. Assessment and Management of Conditions Specifically Related to Stress: MhGAP Intervention Guide Mode; World Health Organization: Geneva, Switzerland, 2013; ISBN 978-92-4-150593-2. [Google Scholar]
- Birur, B.; Moore, N.C.; Davis, L.L. An Evidence-Based Review of Early Intervention and Prevention of Posttraumatic Stress Disorder. Community Ment. Health J. 2017, 53, 183–201. [Google Scholar] [CrossRef]
- Frijling, J.; Olff, M.; van Zuiden, M. Pharmacological Prevention of PTSD: Current Evidence for Clinical Practice. Psychiatr. Ann. 2019, 49, 307–313. [Google Scholar] [CrossRef]
- Williams, T.; Phillips, N.J.; Stein, D.J.; Ipser, J.C. Pharmacotherapy for Post Traumatic Stress Disorder (PTSD). Cochrane Database Syst. Rev. 2022, 3, CD002795. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, M.; Capehart, B.; Friedman, M.J. Pharmacotherapy for Posttraumatic Stress Disorder: Review with Clinical Applications. J. Rehabil. Res. Dev. 2012, 49, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Puetz, T.W.; Youngstedt, S.D.; Herring, M.P. Effects of Pharmacotherapy on Combat-Related PTSD, Anxiety, and Depression: A Systematic Review and Meta-Regression Analysis. PLoS ONE 2015, 10, e0126529. [Google Scholar] [CrossRef]
- Jain, S.; Greenbaum, M.A.; Rosen, C. Concordance between Psychotropic Prescribing for Veterans with PTSD and Clinical Practice Guidelines. Psychiatr. Serv. 2012, 63, 154–160. [Google Scholar] [CrossRef]
- Vergoulis, T.; Vlachos, I.S.; Alexiou, P.; Georgakilas, G.; Maragkakis, M.; Reczko, M.; Gerangelos, S.; Koziris, N.; Dalamagas, T.; Hatzigeorgiou, A.G. TarBase 6.0: Capturing the Exponential Growth of MiRNA Targets with Experimental Support. Nucleic Acids Res. 2012, 40, D222–D229. [Google Scholar] [CrossRef]
- Kuan, P.-F.; Waszczuk, M.A.; Kotov, R.; Clouston, S.; Yang, X.; Singh, P.K.; Glenn, S.T.; Cortes Gomez, E.; Wang, J.; Bromet, E.; et al. Gene Expression Associated with PTSD in World Trade Center Responders: An RNA Sequencing Study. Transl. Psychiatry 2017, 7, 1297. [Google Scholar] [CrossRef]
- Griffin, G.D.; Charron, D.; Al-Daccak, R. Post-Traumatic Stress Disorder: Revisiting Adrenergics, Glucocorticoids, Immune System Effects and Homeostasis. Clin. Transl. Immunol. 2014, 3, e27. [Google Scholar] [CrossRef]
- Black, N.; Stockings, E.; Campbell, G.; Tran, L.T.; Zagic, D.; Hall, W.D.; Farrell, M.; Degenhardt, L. Cannabinoids for the Treatment of Mental Disorders and Symptoms of Mental Disorders: A Systematic Review and Meta-Analysis. Lancet Psychiatry 2019, 6, 995–1010. [Google Scholar] [CrossRef]
- O’Neil, M.E.; Nugent, S.M.; Morasco, B.J.; Freeman, M.; Low, A.; Kondo, K.; Zakher, B.; Elven, C.; Motu’apuaka, M.; Paynter, R.; et al. Benefits and Harms of Plant-Based Cannabis for Posttraumatic Stress Disorder: A Systematic Review. Ann. Intern. Med. 2017, 167, 332–340. [Google Scholar] [CrossRef]
- Bonn-Miller, M.O.; Sisley, S.; Riggs, P.; Yazar-Klosinski, B.; Wang, J.B.; Loflin, M.J.E.; Shechet, B.; Hennigan, C.; Matthews, R.; Emerson, A.; et al. The Short-Term Impact of 3 Smoked Cannabis Preparations versus Placebo on PTSD Symptoms: A Randomized Cross-over Clinical Trial. PLoS ONE 2021, 16, e0246990. [Google Scholar] [CrossRef]
