Abstract
Proteases play a pivotal role in cancer progression, facilitating processes such as extracellular matrix degradation, angiogenesis, and metastasis. Consequently, protease inhibitors have emerged as promising therapeutic agents in oncology. This review provides a comprehensive overview of the mechanisms by which protease inhibitors modulate cancer biology, categorizing inhibitors by their target protease classes, including matrix metalloproteinases, cysteine proteases, and serine proteases. We discuss the therapeutic potential of both synthetic and natural protease inhibitors, highlighting their applications in preclinical and clinical settings. Furthermore, challenges such as specificity, toxicity, and resistance mechanisms are addressed, alongside strategies to overcome these limitations through innovative drug designs and combination therapies. The future of protease inhibitors in cancer treatment lies in precision medicine, leveraging proteomic profiling to tailor therapies to individual tumors. This review underscores the importance of ongoing research and the development of novel approaches to harness protease inhibitors effectively for cancer management.
1. Introduction
Cancer is a multifaceted disease characterized by uncontrolled cell proliferation, the evasion of apoptosis, sustained angiogenesis, and the ability to invade surrounding tissues and metastasize to distant organs [1]. These hallmarks of cancer are supported by intricate biochemical and molecular pathways, many of which involve proteases—enzymes that catalyze the breakdown of proteins by hydrolyzing peptide bonds [2]. Initially recognized for their role in protein turnover and extracellular matrix remodeling, proteases are now understood to be key regulators of several processes integral to cancer progression, including tumor invasion, angiogenesis, immune modulation, and the activation of pro-tumorigenic signaling pathways [3].
Proteases are classified into distinct families based on their catalytic mechanisms (Figure 1), including serine proteases, cysteine proteases, aspartic proteases, and metalloproteases [2,4]. These enzymes are often dysregulated in cancer, with their overexpression or aberrant activity contributing to tumor growth and metastasis [5]. For instance, matrix metalloproteinases (MMPs) degrade components of the extracellular matrix, creating a permissive environment for cancer cells to invade and metastasize [6]. Similarly, cysteine proteases such as cathepsins facilitate tumor invasion and modulate immune responses. These observations underscore the pivotal role of proteases in the tumor microenvironment and their potential as therapeutic targets.
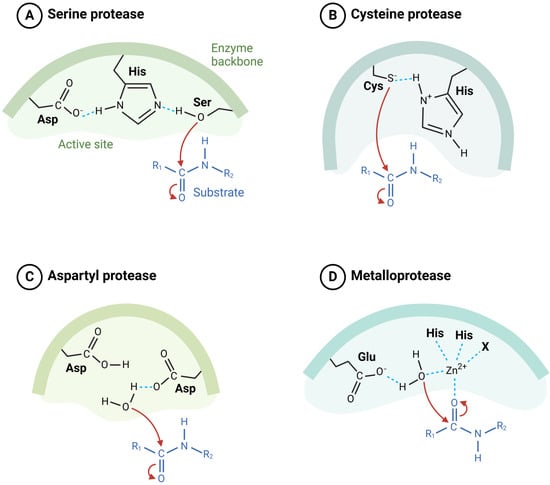
Figure 1.
Classification of proteases based on their catalytic mechanisms. (A) Serine proteases utilize a catalytic triad consisting of Asp, His, and Ser to hydrolyze peptide bonds. (B) Cysteine proteases employ a catalytic dyad of Cys and His for the cleavage of substrate. (C) Aspartyl proteases use two Asp residues in the active site to mediate catalysis. (D) Metalloproteases require a metal ion, typically Zn2⁺, coordinated by His and Glu residues to facilitate peptide bond hydrolysis (illustration created with BioRender.com) (https://app.biorender.com accessed on 2 March 2025).
