

Chirality in Modern Antidepressants: A Comprehensive Review of Stereochemical Impacts on Pharmacology and Therapeutics



Abstract
1. Introduction
- Major depressive disorder—the most common type of depression—involves a persistent depressed mood or loss of interest lasting for at least two weeks, significantly interfering with daily activities, and characterized by feelings of sadness and hopelessness;
- Persistent depressive disorder (dysthymia or dysthymic disorder)—a chronic form of depression with less severe symptoms lasting for at least two years, where a person feels depressed most of the day on most days;
- Perinatal depression (prenatal and postpartum depression)—occurs during or after pregnancy and involves sadness, anxiety, and exhaustion, affecting a mother’s ability to care for herself or her baby;
- Seasonal affective disorder—a type of depression that occurs at specific times of the year, typically beginning in late fall or early winter and resolving in spring or summer, often due to reduced natural sunlight;
- Psychotic depression—a severe form of depression that includes symptoms of psychosis, such as delusions or hallucinations;
- Bipolar disorders (manic depression)—involves alternating episodes of depression and mania, marked by high energy and risky behaviors.
2. Chiral Antidepressants
- Tricyclic antidepressants (TCAs): dibenzazepines (imipramine, clomipramine, trimipramine, desipramine), dibenzocycloheptadienes (amitriptyline, nortriptyline), dibenzoxepines (doxepine);
- Tetracyclic antidepressants: maprotiline, mianserin, mirtazapine;
- Selective serotonin reuptake inhibitors (SSRIs): citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline;
- Serotonin–norepinephrine reuptake inhibitors (SNRIs): desvenlafaxine, duloxetine, milnacipran, venlafaxine;
- Serotonin antagonist and reuptake inhibitors (SARIs): nefazodone, trazodone;
- Norepinephrine reuptake inhibitors (NRIs): reboxetine, teniloxazine, viloxazine;
- Norepinephrine–dopamine reuptake inhibitors (NDRIs): bupropion;
- Monoamine oxidase inhibitors (MAOIs): irreversible (isocarboxazid, phenelzine, tranylcypromine), reversible (metralindole, moclobemide, pirlindole);
- Other antidepressants: agomelatine, esketamine, indeloxazine.
2.1. Selective Serotonin Reuptake Inhibitors (SSRIs)
2.2. Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs)
2.3. Norepinephrine Reuptake Inhibitors (NRIs)
2.4. Norepinephrine–Dopamine Reuptake Inhibitors (NDRIs)
2.5. Tetracyclic Antidepressants
2.6. Other Antidepressants