- Orsolini, L.; Chiappini, S.; Volpe, U.; Berardis, D.D.; Latini, R.; Papanti, G.D.; Corkery, A.J.M. Use of Medicinal Cannabis and Synthetic Cannabinoids in Post-Traumatic Stress Disorder (PTSD): A Systematic Review. Medicina 2019, 55, 525. [Google Scholar] [CrossRef]
- Sbarski, B.; Akirav, I. Cannabinoids as Therapeutics for PTSD. Pharmacol. Ther. 2020, 211, 107551. [Google Scholar] [CrossRef]
- Stanciu, C.N.; Brunette, M.F.; Teja, N.; Budney, A.J. Evidence for Use of Cannabinoids in Mood Disorders, Anxiety Disorders, and PTSD: A Systematic Review. Psychiatr. Serv. 2021, 72, 429–436. [Google Scholar] [CrossRef]
- Krediet, E.; Bostoen, T.; Breeksema, J.; van Schagen, A.; Passie, T.; Vermetten, E. Reviewing the Potential of Psychedelics for the Treatment of PTSD. Int. J. Neuropsychopharmacol. 2020, 23, 385–400. [Google Scholar] [CrossRef]
- Babson, K.A.; Sottile, J.; Morabito, D. Cannabis, Cannabinoids, and Sleep: A Review of the Literature. Curr. Psychiatry Rep. 2017, 19, 23. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I. Medical Use of Cannabinoids. Drugs 2018, 78, 1665–1703. [Google Scholar] [CrossRef]
- Loflin, M.J.; Babson, K.A.; Bonn-Miller, M.O. Cannabinoids as Therapeutic for PTSD. Curr. Opin. Psychol. 2017, 14, 78–83. [Google Scholar] [CrossRef]
- Garakani, A.; Murrough, J.W.; Freire, R.C.; Thom, R.P.; Larkin, K.; Buono, F.D.; Iosifescu, D.V. Pharmacotherapy of Anxiety Disorders: Current and Emerging Treatment Options. Front. Psychiatry 2020, 11, 595584. [Google Scholar] [CrossRef]
- Lowe, D.J.E.; Sasiadek, J.D.; Coles, A.S.; George, T.P. Cannabis and Mental Illness: A Review. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 107–120. [Google Scholar] [CrossRef]
- Betthauser, K.; Pilz, J.; Vollmer, L.E. Use and Effects of Cannabinoids in Military Veterans with Posttraumatic Stress Disorder. Am. J. Health Syst. Pharm. 2015, 72, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Yarnell, S. The Use of Medicinal Marijuana for Posttraumatic Stress Disorder: A Review of the Current Literature. Prim. Care Companion CNS Disord. 2015, 17, PCC.15r01786. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, V.K.; Merchant, J.J.; Smith, C.; Garibaldi, A. Medical Cannabis Use in the United States: A Retrospective Database Study. J. Cannabis Res. 2020, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, M.B.; Abazia, D.T. Medicinal Cannabis: History, Pharmacology, And Implications for the Acute Care Setting. P&T 2017, 42, 180–188. [Google Scholar]
- Shishko, I.; Oliveira, R.; Moore, T.A.; Almeida, K. A Review of Medical Marijuana for the Treatment of Posttraumatic Stress Disorder: Real Symptom Re-Leaf or Just High Hopes? Ment. Health Clin. 2018, 8, 86–94. [Google Scholar] [CrossRef]
- Wilkinson, S.T.; Yarnell, S.; Radhakrishnan, R.; Ball, S.A.; D’Souza, D.C. Marijuana Legalization: Impact on Physicians and Public Health. Annu. Rev. Med. 2016, 67, 453–466. [Google Scholar] [CrossRef]
- Belendiuk, K.A.; Baldini, L.L.; Bonn-Miller, M.O. Narrative Review of the Safety and Efficacy of Marijuana for the Treatment of Commonly State-Approved Medical and Psychiatric Disorders. Addict. Sci. Clin. Pract. 2015, 10, 10. [Google Scholar] [CrossRef]