Protease inhibitors, a diverse group of molecules that block protease activity, have garnered significant interest as anticancer agents; they can be classified based on their structure, origin, or mode of action and include small-molecule inhibitors, monoclonal antibodies, and endogenous protease inhibitors [7]. The therapeutic promise of protease inhibitors lies in their ability to selectively target pathological protease activity, thereby impeding processes such as tumor invasion, angiogenesis, and metastasis [8]. However, the clinical development of protease inhibitors has faced significant challenges, including off-target effects, toxicity, and the development of resistance mechanisms by cancer cells [9].
Protease inhibitors, once heralded as transformative cancer therapies, have faced considerable setbacks in clinical development due to challenges in balancing efficacy and safety [10]. MMP inhibitors exemplify these challenges. Designed to target MMPs critical for tumor invasion and metastasis, these inhibitors showed promise in preclinical studies; however, clinical trials revealed limited therapeutic efficacy in many cancers, as tumors adapted by activating compensatory pathways or utilizing non-MMP proteases for progression [11]. Furthermore, the broad-spectrum nature of many early MMP inhibitors led to significant off-target effects, including musculoskeletal toxicity, which manifested as joint stiffness and pain in patients, and this toxicity often required dose reductions, compromising the inhibitors’ therapeutic potential and ultimately resulting in the discontinuation of several candidates [12].
Another factor contributing to the failure of protease inhibitors in cancer therapy is the complexity of protease function in tumor biology. Proteases such as MMPs, urokinase plasminogen activators (uPAs), and cathepsins have dual roles in promoting and suppressing tumors, depending on the tumor microenvironment, and this functional duality made it challenging to predict patient responses and stratify those who might benefit from protease-targeted treatments [13]. In addition, early protease inhibitors were developed without a precise understanding of the protease profiles unique to different cancers, leading to suboptimal outcomes [14]. Advances in precision medicine and biomarker-driven approaches are now guiding the development of next-generation protease inhibitors, aiming to overcome the limitations that plagued earlier efforts [15]. Nonetheless, the history of protease inhibitors underscores the importance of specificity, biomarker identification, and a deeper understanding of protease networks in successful cancer therapy development.
This review provides a detailed examination of the role of proteases in cancer biology and the therapeutic potential of protease inhibitors. It explores the molecular mechanisms underlying protease activity in cancer, the design and classification of protease inhibitors, and their current status in clinical and preclinical research. Furthermore, this review addresses the limitations associated with protease inhibitors, including issues of selectivity and toxicity, and highlights emerging strategies to overcome these obstacles, such as the use of advanced drug delivery systems and combination therapies. Finally, the potential for precision medicine to tailor protease inhibitor therapies to individual tumor profiles is discussed, offering a roadmap for future advancements in this field.
2. Proteases in Cancer Biology
Proteases, also known as proteinases or peptidases, are enzymes responsible for the hydrolysis of peptide bonds in proteins, playing essential roles in a variety of physiological and pathological processes [16]. In cancer, these enzymes are key regulators of tumor development and progression, contributing to processes such as extracellular matrix (ECM) remodeling, angiogenesis, immune modulation, apoptosis evasion, and metastasis [5]. Dysregulated protease activity is a hallmark of many cancers, with elevated protease expression or activity often correlating with aggressive tumor behavior and poor prognosis [17].
Proteases are classified into major families based on their catalytic mechanisms and active site residues. Serine proteases, which utilize a serine residue for catalysis, are involved in tissue remodeling, the activation of growth factors, and ECM degradation [18]. Examples include urokinase plasminogen activator (uPA) [19] and kallikreins [20], both of which have well-established roles in cancer progression. Cysteine proteases, characterized by a cysteine residue at their active site, include enzymes such as cathepsins which are implicated in tumor invasion, angiogenesis, and immune modulation, with cathepsins B and L frequently overexpressed in various malignancies [21]. Metalloproteases, which depend on a metal ion such as zinc for activity, represent another key protease family in cancer which includes matrix metalloproteinases (MMPs), the most studied metalloproteases, and are central to ECM degradation and metastatic dissemination [22]. Finally, aspartic proteases, which use aspartic acid residues in their catalytic site, include cathepsin D, an enzyme involved in tumor invasion and metastasis [23].