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bernard, J.E.R. Depression: A review of its definition. MOJ Addict. Med. Ther. 2018, 5, 6–7. [Google Scholar]
- Richards, D. Prevalence and clinical course of depression: A review. Clin. Psychol. Rev. 2011, 31, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.; Martinez, J.H.; Young, D.; Chelminski, I.; Dalrymple, K. Severity classification on the Hamilton depression rating scale. J. Affect. Disord. 2013, 150, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Durisko, Z.; Mulsant, B.H.; Andrews, P.W. An adaptationist perspective on the etiology of depression. J. Affect. Disord. 2015, 172, 315–323. [Google Scholar] [CrossRef]
- Saveanu, R.V.; Nemeroff, C.B. Etiology of depression: Genetic and environmental factors. Psychiatr. Clin. 2012, 35, 51–71. [Google Scholar] [CrossRef]
- Lépine, J.P.; Briley, M. The increasing burden of depression. Neuropsychiatr. Dis. Treat. 2011, 7 (Suppl. S1), 3–7. [Google Scholar]
- Fried, E.I.; Nesse, R.M. Depression sum-scores don’t add up: Why analyzing specific depression symptoms is essential. BMC Med. 2015, 13, 72. [Google Scholar] [CrossRef]
- Cuijpers, P.; van Straten, A.; Schuurmans, J.; van Oppen, P.; Hollon, S.D.; Andersson, G. Psychotherapy for chronic major depression and dysthymia: A meta-analysis. Clin. Psychol. Rev. 2010, 30, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, P.; Huibers, M.; Ebert, D.D.; Koole, S.L.; Andersson, G. How much psychotherapy is needed to treat depression? A meta-regression analysis. J. Affect. Disord. 2013, 149, 1–13. [Google Scholar] [CrossRef]
- Pigott, H.E.; Leventhal, A.M.; Alter, G.S.; Boren, J.J. Efficacy and effectiveness of antidepressants: Current status of research. Psychother. Psychosom. 2010, 79, 267–279. [Google Scholar] [CrossRef]
- Faquih, A.E.; Memon, R.I.; Hafeez, H.; Zeshan, M.; Naveed, S. A review of novel antidepressants: A guide for clinicians. Cureus 2019, 11, e4185. [Google Scholar] [CrossRef] [PubMed]
- Penn, E.; Tracy, D.K. The drugs don’t work? Antidepressants and the current and future pharmacological management of depression. Ther. Adv. Psychopharmacol. 2012, 2, 179–188. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Marathe, S.V.; D’almeida, P.L.; Virmani, G.; Bathini, P.; Alberi, L. Effects of monoamines and antidepressants on astrocyte physiology: Implications for monoamine hypothesis of depression. J. Exp. Neurosci. 2018, 12, 1179069518789149. [Google Scholar] [CrossRef]