- Borisov, N.; Ilnytskyy, Y.; Byeon, B.; Kovalchuk, O.; Kovalchuk, I. System, Method and Software for Calculation of a Cannabis Drug Efficiency Index for the Reduction of Inflammation. Int. J. Mol. Sci. 2020, 22, 388. [Google Scholar] [CrossRef]
- Tkachev, V.; Sorokin, M.; Garazha, A.; Borisov, N.; Buzdin, A. Oncobox Method for Scoring Efficiencies of Anticancer Drugs Based on Gene Expression Data. In Nucleic Acid Detection and Structural Investigations; Astakhova, K., Bukhari, S.A., Eds.; Springer: New York, NY, USA, 2020; Volume 2063, pp. 235–255. ISBN 978-1-07-160137-2. [Google Scholar]
- Wang, Y.; Chu, F.; Lin, J.; Li, Y.; Johnson, N.; Zhang, J.; Gai, C.; Su, Z.; Cheng, H.; Wang, L.; et al. Erianin, the Main Active Ingredient of Dendrobium Chrysotoxum Lindl, Inhibits Precancerous Lesions of Gastric Cancer (PLGC) through Suppression of the HRAS-PI3K-AKT Signaling Pathway as Revealed by Network Pharmacology and in Vitro Experimental Verification. J. Ethnopharmacol. 2021, 279, 114399. [Google Scholar] [CrossRef]
- Khojasteh Poor, F.; Keivan, M.; Ramazii, M.; Ghaedrahmati, F.; Anbiyaiee, A.; Panahandeh, S.; Khoshnam, S.E.; Farzaneh, M. Mini Review: The FDA-Approved Prescription Drugs That Target the MAPK Signaling Pathway in Women with Breast Cancer. Breast Dis. 2021, 40, 51–62. [Google Scholar] [CrossRef]
- Yuan, L.; Zhou, M.; Huang, D.; Wasan, H.S.; Zhang, K.; Sun, L.; Huang, H.; Ma, S.; Shen, M.; Ruan, S. Resveratrol Inhibits the Invasion and Metastasis of Colon Cancer through Reversal of Epithelial- Mesenchymal Transition via the AKT/GSK-3β/Snail Signaling Pathway. Mol. Med. Rep. 2019, 20, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-X.; Li, R.-Z.; Sun, A.; Zhou, H.; Neher, E.; Yang, J.-S.; Huang, J.-M.; Zhang, Y.-Z.; Jiang, Z.-B.; Liang, T.-L.; et al. Metabolomics and Integrated Network Pharmacology Analysis Reveal Tricin as the Active Anti-Cancer Component of Weijing Decoction by Suppression of PRKCA and Sphingolipid Signaling. Pharmacol. Res. 2021, 171, 105574. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Huang, J.-H.; Zhang, Z.-Y.; Du, Q.; Peng, W.-J.; Yu, R.; Zhang, S.-F.; Zhang, S.-H.; Qin, Y.-H. A Network Pharmacology-Based Strategy for Predicting Active Ingredients and Potential Targets of LiuWei DiHuang Pill in Treating Type 2 Diabetes Mellitus. Drug Des. Dev. Ther. 2019, 13, 3989–4005. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Dong, J.; Xue, C.; Li, X.; Liu, K.; Liu, B.; Cheng, J.; Huang, F. Liuwei Dihuang Pills Alleviate the Polycystic Ovary Syndrome with Improved Insulin Sensitivity through PI3K/Akt Signaling Pathway. J. Ethnopharmacol. 2020, 250, 111965. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sang, X.; Shao, R.; Qin, H.; Chen, X.; Xue, Z.; Li, L.; Wang, Y.; Zhu, Y.; Chang, Y.; et al. Xuanfei Baidu Decoction Protects against Macrophages Induced Inflammation and Pulmonary Fibrosis via Inhibiting IL-6/STAT3 Signaling Pathway. J. Ethnopharmacol. 2022, 283, 114701. [Google Scholar] [CrossRef]
- Li, X.; Shan, C.; Wu, Z.; Yu, H.; Yang, A.; Tan, B. Emodin Alleviated Pulmonary Inflammation in Rats with LPS-Induced Acute Lung Injury through Inhibiting the MTOR/HIF-1α/VEGF Signaling Pathway. Inflamm. Res. 2020, 69, 365–373. [Google Scholar] [CrossRef]