The functional roles of proteases in cancer biology are diverse and integral to tumor progression. Proteases such as MMPs are critical for ECM degradation, allowing cancer cells to invade surrounding tissues and migrate to distant sites [24]. In addition to facilitating invasion, proteases promote angiogenesis by releasing and activating pro-angiogenic factors, including vascular endothelial growth factor (VEGF) [25]. Furthermore, proteases contribute to apoptosis evasion by degrading apoptotic mediators or activating survival pathways, enabling cancer cells to resist programmed cell death [26]. Proteases also play a pivotal role in metastasis, aiding in the detachment of tumor cells from the primary tumor, intravasation into blood vessels, and colonization of distant organs [27]. Additionally, proteases modulate the immune response, often suppressing immune surveillance by cleaving immune regulatory molecules, thus enhancing tumor immune evasion [28].
Proteases play a critical role in modulating apoptotic pathways by interacting with key regulators such as Bcl-2, p53, and caspases [26]. For instance, MMP-7 has been implicated in the cleavage of Fas ligands, thereby inhibiting Fas-mediated apoptosis in cancer cells [29]. Similarly, cathepsin B can degrade pro-apoptotic proteins, enhancing tumor cell survival under stress conditions [30]. The intricate relationship between proteases and apoptotic regulators suggests that combining protease inhibitors with apoptosis-inducing agents, such as Bcl-2 antagonists, may enhance therapeutic efficacy in resistant cancers [31].
Several proteases are particularly noteworthy for their roles in cancer. MMPs, such as MMP-2 and MMP-9, degrade type IV collagen in the basement membrane, a critical step in tumor invasion and metastasis, and the overexpression of these proteases is associated with advanced stages of cancer and poor clinical outcomes [32]. Cathepsins, another prominent group, are involved in ECM remodeling and the activation of pro-angiogenic factors and cathepsin B and cathepsin L are frequently upregulated in aggressive cancers [33,34], while cathepsin D, an aspartic protease, is implicated in enhanced metastatic potential, particularly in breast cancer [35]. The uPA, a serine protease, converts plasminogen to plasmin, a protease that degrades fibrin and ECM components, and is closely linked to cancer cell invasion and metastasis [36]. The overactivation of these and other proteases underscores their critical role in cancer biology and highlights their potential as therapeutic targets.
The tumor microenvironment (TME) plays a critical role in determining whether proteases act as tumor-promoting or tumor-suppressing factors [37]. In hypoxic conditions, certain proteases such as MMPs are upregulated to facilitate angiogenesis and invasion [38]. Conversely, in a more immune-infiltrated microenvironment, proteases like MMP-12 may enhance anti-tumor immunity by promoting macrophage-mediated responses [39]. Additionally, cancer-associated fibroblasts can alter extracellular pH, influencing protease activity and shifting their role toward either matrix degradation or immune suppression [40]. Understanding these context-dependent functions is crucial for optimizing the therapeutic application of protease inhibitors
In summary, proteases are central players in the molecular and cellular mechanisms that drive cancer progression. Their diverse functional roles and frequent dysregulation in tumors make them attractive targets for therapeutic intervention. Understanding the complex biology of proteases provides the foundation for developing inhibitors that can mitigate their pro-tumorigenic effects and improve clinical outcomes for cancer patients.