- Hammen, C. Depression and stressful environments: Identifying gaps in conceptualization and measurement. Anxiety Stress Coping 2016, 29, 335–351. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Sekhon, B.S. Exploiting the power of stereochemistry in drugs: An overview of racemic and enantiopure drugs. J. Mod. Med. Chem. 2013, 1, 10–36. [Google Scholar] [CrossRef]
- Peepliwal, A.K.; Bagade, S.B.; Bonde, C.G. A Review: Stereochemical consideration and eudismic ratio in chiral drug development. J. Biomed. Sci. Res. 2010, 2, 29–45. [Google Scholar]
- de la Torre, G.B.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24, 809. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85. [Google Scholar]
- Budău, M.; Hancu, G.; Rusu, A.; Cârcu-Dobrin, M.; Muntean, D.L. Chirality of modern antidepressants: An overview. Adv. Pharm. Bull. 2017, 7, 495. [Google Scholar] [CrossRef]
- Vashistha, V.K.; Sethi, S.; Tyagi, I.; Das, D.K. Chirality of antidepressive drugs: An overview of stereoselectivity. Asian Biomed. 2022, 16, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Alvano, S.A.; Zieher, L.M. An updated classification of antidepressants: A proposal to simplify treatment. Pers. Med. Psychiatry 2020, 19, 100042. [Google Scholar] [CrossRef]
- Rossi, A.; Barraco, A.; Donda, P. Fluoxetine: A review on evidence based medicine. Ann. Gen. Hosp. Psychiatry 2004, 3, 2. [Google Scholar] [CrossRef]
- Wong, D.T.; Perry, K.W.; Bymaster, F.P. The discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discov. 2005, 4, 764–774. [Google Scholar] [CrossRef]
- Perez-Caballero, L.; Torres-Sanchez, S.; Bravo, L.; Mico, J.A.; Berrocoso, E. Fluoxetine: A case history of its discovery and preclinical development. Expert Opin. Drug Discov. 2014, 9, 567–578. [Google Scholar] [CrossRef]
- Mandrioli, R.; Forti, G.C.; Raggi, M.A. Fluoxetine metabolism and pharmacological interactions: The role of cytochrome p450. Curr. Drug Metab. 2006, 7, 127–133. [Google Scholar] [CrossRef]
- Eap, C.B.; Bondolfi, G.; Zullino, D.; Savary-Cosendai, L.; Powell-Golay, K.; Kosel, M.; Baumann, P. Concentrations of the enantiomers of fluoxetine and norfluoxetine after multiple doses of fluoxetine in cytochrome P4502D6 poor and extensive metabolizers. J. Clin. Psychopharmacol. 2001, 21, 330–334. [Google Scholar] [CrossRef]
- Scordo, M.G.; Spina, E.; Dahl, M.L.; Gatti, G.; Perucca, E. Influence of CYP2C9, 2C19 and 2D6 genetic polymorphisms on the steady-state plasma concentrations of the enantiomers of fluoxetine and norfluoxetine. Basic Clin. Pharmacol. Toxicol. 2005, 97, 296–301. [Google Scholar] [CrossRef]