- Zhao, J.; Tian, S.; Lu, D.; Yang, J.; Zeng, H.; Zhang, F.; Tu, D.; Ge, G.; Zheng, Y.; Shi, T.; et al. Systems Pharmacological Study Illustrates the Immune Regulation, Anti-Infection, Anti-Inflammation, and Multi-Organ Protection Mechanism of Qing-Fei-Pai-Du Decoction in the Treatment of COVID-19. Phytomedicine 2021, 85, 153315. [Google Scholar] [CrossRef]
- Li, Y.; Chu, F.; Li, P.; Johnson, N.; Li, T.; Wang, Y.; An, R.; Wu, D.; Chen, J.; Su, Z.; et al. Potential Effect of Maxing Shigan Decoction against Coronavirus Disease 2019 (COVID-19) Revealed by Network Pharmacology and Experimental Verification. J. Ethnopharmacol. 2021, 271, 113854. [Google Scholar] [CrossRef]
- Shatzel, J.J.; Olson, S.R.; Tao, D.L.; McCarty, O.J.T.; Danilov, A.V.; DeLoughery, T.G. Ibrutinib-Associated Bleeding: Pathogenesis, Management and Risk Reduction Strategies. J. Thromb. Haemost. 2017, 15, 835–847. [Google Scholar] [CrossRef]
- Wu, N.; Yuan, T.; Yin, Z.; Yuan, X.; Sun, J.; Wu, Z.; Zhang, Q.; Redshaw, C.; Yang, S.; Dai, X. Network Pharmacology and Molecular Docking Study of the Chinese Miao Medicine Sidaxue in the Treatment of Rheumatoid Arthritis. Drug Des. Devel. Ther. 2022, 16, 435–466. [Google Scholar] [CrossRef]
- Ozerov, I.V.; Lezhnina, K.V.; Izumchenko, E.; Artemov, A.V.; Medintsev, S.; Vanhaelen, Q.; Aliper, A.; Vijg, J.; Osipov, A.N.; Labat, I.; et al. In Silico Pathway Activation Network Decomposition Analysis (IPANDA) as a Method for Biomarker Development. Nat. Commun. 2016, 7, 13427. [Google Scholar] [CrossRef]
- Lashmanova, E.; Zemskaya, N.; Proshkina, E.; Kudryavtseva, A.; Volosnikova, M.; Marusich, E.; Leonov, S.; Zhavoronkov, A.; Moskalev, A. The Evaluation of Geroprotective Effects of Selected Flavonoids in Drosophila Melanogaster and Caenorhabditis Elegans. Front. Pharmacol. 2017, 8, 884. [Google Scholar] [CrossRef]
- Borisov, N.; Sorokin, M.; Garazha, A.; Buzdin, A. Quantitation of Molecular Pathway Activation Using RNA Sequencing Data. In Nucleic Acid Detection and Structural Investigations; Astakhova, K., Bukhari, S.A., Eds.; Springer: New York, NY, USA, 2020; Volume 2063, pp. 189–206. ISBN 978-1-07-160137-2. [Google Scholar]
- Gao, S.; Wang, X. TAPPA: Topological Analysis of Pathway Phenotype Association. Bioinformics 2007, 23, 3100–3102. [Google Scholar] [CrossRef]
- Ibrahim, M.A.-H.; Jassim, S.; Cawthorne, M.A.; Langlands, K. A Topology-Based Score for Pathway Enrichment. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 563–573. [Google Scholar] [CrossRef]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A Systems Biology Approach for Pathway Level Analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A Novel Signaling Pathway Impact Analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Nakaya, A.; Katayama, T.; Itoh, M.; Hiranuka, K.; Kawashima, S.; Moriya, Y.; Okuda, S.; Tanaka, M.; Tokimatsu, T.; Yamanishi, Y.; et al. KEGG OC: A Large-Scale Automatic Construction of Taxonomy-Based Ortholog Clusters. Nucleic Acids Res. 2013, 41, D353–D357. [Google Scholar] [CrossRef]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational Prediction of Human Metabolic Pathways from the Complete Human Genome. Genome Biol. 2004, 6, R2. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Li, C.; Marcu, A.; Badran, H.; Pon, A.; Budinski, Z.; Patron, J.; Lipton, D.; Cao, X.; Oler, E.; et al. PathBank: A Comprehensive Pathway Database for Model Organisms. Nucleic Acids Res. 2020, 48, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B. Preprocessing and Normalization for Affymetrix GeneChip Expression Microarrays. In Methods in Microarray Normalization; Stafford, P., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 41–59. ISBN 978-1-4200-5278-7. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Segman, R.H.; Shefi, N.; Goltser-Dubner, T.; Friedman, N.; Kaminski, N.; Shalev, A.Y. Peripheral Blood Mononuclear Cell Gene Expression Profiles Identify Emergent Post-Traumatic Stress Disorder among Trauma Survivors. Mol. Psychiatry 2005, 10, 425, 500–513. [Google Scholar] [CrossRef]
- Breen, M.S.; Maihofer, A.X.; Glatt, S.J.; Tylee, D.S.; Chandler, S.D.; Tsuang, M.T.; Risbrough, V.B.; Baker, D.G.; O’Connor, D.T.; Nievergelt, C.M.; et al. Gene Networks Specific for Innate Immunity Define Post-Traumatic Stress Disorder. Mol. Psychiatry 2015, 20, 1538–1545. [Google Scholar] [CrossRef]
- Rusch, H.L.; Robinson, J.; Yun, S.; Osier, N.D.; Martin, C.; Brewin, C.R.; Gill, J.M. Gene Expression Differences in PTSD Are Uniquely Related to the Intrusion Symptom Cluster: A Transcriptome-Wide Analysis in Military Service Members. Brain Behav. Immun. 2019, 80, 904–908. [Google Scholar] [CrossRef]
- Kuan, P.-F.; Ren, X.; Clouston, S.; Yang, X.; Jonas, K.; Kotov, R.; Bromet, E.; Luft, B.J. PTSD Is Associated with Accelerated Transcriptional Aging in World Trade Center Responders. Transl. Psychiatry 2021, 11, 311. [Google Scholar] [CrossRef]
- Cathomas, F.; Bevilacqua, L.; Ramakrishnan, A.; Kronman, H.; Costi, S.; Schneider, M.; Chan, K.L.; Li, L.; Nestler, E.J.; Shen, L.; et al. Whole Blood Transcriptional Signatures Associated with Rapid Antidepressant Response to Ketamine in Patients with Treatment Resistant Depression. Transl. Psychiatry 2022, 12, 12. [Google Scholar] [CrossRef]
- Sorokin, M.; Borisov, N.; Kuzmin, D.; Gudkov, A.; Zolotovskaia, M.; Garazha, A.; Buzdin, A. Algorithmic Annotation of Functional Roles for Components of 3,044 Human Molecular Pathways. Front. Genet. 2021, 12, 617059. [Google Scholar] [CrossRef]
- Aliper, A.M.; Korzinkin, M.B.; Kuzmina, N.B.; Zenin, A.A.; Venkova, L.S.; Smirnov, P.Y.; Zhavoronkov, A.A.; Buzdin, A.A.; Borisov, N.M. Mathematical Justification of Expression-Based Pathway Activation Scoring (PAS). Methods Mol. Biol. 2017, 1613, 31–51. [Google Scholar] [CrossRef]
- Kuzmina, N.B.; Borisov, N.M. Handling Complex Rule-Based Models of Mitogenic Cell Signaling (On the Example of ERK Activation upon EGF Stimulation). Int. Proc. Chem. Biol. Environ. Eng. 2011, 5, 76–82. [Google Scholar]
- Zolotovskaia, M.A.; Tkachev, V.S.; Guryanova, A.A.; Simonov, A.M.; Raevskiy, M.M.; Efimov, V.V.; Wang, Y.; Sekacheva, M.I.; Garazha, A.V.; Borisov, N.M.; et al. OncoboxPD: Human 51 672 Molecular Pathways Database with Tools for Activity Calculating and Visualization. Comput. Struct. Biotechnol. J. 2022, 20, 2280–2291. [Google Scholar] [CrossRef]