- McConathy, J.; Owens, M.J. Stereochemistry in drug action. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.J.; Ahmed, F.; Findley, L.J.; MacGregor, E.A.; Wilkinson, M. S-fluoxetine in the prophylaxis of migraine: A phase II double-blind randomized placebo-controlled study. Cephalalgia 1998, 18, 283–286. [Google Scholar] [CrossRef]
- Bezchlibnyk-Butler, K.; Aleksic, I.; Kennedy, S.H. Citalopram—A review of pharmacological and clinical effects. J. Psychiatry Neurosci. 2000, 25, 241. [Google Scholar]
- Pollock, B.G. Citalopram: A comprehensive review. Expert Opin. Pharmacother. 2001, 2, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Hyttel, J.; Bøgesø, K.P.; Perregaard, J.; Sanchez, C. The pharmacological effect of citalopram resides in the (S)-(+)-enantiomer. J. Neural Transm. Gen. Sect. JNT 1992, 88, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, J.; Priskorn, M.; Poulsen, M.; Segonzac, A.; Grollier, G.; Larsen, F. Steady-state pharmacokinetics of the enantiomers of citalopram and its metabolites in humans. Chirality 1997, 9, 686–692. [Google Scholar] [CrossRef]
- Baumann, P. Pharmacology and pharmacokinetics of citalopram and other SSRIs. Int. Clin. Psychopharmacol. 1996, 11, 5–12. [Google Scholar] [CrossRef]
- Brøsen, K.; Naranjo, C.A. Review of pharmacokinetic and pharmacodynamic interaction studies with citalopram. Eur. Neuropsychopharmacol. 2001, 11, 275–283. [Google Scholar] [CrossRef]
- Sánchez, C. The pharmacology of citalopram enantiomers: The antagonism by R-citalopram on the effect of S-citalopram. Basic Clin. Pharmacol. Toxicol. 2006, 99, 91–95. [Google Scholar] [CrossRef]
- Sánchez, C.; Bøgesø, K.P.; Ebert, B.; Reines, E.H.; Braestrup, C. Escitalopram versus citalopram: The surprising role of the R-enantiomer. Psychopharmacology 2004, 174, 163–176. [Google Scholar] [CrossRef]
- Montgomery, S.A.; Loft, H.; Sánchez, C.; Reines, E.H.; Papp, M. Escitalopram (S-enantiomer of citalopram): Clinical efficacy and onset of action predicted from a rat model. Pharmacol. Toxicol. 2001, 88, 282–286. [Google Scholar] [CrossRef] [PubMed]
- McRae, A.L.; Brady, K.T. Review of sertraline and its clinical applications in psychiatric disorders. Expert Opin. Pharmacother. 2001, 2, 883–892. [Google Scholar] [CrossRef]
- Doogan, D.P.; Caillard, V. Sertraline in the prevention of depression. Br. J. Psychiatry 1992, 160, 217–222. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, G.; Born, L.; Steiner, M. The selective serotonin reuptake inhibitor sertraline: Its profile and use in psychiatric disorders. CNS Drug Rev. 2001, 7, 1–24. [Google Scholar] [CrossRef]