- Borisov, N.; Suntsova, M.; Sorokin, M.; Garazha, A.; Kovalchuk, O.; Aliper, A.; Ilnitskaya, E.; Lezhnina, K.; Korzinkin, M.; Tkachev, V.; et al. Data Aggregation at the Level of Molecular Pathways Improves Stability of Experimental Transcriptomic and Proteomic Data. Cell Cycle 2017, 16, 1810–1823. [Google Scholar] [CrossRef]
- Tkachev, V.; Sorokin, M.; Mescheryakov, A.; Simonov, A.; Garazha, A.; Buzdin, A.; Muchnik, I.; Borisov, N. FLOating-Window Projective Separator (FloWPS): A Data Trimming Tool for Support Vector Machines (SVM) to Improve Robustness of the Classifier. Front. Genet. 2019, 9, 717. [Google Scholar] [CrossRef]
- Tkachev, V.; Sorokin, M.; Borisov, C.; Garazha, A.; Buzdin, A.; Borisov, N. Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology. Int. J. Mol. Sci. 2020, 21, 713. [Google Scholar] [CrossRef]
- Obuchowski, N.A. Receiver Operating Characteristic Curves and Their Use in Radiology. Radiology 2003, 229, 3–8. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Borisov, N.; Sergeeva, A.; Suntsova, M.; Raevskiy, M.; Gaifullin, N.; Mendeleeva, L.; Gudkov, A.; Nareiko, M.; Garazha, A.; Tkachev, V.; et al. Machine Learning Applicability for Classification of PAD/VCD Chemotherapy Response Using 53 Multiple Myeloma RNA Sequencing Profiles. Front. Oncol. 2021, 11, 652063. [Google Scholar] [CrossRef]
- Zhou, R.; Wen, Z.; Liao, Y.; Wu, J.; Xi, S.; Zeng, D.; Sun, H.; Wu, J.; Shi, M.; Bin, J.; et al. Evaluation of Stromal Cell Infiltration in the Tumor Microenvironment Enable Prediction of Treatment Sensitivity and Prognosis in Colon Cancer. Comput. Struct. Biotechnol. J. 2022, 20, 2153–2168. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef]
- Malas, T.B.; Leonhard, W.N.; Bange, H.; Granchi, Z.; Hettne, K.M.; Van Westen, G.J.P.; Price, L.S.; ’t Hoen, P.A.C.; Peters, D.J.M. Prioritization of Novel ADPKD Drug Candidates from Disease-Stage Specific Gene Expression Profiles. EBioMedicine 2020, 51, 102585. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.C.; Kraler, S.; Lüscher, T.F.; Aikawa, E. Multi-Omics Approaches to Define Calcific Aortic Valve Disease Pathogenesis. Circ. Res. 2021, 128, 1371–1397. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Luo, H.; Xi, Z.; Rogaeva, E. Drug Repositioning for Diabetes Based on “omics” Data Mining. PLoS ONE 2015, 10, e0126082. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Klein, A.; Madiehe, A.; Meyer, M. Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease. Int. J. Mol. Sci. 2021, 22, 9431. [Google Scholar] [CrossRef]
- Suratanee, A.; Plaimas, K. Heterogeneous Network Model to Identify Potential Associations Between Plasmodium Vivax and Human Proteins. Int. J. Mol. Sci. 2020, 21, 1310. [Google Scholar] [CrossRef]
- Danieli, M.G.; Tonacci, A.; Paladini, A.; Longhi, E.; Moroncini, G.; Allegra, A.; Sansone, F.; Gangemi, S. A Machine Learning Analysis to Predict the Response to Intravenous and Subcutaneous Immunoglobulin in Inflammatory Myopathies. A Proposal for a Future Multi-Omics Approach in Autoimmune Diseases. Autoimmun. Rev. 2022, 21, 103105. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Chumnanpuen, P.; Schaduangrat, N.; Lio’, P.; Moni, M.A.; Shoombuatong, W. Improved Prediction and Characterization of Blood-Brain Barrier Penetrating Peptides Using Estimated Propensity Scores of Dipeptides. J. Comput. Aided. Mol. Des. 2022, 36, 781–796. [Google Scholar] [CrossRef]