- De Vane, C.L.; Liston, H.L.; Markowitz, J.S. Clinical pharmacokinetics of sertraline. Clin. Pharmacokinet. 2002, 41, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
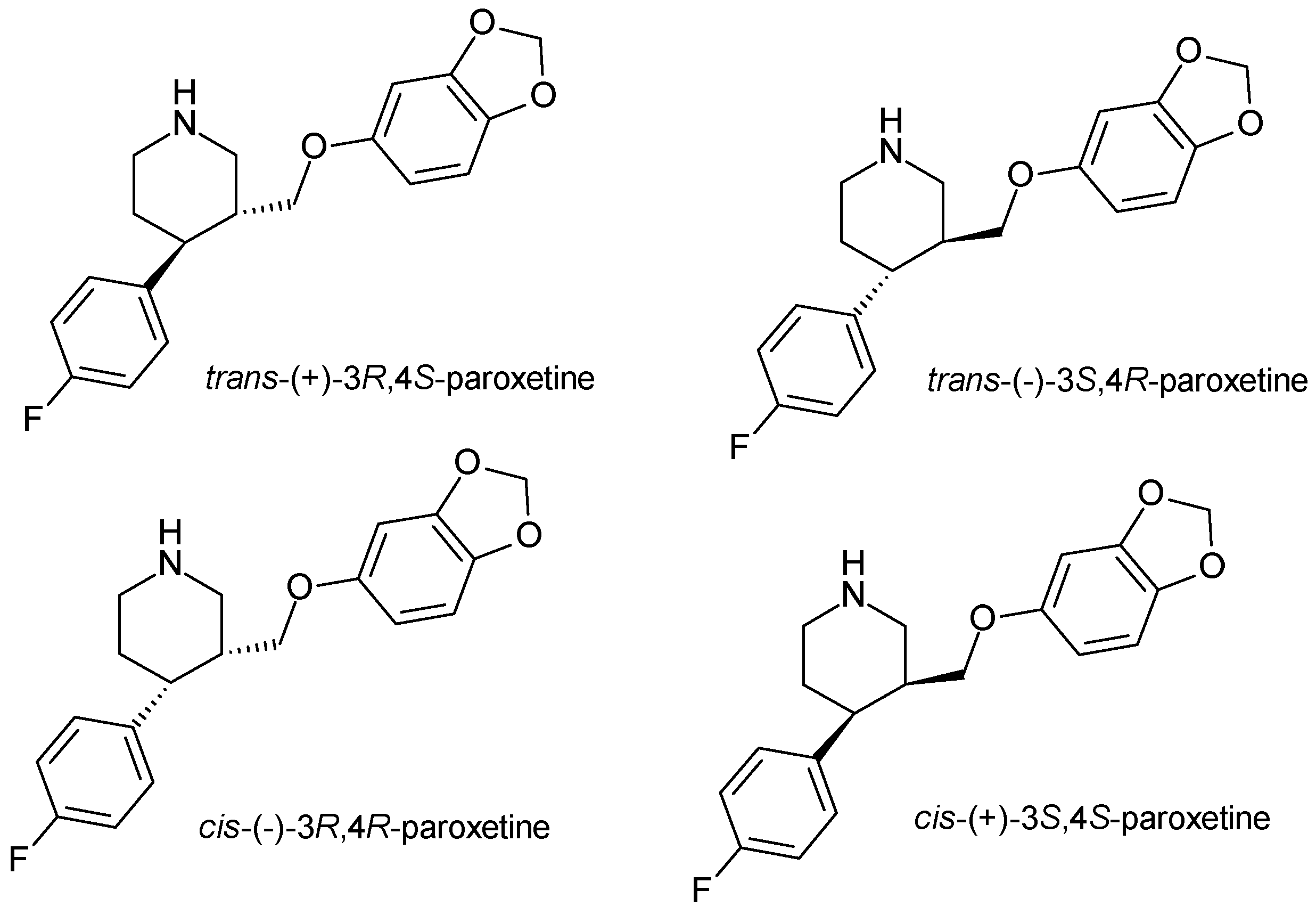
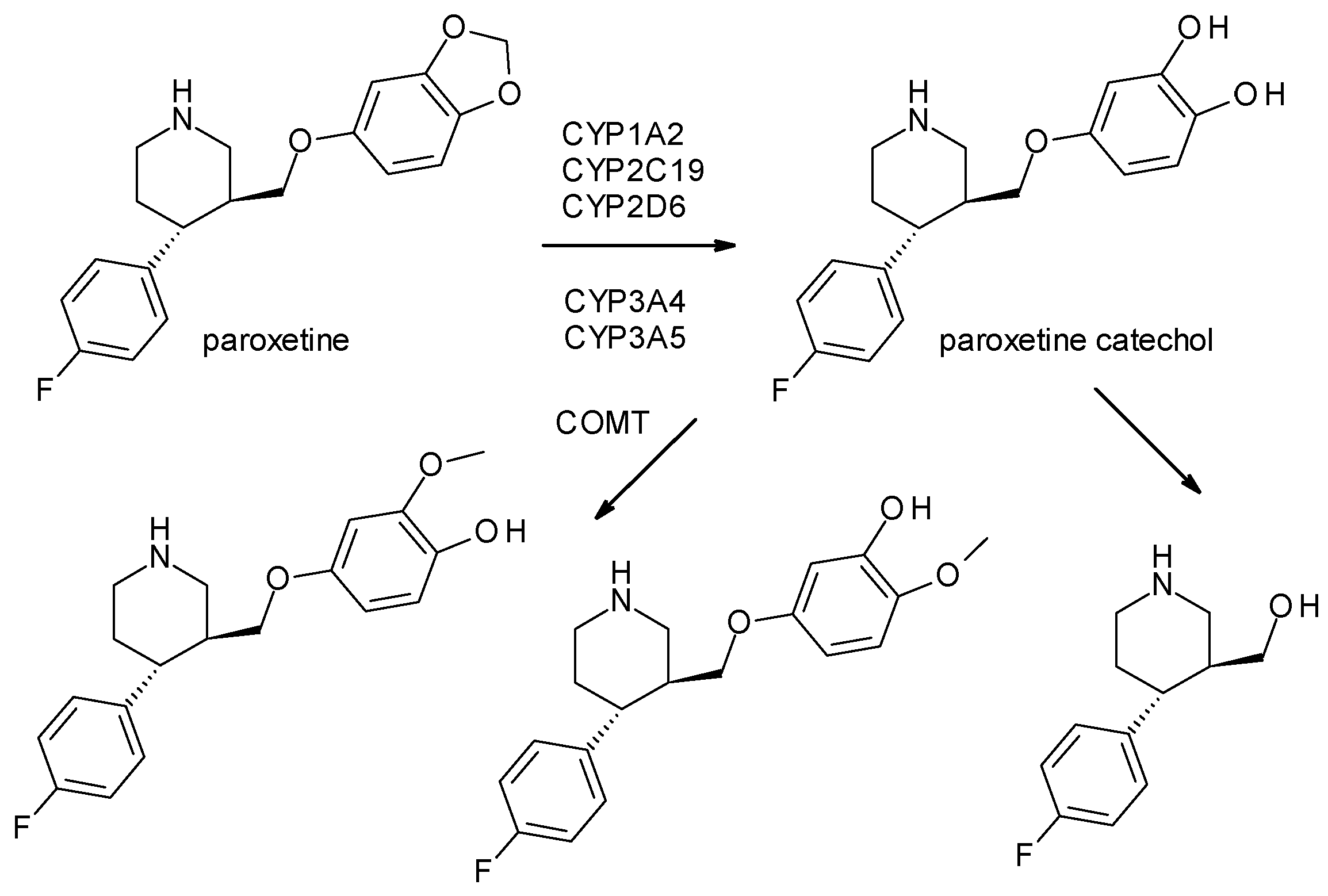
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A review. CNS Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef]
- Tang, S.W.; Helmeste, D. Paroxetine. Expert Opin. Pharmacother. 2008, 9, 787–794. [Google Scholar] [CrossRef]
- Kaye, C.M.; Haddock, R.E.; Langley, P.F.; Mellows, G.; Tasker, T.C.G.; Zussman, B.D.; Greb, W.H. A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr. Scand. 1989, 80, 60–75. [Google Scholar] [CrossRef]
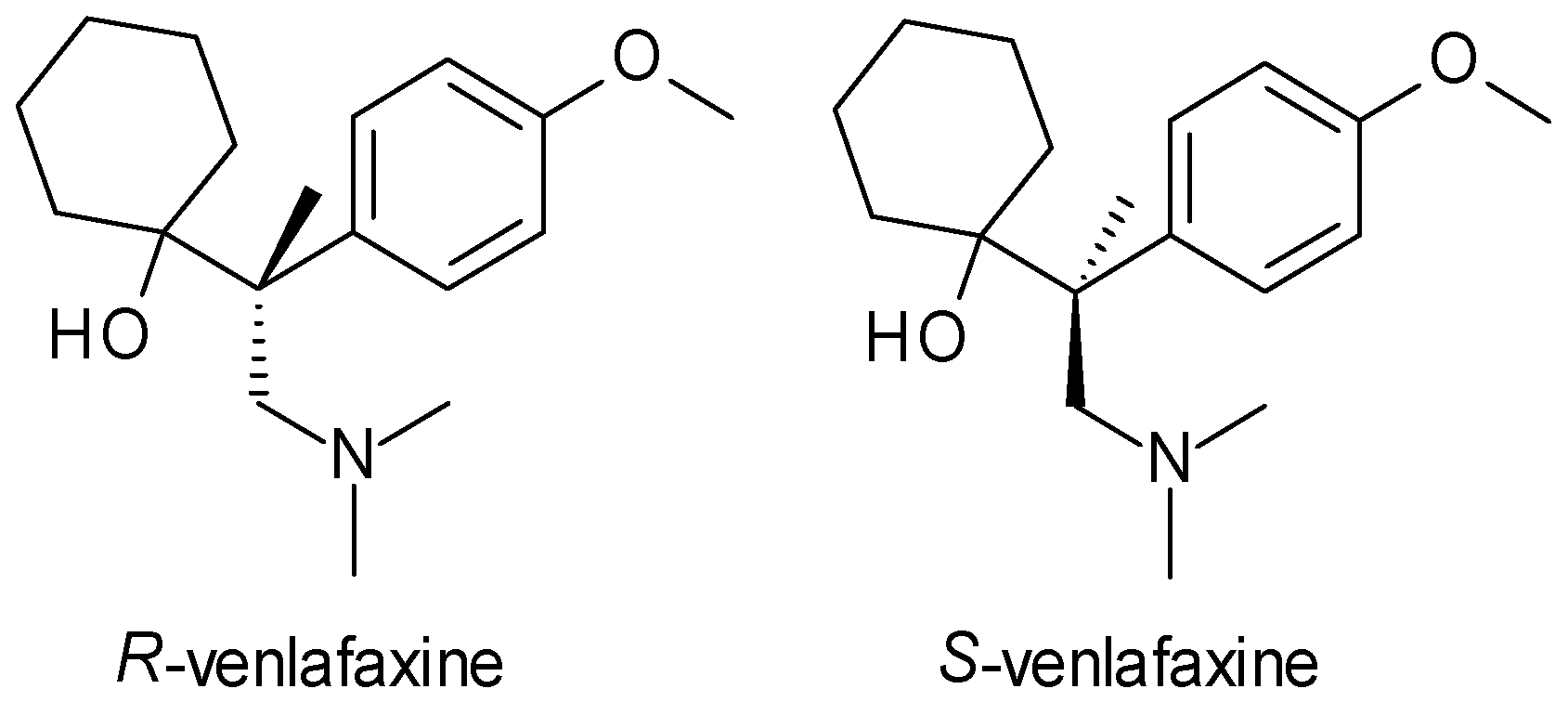
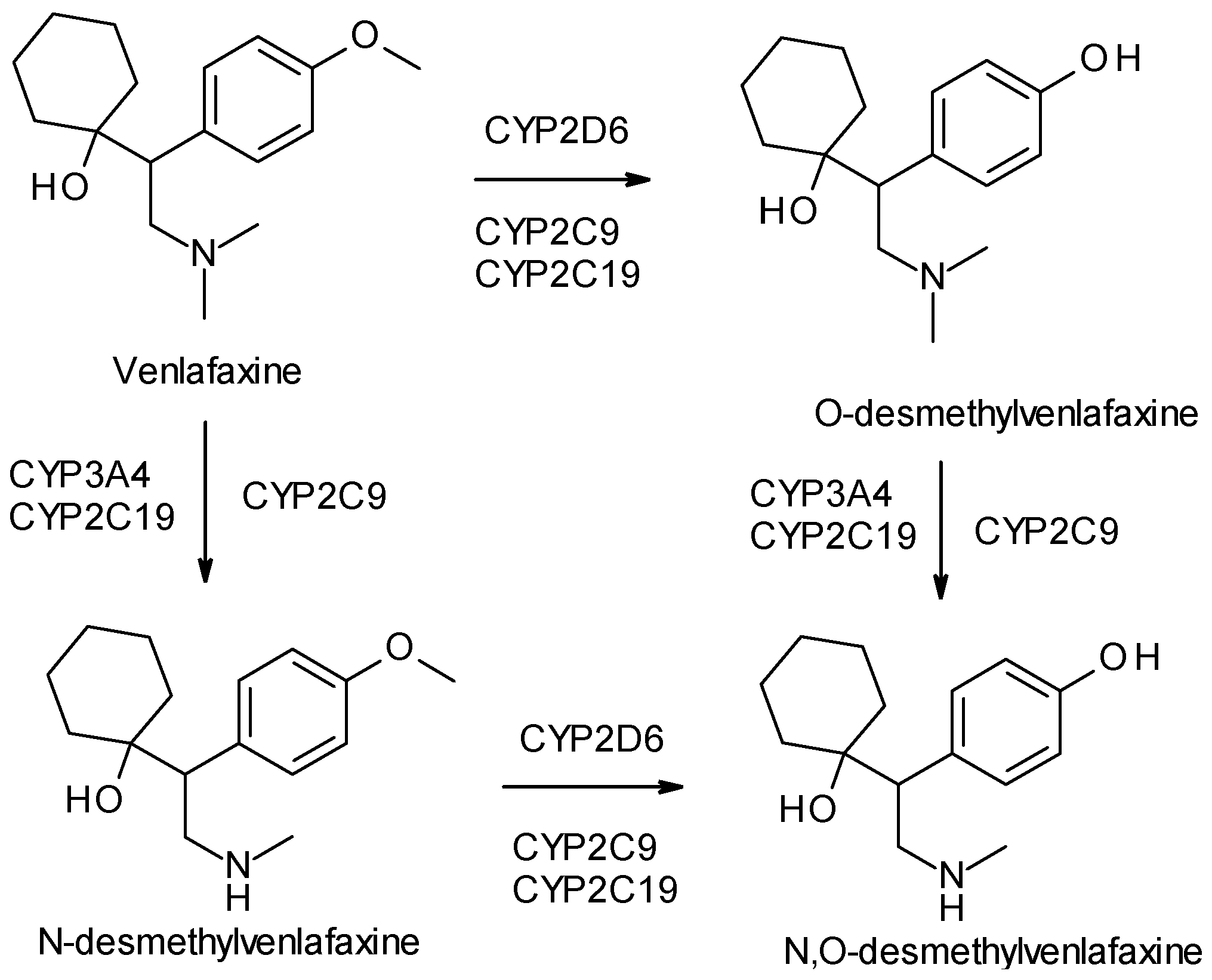
- Gutierrez, M.A.; Stimmel, G.L.; Aiso, J.Y. Venlafaxine: A 2003 update. Clin. Ther. 2003, 25, 2138–2154. [Google Scholar] [CrossRef]
- Roseboom, P.H.; Kalin, N.H. Neuropharmacology of venlafaxine. Depress. Anxiety 2000, 12, 20–29. [Google Scholar] [CrossRef]
- Harvey, A.T.; Rudolph, R.L.; Preskorn, S.H. Evidence of the dual mechanisms of action of venlafaxine. Arch. Gen. Psychiatry 2000, 57, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Ereshefsky, L.; Dugan, D. Review of the pharmacokinetics, pharmacogenetics, and drug interaction potential of antidepressants: Focus on venlafaxine. Depress. Anxiety 2000, 12, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, P.; Alves, G.; Llerena, A.; Falcão, A. Venlafaxine pharmacokinetics focused on drug metabolism and potential biomarkers. Drug Metab. Drug Interact. 2014, 29, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Deecher, D.C.; Beyer, C.E.; Johnston, G.; Bray, J.; Shah, S.; Abou-Gharbia, M.; Andree, T.H. Desvenlafaxine succinate: A new serotonin and norepinephrine reuptake inhibitor. J. Pharmacol. Exp. Ther. 2006, 318, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B.; Schatzberg, A.F.; Goldstein, D.J.; Detke, M.J.; Mallinckrodt, C.; Lu, Y.; Tran, P.V. Duloxetine for the treatment of major depressive disorder. Psychopharmacol. Bull. 2002, 36, 106–132. [Google Scholar]
- Rodrigues-Amorim, D.; Olivares, J.M.; Spuch, C.; Rivera-Baltanás, T. A systematic review of efficacy, safety, and tolerability of duloxetine. Front. Psychiatry 2020, 11, 554899. [Google Scholar] [CrossRef]
- Knadler, M.P.; Lobo, E.; Chappell, J.; Bergstrom, R. Duloxetine: Clinical pharmacokinetics and drug interactions. Clin. Pharmacokinet. 2011, 50, 281–294. [Google Scholar] [CrossRef]
- Pae, C.U.; Marks, D.M.; Shah, M.; Han, C.; Ham, B.J.; Patkar, A.A.; Masand, P.S. Milnacipran: Beyond a role of antidepressant. Clin. Neuropharmacol. 2009, 32, 355–363. [Google Scholar] [CrossRef]
- Kasper, S.; Pail, G. Milnacipran: A unique antidepressant? Neuropsychiatr. Dis. Treat. 2010, 6 (Suppl. S1), 23–31. [Google Scholar]
- Lopez-Ibor, J.; Guelfi, J.D.; Pletan, Y.; Tournoux, A.; Prost, J.F. Milnacipran and selective serotonin reuptake inhibitors in major depression. Int. Clin. Psychopharmacol. 1996, 11, 41–46. [Google Scholar] [CrossRef]
- Puozzo, C.; Panconi, E.; Deprez, D. Pharmacology and pharmacokinetics of milnacipran. Int. Clin. Psychopharmacol. 2002, 17 (Suppl. S1), S25–S35. [Google Scholar] [CrossRef] [PubMed]
- Hindmarch, I.; Rigney, U.; Stanley, N.; Briley, M. Pharmacodynamics of milnacipran in young and elderly volunteers. Br. J. Clin. Pharmacol. 2000, 49, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Hajós, M.; Fleishaker, J.C.; Filipiak-Reisner, J.K.; Brown, M.T.; Wong, E.H. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: Pharmacological and clinical profile. CNS Drug Rev. 2004, 10, 23–44. [Google Scholar] [CrossRef]
- Schatzberg, A.F. Clinical efficacy of reboxetine in major depression. J. Clin. Psychiatry 2000, 61 (Suppl. S10), 31–38. [Google Scholar]
- Fleishaker, J.C. Clinical pharmacokinetics of reboxetine, a selective norepinephrine reuptake inhibitor for the treatment of patients with depression. Clin. Pharmacokinet. 2000, 39, 413–427. [Google Scholar] [CrossRef]
- Foley, K.F.; DeSanty, K.P.; Kast, R.E. Bupropion: Pharmacology and therapeutic applications. Expert Rev. Neurother. 2006, 6, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.; Yang, L.P.; Curran, M.P. Bupropion: A review of its use in the management of major depressive disorder. Drugs 2008, 68, 653–689. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, J.W.; Pradko, J.F.; Muir, K.T. Bupropion for major depressive disorder: Pharmacokinetic and formulation considerations. Clin. Ther. 2005, 27, 1685–1695. [Google Scholar] [CrossRef]
- Montgomery, S.A.; Bullock, T.; Pinder, R.M. The clinical profile of mianserin. Nord. Psykiatr. Tidsskr. 1991, 45 (Suppl. S24), 27–35. [Google Scholar] [CrossRef]
- Pinder, R.M. Mianserin: Pharmacological and clinical correlates. Nord. Psykiatr. Tidsskr. 1991, 45 (Suppl. S24), 13–26. [Google Scholar] [CrossRef]
- Gorman, J.M. Mirtazapine: Clinical overview. J. Clin. Psychiatry 1999, 60, 9–13. [Google Scholar] [PubMed]