- Aliper, A.M.; Csoka, A.B.; Buzdin, A.; Jetka, T.; Roumiantsev, S.; Moskalev, A.; Zhavoronkov, A. Signaling Pathway Activation Drift during Aging: Hutchinson-Gilford Progeria Syndrome Fibroblasts Are Comparable to Normal Middle-Age and Old-Age Cells. Aging 2015, 7, 26–37. [Google Scholar] [CrossRef]
- Makarev, E.; Cantor, C.; Zhavoronkov, A.; Buzdin, A.; Aliper, A.; Csoka, A.B. Pathway Activation Profiling Reveals New Insights into Age-Related Macular Degeneration and Provides Avenues for Therapeutic Interventions. Aging 2014, 6, 1064–1075. [Google Scholar] [CrossRef][Green Version]
- Poddubskaya, E.; Buzdin, A.; Garazha, A.; Sorokin, M.; Glusker, A.; Aleshin, A.; Allina, D.; Moiseev, A.; Sekacheva, M.; Suntsova, M.; et al. Oncobox, Gene Expression-Based Second Opinion System for Predicting Response to Treatment in Advanced Solid Tumors. J. Clin. Oncol. 2019, 37, e13143. [Google Scholar] [CrossRef]
- Neigh, G.N.; Ali, F.F. Co-Morbidity of PTSD and Immune System Dysfunction: Opportunities for Treatment. Curr. Opin. Pharmacol. 2016, 29, 104–110. [Google Scholar] [CrossRef]
- Monsour, M.; Ebedes, D.; Borlongan, C.V. A Review of the Pathology and Treatment of TBI and PTSD. Exp. Neurol. 2022, 351, 114009. [Google Scholar] [CrossRef]
- Borisov, N.; Buzdin, A. Transcriptomic Harmonization as the Way for Suppressing Cross-Platform Bias and Batch Effect. Biomedicines 2022, 10, 2318. [Google Scholar] [CrossRef]






| Reference | [80] | [81] | [81] | [82] | [30,83] | [84] |
|---|---|---|---|---|---|---|
| GSE ID | GSE860 | GSE64814-GPL6244 | GSE64814-GPL11154 | GSE81761 | GSE97356 | GSE185855 |
| Cases | 17 | 48 | 94 | 50 | 91 | 46 |
| Controls | 16 | 48 | 94 | 33 | 233 | 21 |
| Number of genes | 9068 | 12,631 | 10,184 | 22,878 | 25,830 | 55,178 |
| GSE ID, Clinical Remarks | GSE860 | GSE64814-GPL6244 (MH) | GSE64814-GPL11154 (NGS) | GSE81761 No Improvement; Non-Responders | GSE81761 Improvement; Responders | GSE97356 | GSE185855 TRD: Non-Responders | GSE185855 TRD: Responders | |
|---|---|---|---|---|---|---|---|---|---|
| SPIA with differential expression filter [51] | t(U) | −5.27 | −1.38 | 0.00 | 2.17 | 2.17 | −2.29 | −2.48 | −0.53 |
| t(T) | 3.35 | 0.49 | 0.00 | −1.18 | −2.12 | 0.49 | −1.21 | −1.89 | |
| DEI | 0.22 | 0.47 | 0.00 | 0.30 | 0.01 | 0.65 | 0.35 | −0.56 | |
| DEIm | 0.69 | 0.51 | 0.00 | 0.63 | 0.98 | 0.43 | 0.15 | −0.39 | |
| DEIb | 0.46 | 0.49 | 0.00 | 0.46 | 0.50 | 0.54 | 0.25 | −0.47 | |
| p-value | 5 × 10−8 | 0.11 | 0 | 0.007 | 0.008 | 0.16 | 0.29 | 0.33 | |
| SPIA without differential expression filter [51] | t(U) | −0.19 | 5.50 | −3.71 | 2.96 | 2.96 | −1.20 | −1.08 | −0.56 |
| t(T) | 5.69 | −7.83 | 3.29 | 2.50 | −3.71 | −1.24 | −1.56 | −3.52 | |
| DEI | −0.94 | −0.17 | 0.06 | 0.08 | −0.11 | −0.02 | −0.19 | −0.73 | |
| DEIm | −0.87 | 0.65 | 0.89 | 0.04 | 0.78 | −0.01 | −0.10 | −0.57 | |
| DEIb | −0.90 | 0.24 | 0.48 | 0.06 | 0.33 | −0.01 | −0.14 | −0.65 | |