- Molero, P.; Ramos-Quiroga, J.A.; Martin-Santos, R.; Calvo-Sánchez, E.; Gutiérrez-Rojas, L.; Meana, J.J. Antidepressant efficacy and tolerability of ketamine and esketamine: A critical review. CNS Drugs 2018, 32, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Nikayin, S.; Murphy, E.; Krystal, J.H.; Wilkinson, S.T. Long-term safety of ketamine and esketamine in treatment of depression. Expert Opin. Drug Saf. 2022, 21, 777–787. [Google Scholar] [CrossRef]
- Bahji, A.; Vazquez, G.H.; Zarate, C.A., Jr. Comparative efficacy of racemic ketamine and esketamine for depression: A systematic review and meta-analysis. J. Affect. Disord. 2021, 278, 542–555. [Google Scholar] [CrossRef]
- Bozymski, K.M.; Crouse, E.L.; Titus-Lay, E.N.; Ott, C.A.; Nofziger, J.L.; Kirkwood, C.K. Esketamine: A novel option for treatment-resistant depression. Ann. Pharmacother. 2020, 54, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Anttila, S.A.; Leinonen, E.V. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001, 7, 249–264. [Google Scholar] [CrossRef]
- Baker, G.B.; Prior, T.I. Stereochemistry and drug efficacy and development: Relevance of chirality to antidepressant and antipsychotic drugs. Ann. Med. 2002, 34, 537–543. [Google Scholar] [CrossRef]
- DeVane, C.L.; Boulton, D.W. Great expectations in stereochemistry: Focus on antidepressants. CNS Spectr. 2002, 7, 28–33. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Class | Chiral Centers | Form Used in Therapy | Differences in Pharmacological Activity of Enantiomers |
|---|---|---|---|---|
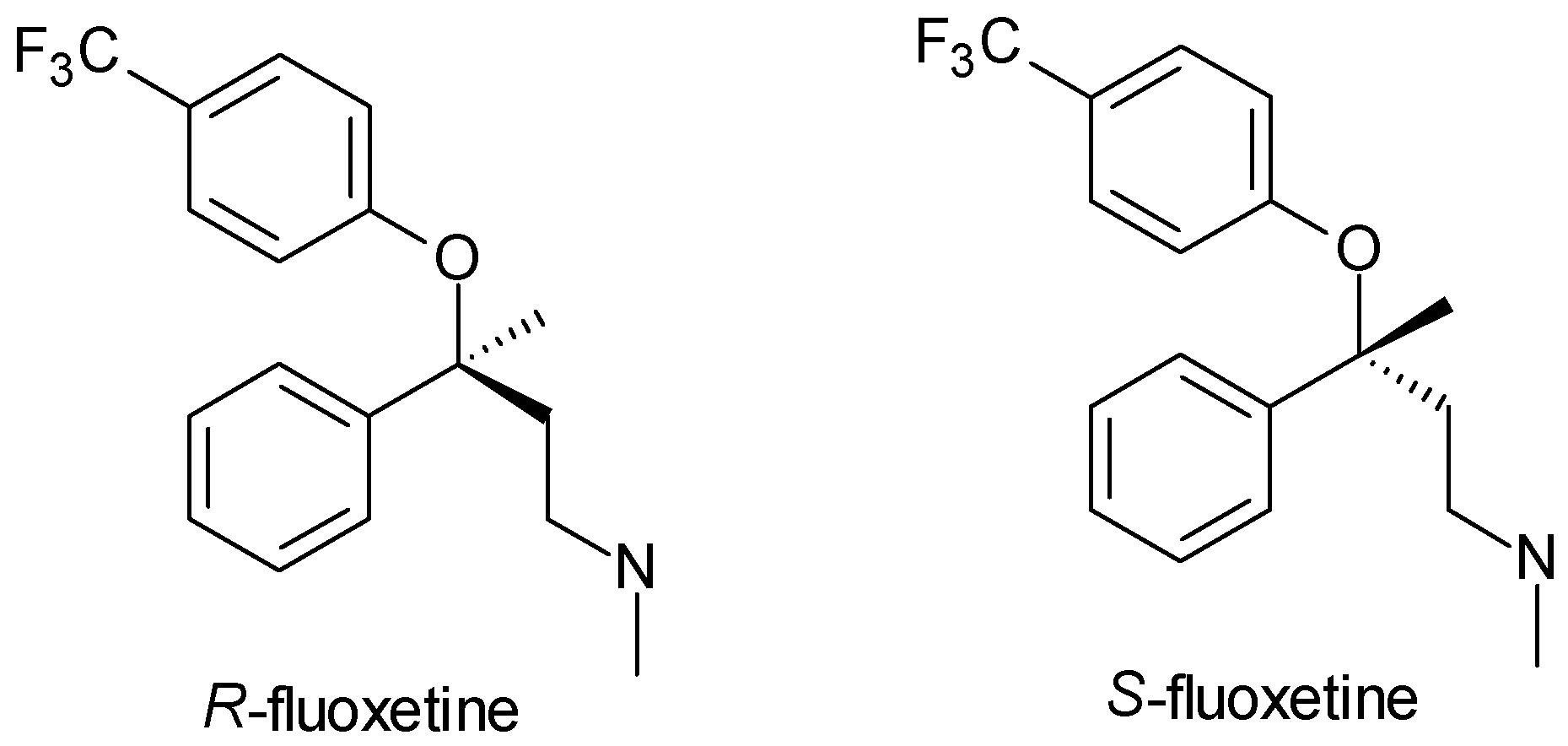
| Fluoxetine | SSRI | 1 | Racemic mixture (R, S-fluoxetine) | Both enantiomers inhibit serotonin reuptake; S-fluoxetine has a longer half-life and higher potency. |
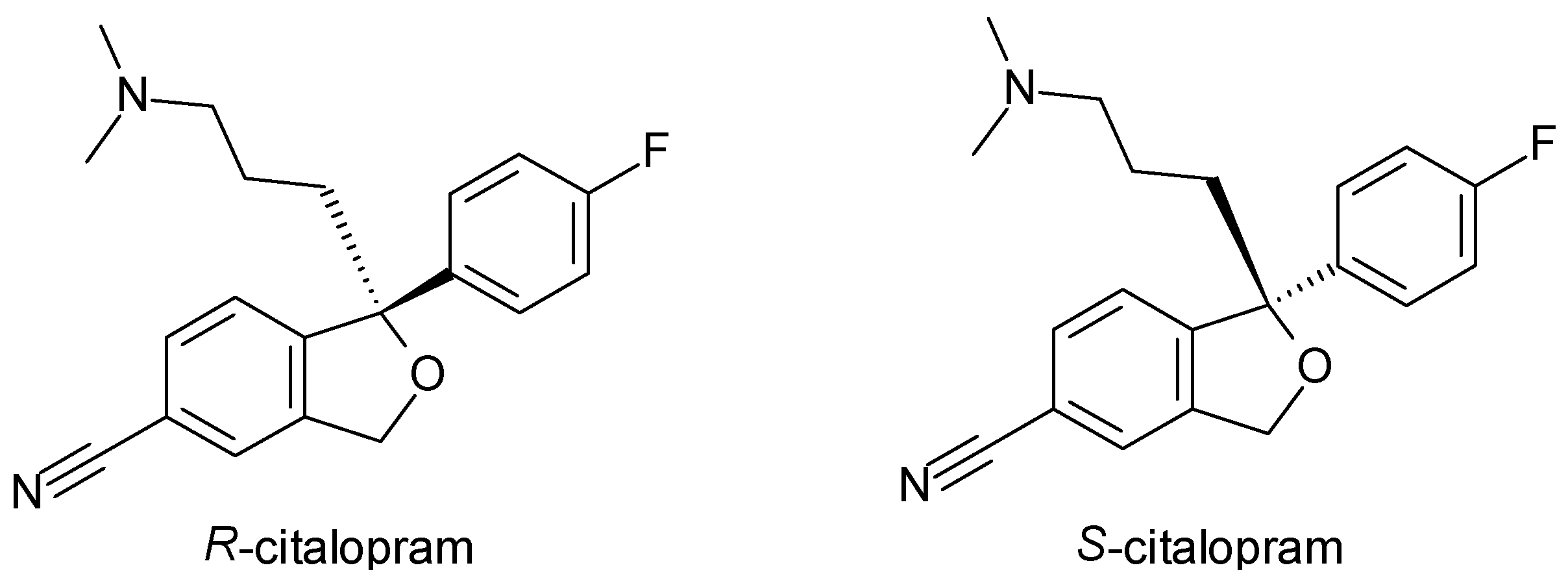
| Citalopram | SSRI | 1 | Racemic mixture (R, S-citalopram), pure enantiomer (S-citalopram) | S-citalopram is 30 times more potent at inhibiting serotonin reuptake than R-citalopram. |
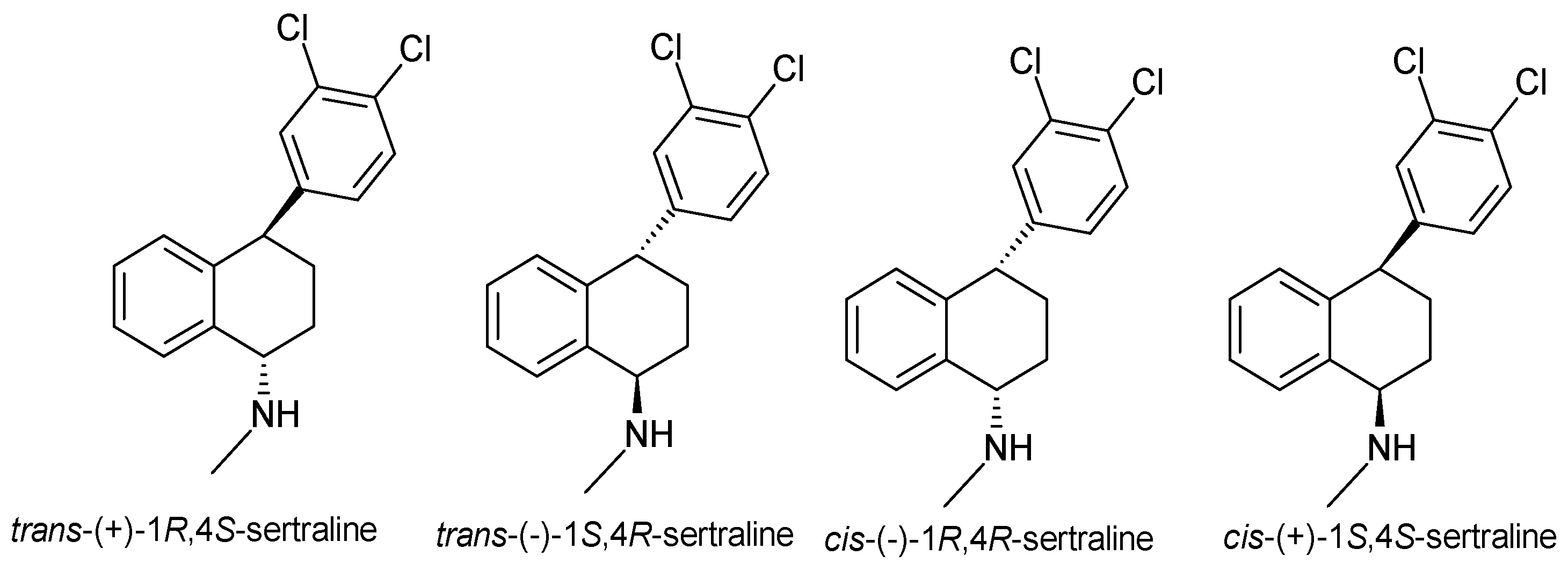
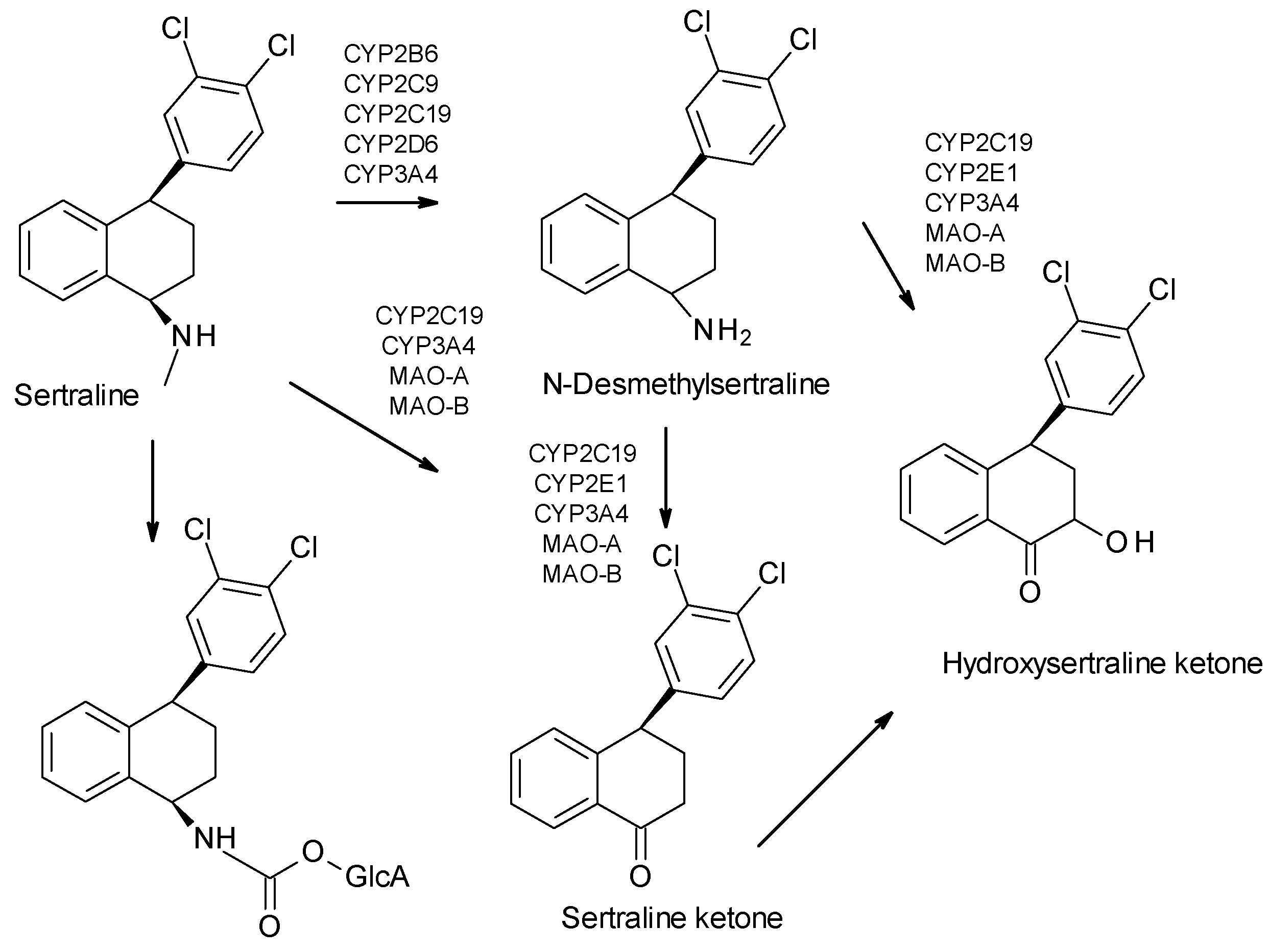
| Sertraline | SSRI | 2 | Pure enantiomer (cis-1S, 4S-sertraline) | cis-1S, 4S-sertraline is the most selective at inhibiting serotonin uptake. |
| Paroxetine | SSRI | 2 | Pure enantiomer (trans-3S, 4R-paroxetine) | trans-3S, 4R-paroxetine is the most selective for serotonin transporters. |
| Venlafaxine | SNRI | 1 | Racemic mixture (R, S-venlafaxine) | R-venlafaxine inhibits both serotonin and norepinephrine reuptake, while S-venlafaxine is more selective for serotonin. |
| Duloxetine | SNRI | 1 | Pure enantiomer (S-duloxetine) | S-duloxetine is more potent as serotonin and norepinephrine reuptake inhibitor. |
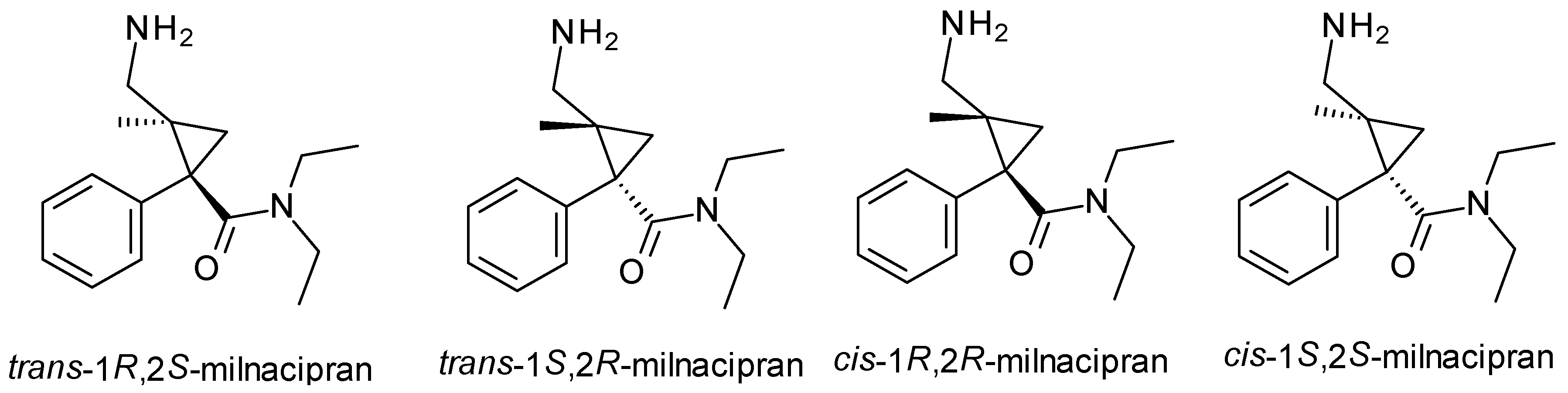
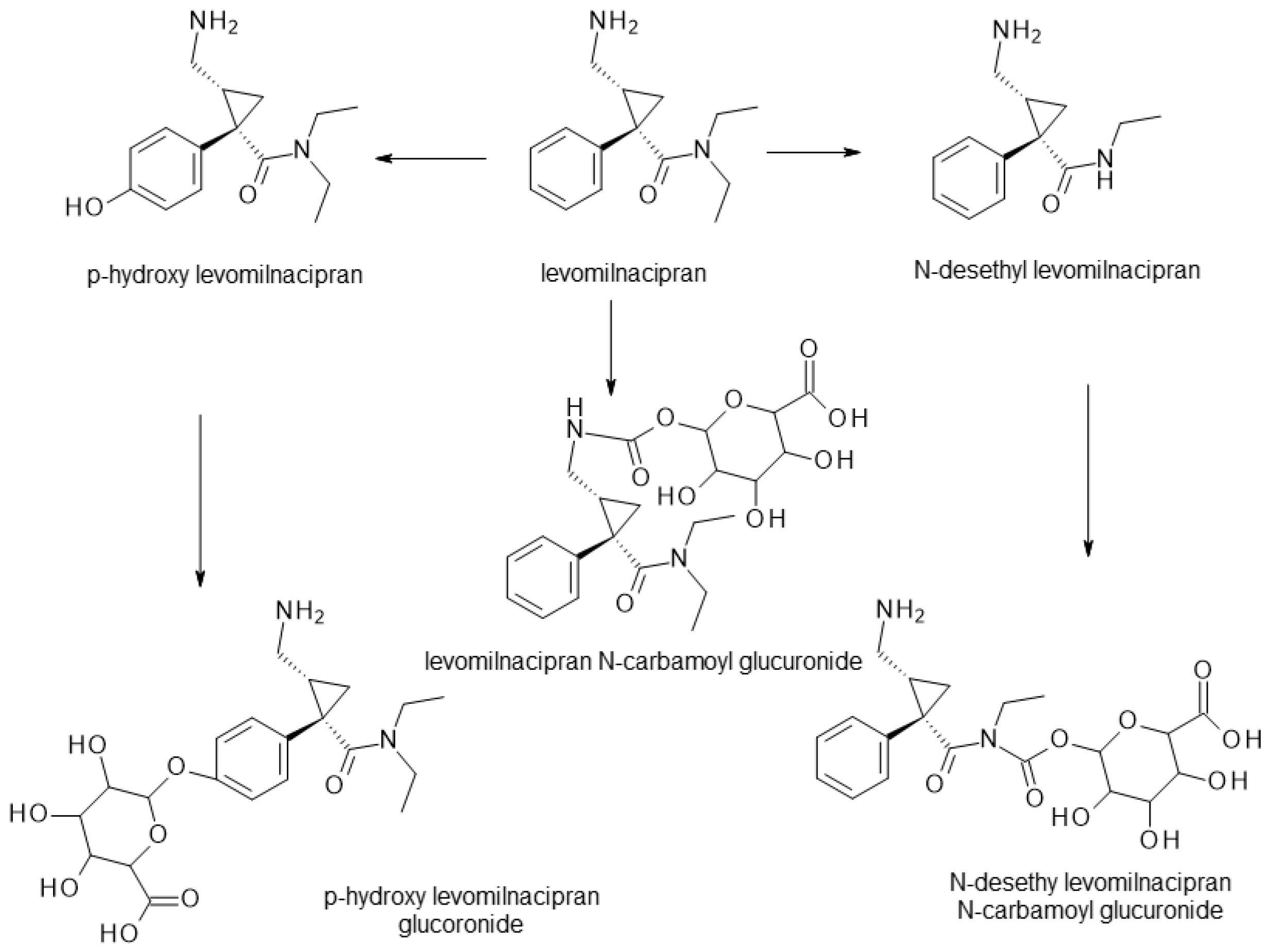
| Milnacipran | SNRI | 2 | Mixture (cis-1R, 2S-milnacipran, cis-1S, 2R-milnacipran), pure enantiomer (levomilancipran) | Levomilnacipran (1S, 2R) is more effective at inhibiting serotonin and norepinephrine reuptake. |
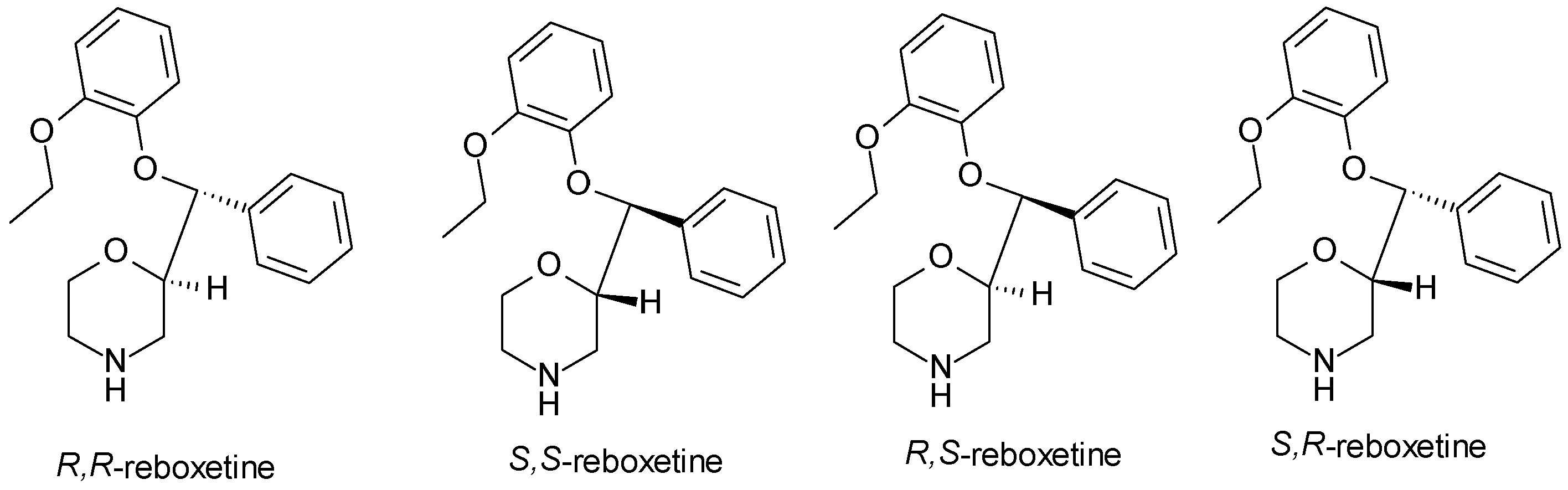
| Reboxetine | NRI | 2 | Mixture (R, R-reboxetine, S, S-reboxetine) | S, S-reboxetine is more effective at inhibiting norepinephrine reuptake |
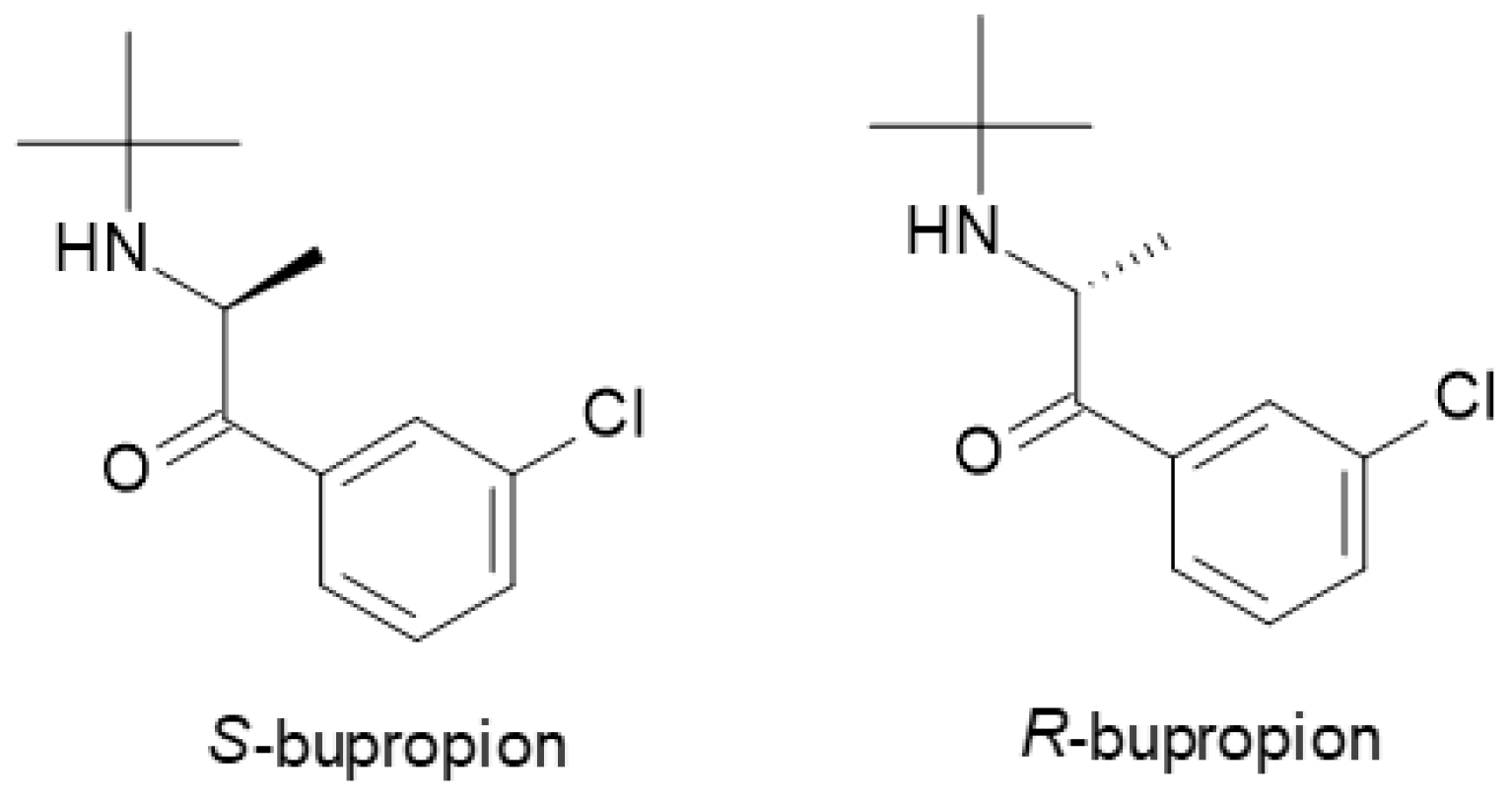
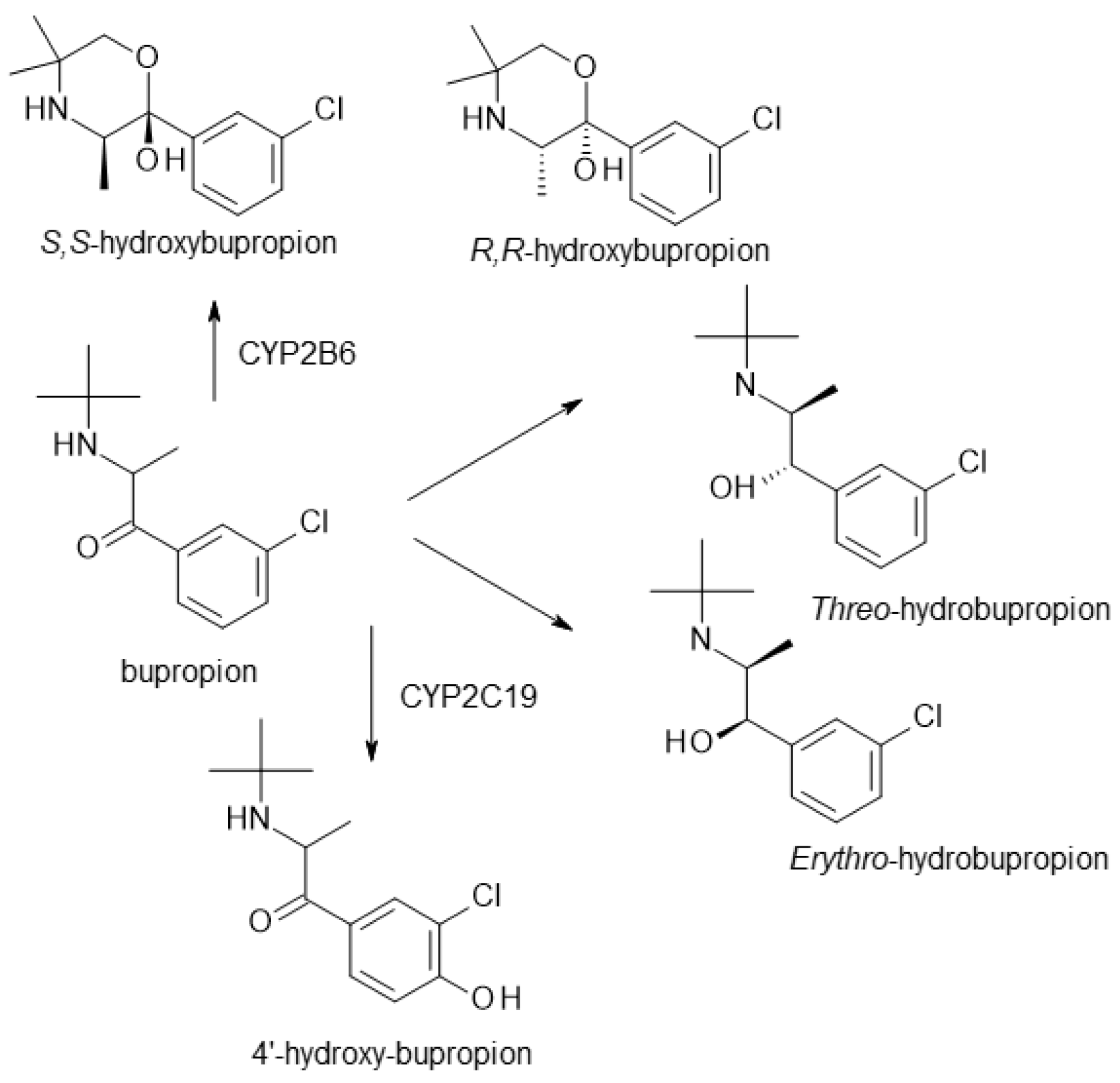
| Bupropion | NDRI | 1 | Racemic mixture (R-bupropion, S-bupropion) | R-bupropion is more potent in inhibiting norepinephrine and dopamine reuptake, longer half-life than S-bupropion. |
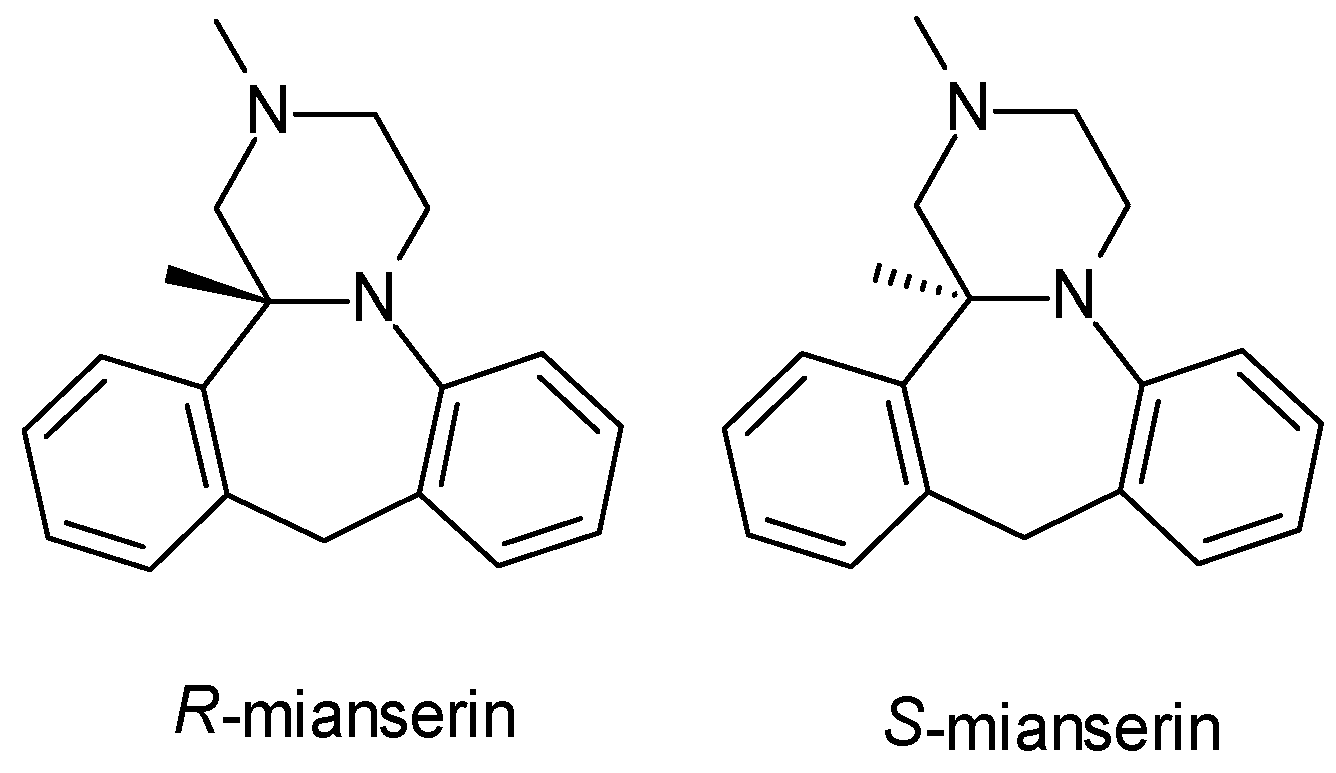
| Mianserin | TA | 1 | Racemic mixture (R-mianserin, S-mianserin) | S-mianserin is more potent in inhibiting norepinephrine reuptake; both enantiomers exhibit sedative effects through histamine and serotonin receptor antagonism. |
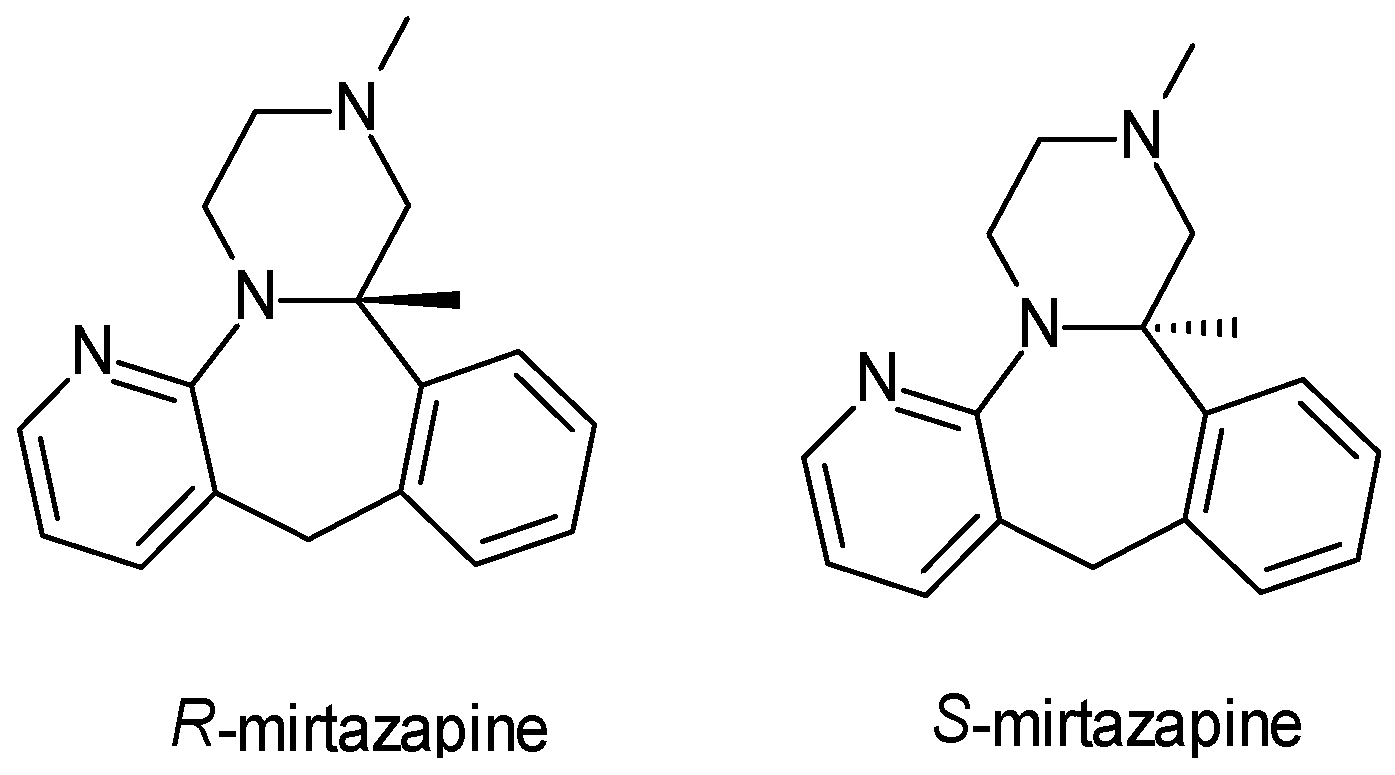
| Mirtazepin | TA | 1 | Racemic mixture (R-mirtazepin, S-mirtazepin) | S-mirtazapine is more potent in α2-autoreceptor inhibition and antagonism of 5-HT2 receptors. |
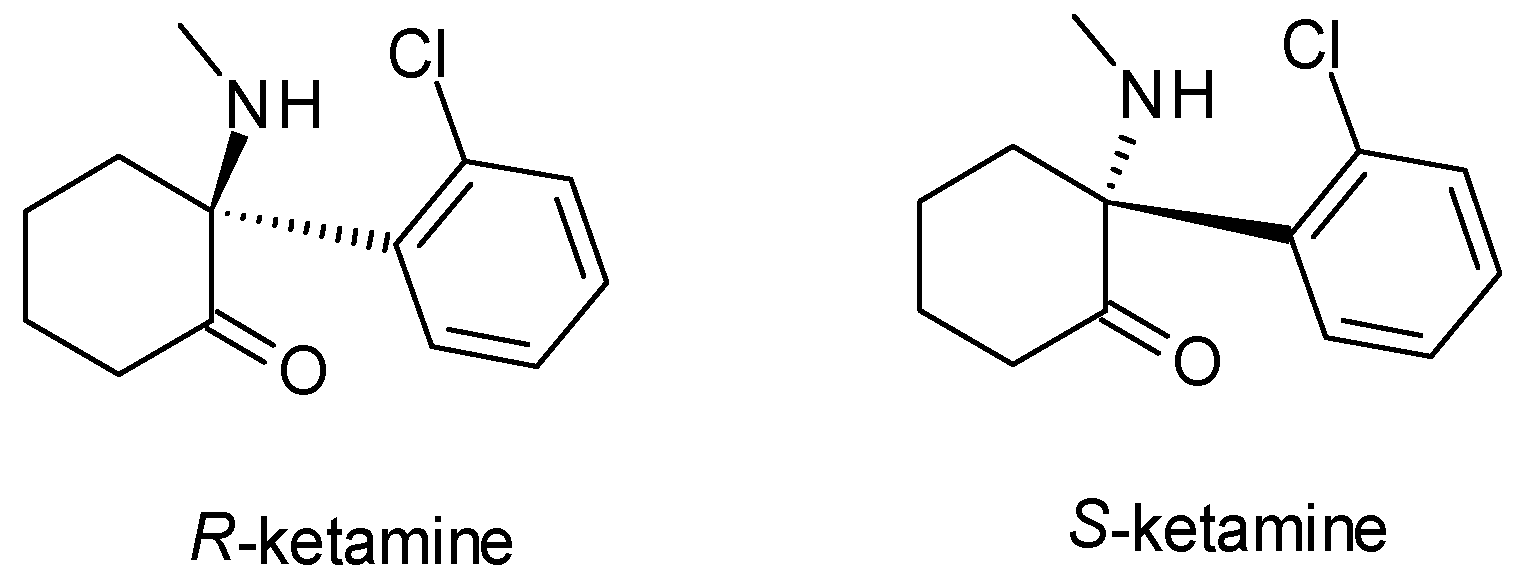
| Esketamine | Other antidepressants | 1 | Pure enantiomer (S-ketamine) | S-ketamine is more potent in binding to NMDA receptors, stronger antidepressant effects at lower doses compared to R-ketamine. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hancu, G.; Uilăcan, A.; Blebea, N.M. Chirality in Modern Antidepressants: A Comprehensive Review of Stereochemical Impacts on Pharmacology and Therapeutics. Drugs Drug Candidates 2024, 3, 654-673. https://doi.org/10.3390/ddc3040037
Hancu G, Uilăcan A, Blebea NM. Chirality in Modern Antidepressants: A Comprehensive Review of Stereochemical Impacts on Pharmacology and Therapeutics. Drugs and Drug Candidates. 2024; 3(4):654-673. https://doi.org/10.3390/ddc3040037
Chicago/Turabian StyleHancu, Gabriel, Alexandra Uilăcan, and Nicoleta Mirela Blebea. 2024. "Chirality in Modern Antidepressants: A Comprehensive Review of Stereochemical Impacts on Pharmacology and Therapeutics" Drugs and Drug Candidates 3, no. 4: 654-673. https://doi.org/10.3390/ddc3040037
APA StyleHancu, G., Uilăcan, A., & Blebea, N. M. (2024). Chirality in Modern Antidepressants: A Comprehensive Review of Stereochemical Impacts on Pharmacology and Therapeutics. Drugs and Drug Candidates, 3(4), 654-673. https://doi.org/10.3390/ddc3040037