| p-value | 9 × 10−6 | 9 × 10−17 | 5 × 10−7 | 0.03 | 1.4 × 10−6 | 0.70 | 0.51 | 0.004 | |
| Oncobox Pathway Database [88] | t(U) | −0.75 | 11.21 | −8.63 | 4.52 | 4.52 | −0.67 | 0.02 | −3.06 |
| t(T) | 15.79 | −16.64 | 7.41 | 4.93 | −5.41 | 2.26 | 0.95 | −11.53 | |
| DEI | −0.91 | −0.20 | 0.08 | −0.04 | −0.09 | −0.54 | −0.95 | −0.58 | |
| DEIm | −0.82 | 0.61 | 0.87 | −0.02 | 0.82 | −0.08 | −0.90 | −0.41 | |
| DEIb | −0.86 | 0.21 | 0.47 | −0.03 | 0.37 | −0.31 | −0.93 | −0.49 | |
| p-value | 3 × 10−38 | 6 × 10−52 | 5 × 10−27 | 0.65 | 4 × 10−11 | 0.03 | 0.30 | 1.0 × 10−14 | |
| Dataset | Exp_D_vs_H | Exp_P_vs_A | Exp_R_vs_N | SPIA_D_vs_H | SPIA_P_vs_A | SPIA_R_vs_N |
|---|---|---|---|---|---|---|
| GSE860 | 12 | 7 | - | 16 | 21 | - |
| GSE64814-GPL6244 | 3 | 21 | - | 0 | 3 | - |
| GSE64814-GPL11154 | 0 | 21 | - | 0 | 0 | - |
| GSE97356 | 0 | 15 | - | 0 | 0 | - |
| GSE81761 | 12 | 7 | 16 | 4 | 18 | 18 |
| GSE185855 | 8 | 9 | 11 | 15 | 28 | 19 |
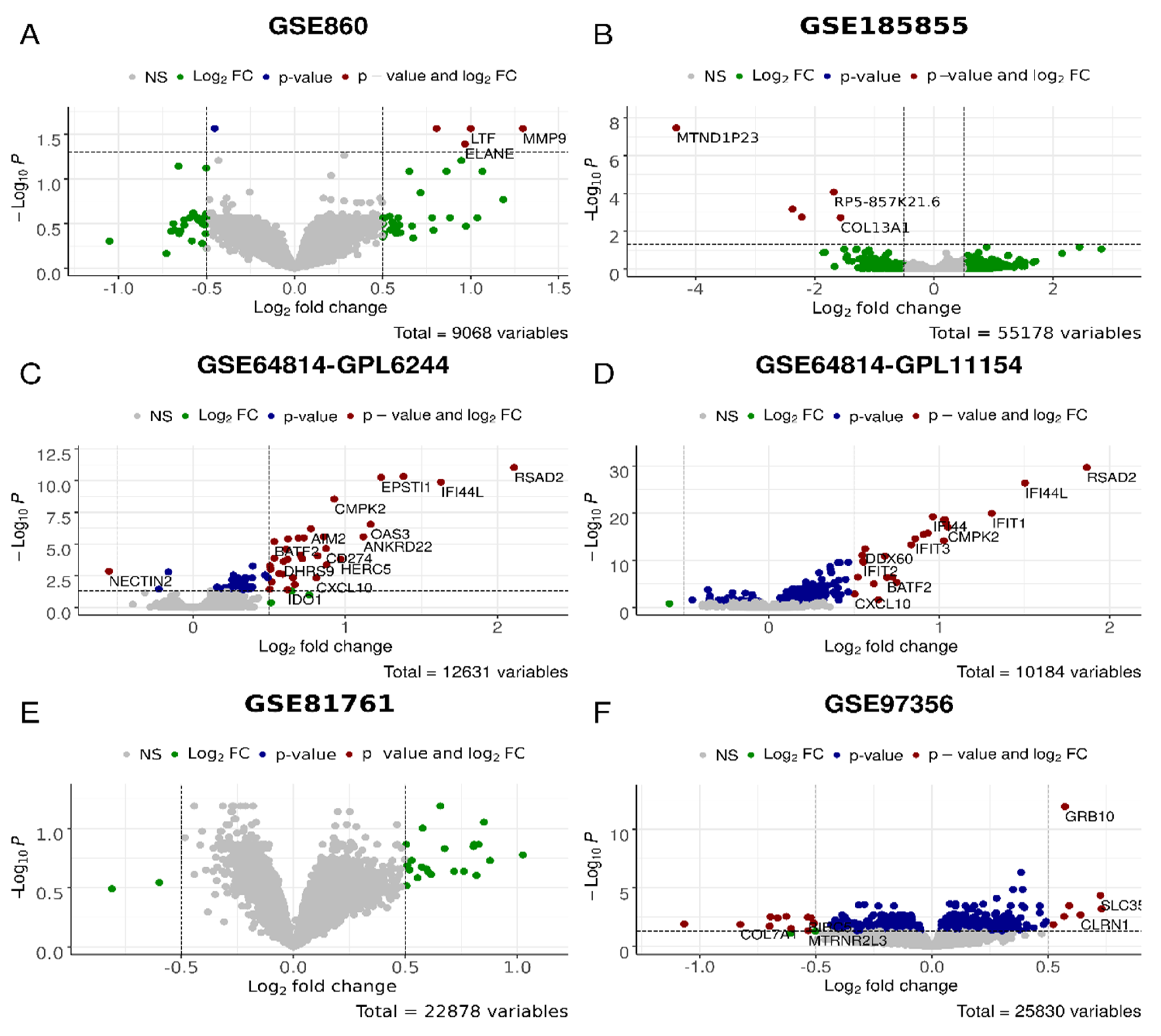
| Cohort | D_vs_H | A_vs_P | R_vs_N | |||
|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | |
| GSE185855 | 5 | 93 | 8 | |||
| GSE81761 | 759 | 16 | 567 | |||
| GSE64814-GPL11154 | 25 | 2 | 4 | |||
| GSE64814-GPL6244 | 33 | 1 | 341 | 75 | ||
| GSE860 | 4 | |||||
| GSE97356 | 6 | 13 | ||||
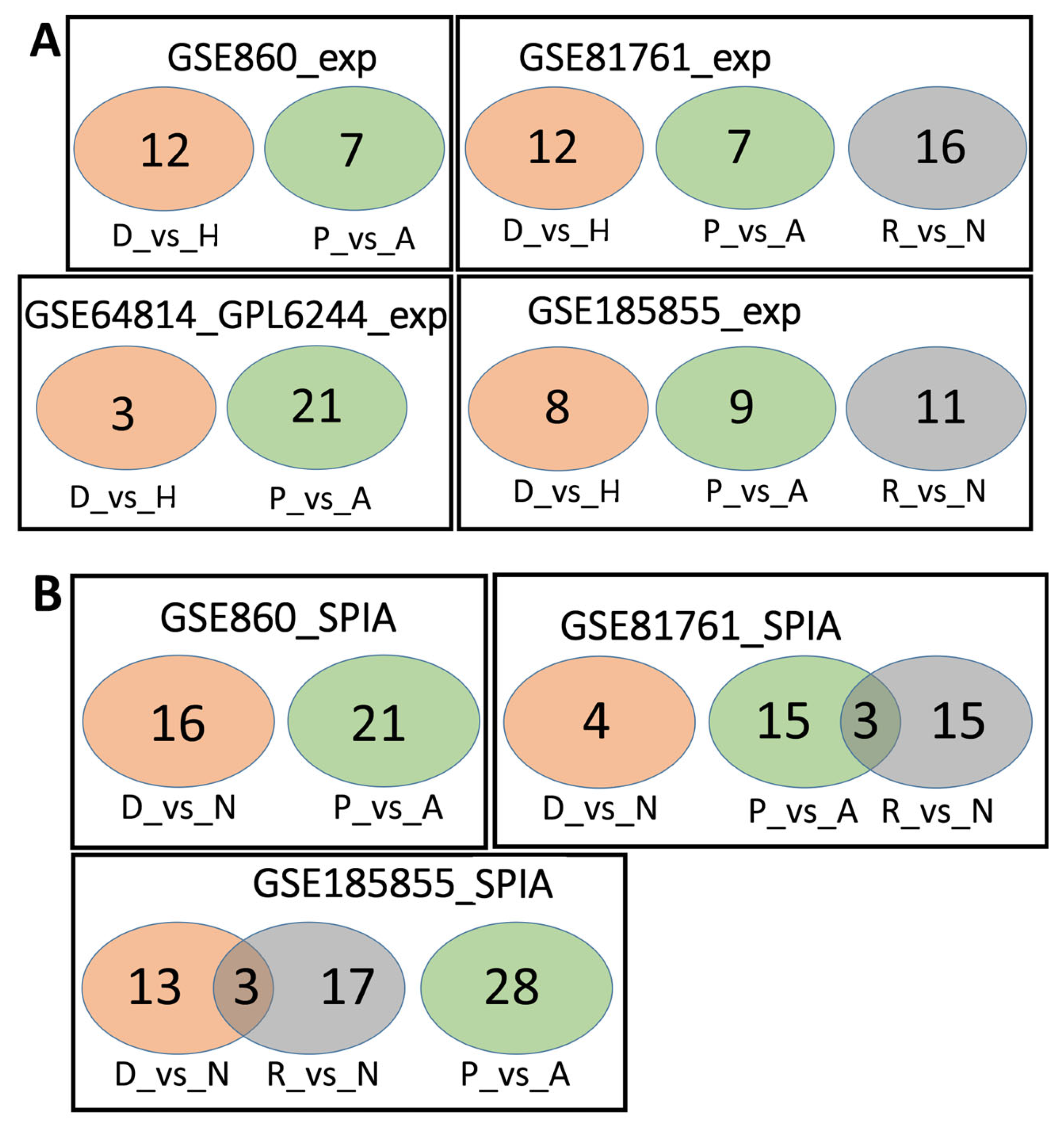
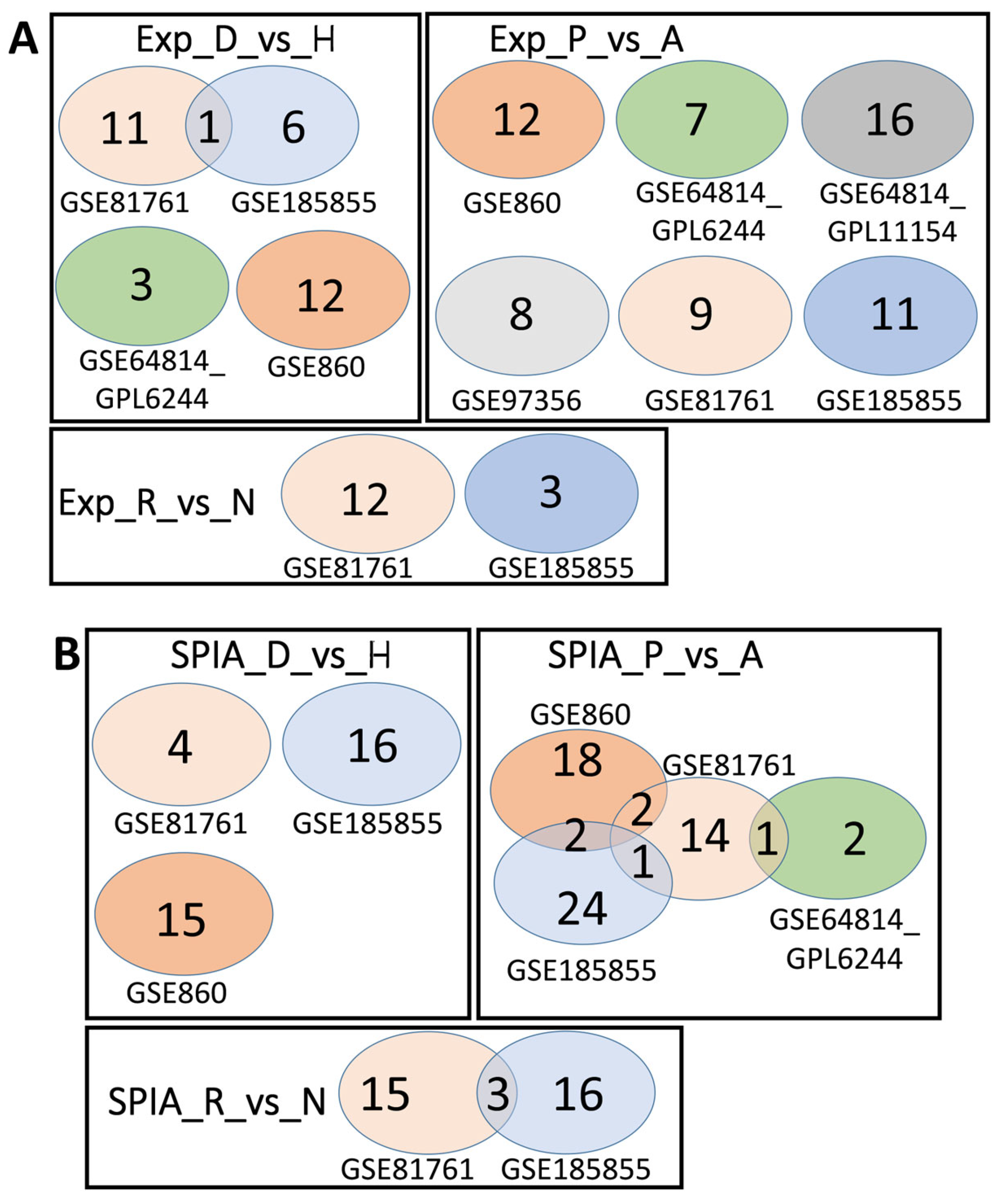
| Dataset | AUC D_vs_H | AUC P_vs_A | AUC R_vs_N | DESeq2 D_vs_H | DESeq2 P_vs_A | DESeq2 R_vs_N | Overlap D_vs_H | Overlap P_vs_A | Overlap R_vs_N |
|---|---|---|---|---|---|---|---|---|---|
| GSE860 | 12 | 7 | - | 4 | 0 | - | 1 | 0 | - |
| GSE64814-GPL6244 | 3 | 21 | - | 34 | 416 | - | 1 | 17 | - |
| GSE64814-GPL11154 | 0 | 21 | - | 25 | 6 | - | 0 | 0 | - |
| GSE97356 | 0 | 15 | - | 19 | 0 | - | 0 | 0 | - |
| GSE81761 | 12 | 7 | 16 | 0 | 759 | 583 | 0 | 0 | 7 |
| GSE185855 | 8 | 9 | 11 | 5 | 0 | 105 | 0 | 0 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borisov, N.; Ilnytskyy, Y.; Byeon, B.; Kovalchuk, O.; Kovalchuk, I. Application of Drug Efficiency Index Metric for Analysis of Post-Traumatic Stress Disorder and Treatment Resistant Depression Gene Expression Profiles. Psychoactives 2023, 2, 92-112. https://doi.org/10.3390/psychoactives2020007
Borisov N, Ilnytskyy Y, Byeon B, Kovalchuk O, Kovalchuk I. Application of Drug Efficiency Index Metric for Analysis of Post-Traumatic Stress Disorder and Treatment Resistant Depression Gene Expression Profiles. Psychoactives. 2023; 2(2):92-112. https://doi.org/10.3390/psychoactives2020007
Chicago/Turabian StyleBorisov, Nicolas, Yaroslav Ilnytskyy, Boseon Byeon, Olga Kovalchuk, and Igor Kovalchuk. 2023. "Application of Drug Efficiency Index Metric for Analysis of Post-Traumatic Stress Disorder and Treatment Resistant Depression Gene Expression Profiles" Psychoactives 2, no. 2: 92-112. https://doi.org/10.3390/psychoactives2020007
APA StyleBorisov, N., Ilnytskyy, Y., Byeon, B., Kovalchuk, O., & Kovalchuk, I. (2023). Application of Drug Efficiency Index Metric for Analysis of Post-Traumatic Stress Disorder and Treatment Resistant Depression Gene Expression Profiles. Psychoactives, 2(2), 92-112. https://doi.org/10.3390/psychoactives2020007