3. Protease Inhibitors Classification and Mechanisms
Protease inhibitors represent a diverse group of molecules designed to modulate the activity of proteases, which are often dysregulated in cancer [41]. These inhibitors are categorized based on their origin, molecular structure, and mode of action. Synthetic inhibitors are chemically designed molecules that are highly specific to their target proteases; for instance, batimastat and marimastat are synthetic inhibitors of MMPs, developed to block the degradation of the extracellular matrix and prevent tumor invasion and metastasis [42]. Natural inhibitors, on the other hand, are derived from biological sources such as endogenous proteins or microbial products and include engineered tissue inhibitors of metalloproteinases (TIMPs) [43], which regulate MMP activity under normal physiological conditions, and compounds such as epoxomicin, a natural cysteine protease inhibitor [44]. Monoclonal antibodies have also emerged as a powerful class of inhibitors, providing specificity and versatility in targeting proteases, where they can bind to proteases with high affinity, and neutralizing their activity; for example, monoclonal antibodies targeting uPA [45] or cathepsin proteins [46] have shown promise in preclinical studies. Small-molecule inhibitors, which are low-molecular-weight compounds, are particularly attractive due to their ability to penetrate tissues and cells effectively. Examples include inhibitors of cysteine proteases such as cathepsins [47] and compounds targeting serine proteases like uPA [48].
Protease inhibitors function through several distinct mechanisms, depending on how they interact with the enzyme’s active or regulatory sites. The three primary reversible inhibition mechanisms include competitive, non-competitive, and uncompetitive inhibition, while some inhibitors act irreversibly by covalently modifying the enzyme. The mechanisms of action of protease inhibitors depend on their interaction with the enzyme’s active or regulatory sites. Competitive inhibition, one of the most common mechanisms, involves the inhibitor binding to the protease’s active site, thereby preventing substrate access and enzymatic activity [49]. This mechanism is exemplified by batimastat and marimastat, which mimic the substrate structure to occupy the active site of MMPs [42]. Non-competitive inhibitors bind to an allosteric site (a region distinct from the active site), inducing conformational changes in the enzyme that reduce its catalytic efficiency. Unlike competitive inhibitors, non-competitive inhibitors do not compete with the substrate, meaning they can inhibit enzyme activity regardless of substrate concentration. This mechanism provides an advantage in maintaining inhibition even when substrate levels fluctuate. An example is the inhibition of certain serine proteases through allosteric modulation, reducing their proteolytic activity [50]. Additionally, irreversible inhibitors form covalent bonds with the enzyme, permanently modifying its active site or regulatory regions. These inhibitors, such as CA-074 as an irreversible inhibitor of cathepsin B, lead to sustained enzyme inactivation, requiring new enzyme synthesis to restore function. This mechanism is advantageous in long-term therapeutic applications, such as cancer treatment. These inhibition strategies highlight the diverse mechanisms through which protease inhibitors regulate enzymatic activity, and a detailed understanding of these mechanisms is essential for designing selective and potent inhibitors for therapeutic applications [51].
Several protease inhibitors have been explored in research and clinical trials for cancer therapy. Batimastat and marimastat, as early MMP inhibitors, demonstrated efficacy in reducing tumor invasion and metastasis in preclinical models but faced limitations in clinical applications due to their toxicity and lack of specificity [52]. Efforts to overcome these challenges have led to the development of more selective inhibitors, such as those targeting specific cysteine proteases like cathepsin B and L, which are critical in tumor invasion and metastasis [8]. Endogenous inhibitors such as TIMPs have also been investigated for therapeutic use, with strategies focusing on enhancing their selectivity and delivery [43]. Small-molecule inhibitors targeting serine proteases, including uPA inhibitors, have shown potential in modulating plasminogen activation pathways critical for cancer cell invasion [53]. Overall, protease inhibitors, with their diverse structures and mechanisms, remain a promising class of therapeutic agents in cancer research, although overcoming the challenges of specificity, toxicity, and resistance remains a key focus for their successful clinical translation.
4. Clinical Applications and Therapeutic Potential
Protease inhibitors have demonstrated significant potential in cancer therapy, with preclinical and clinical studies highlighting their effectiveness in various cancer types [54] (Table 1). Their role in mitigating tumor progression, invasion, and metastasis underscores their therapeutic importance. However, the translation of these findings into clinical practice has been met with both successes and challenges, necessitating further refinement and innovation [10]. Protease inhibitors have been evaluated across multiple cancer types, with varying degrees of success. In breast cancer, inhibitors of MMPs such as marimastat have shown promise in reducing metastatic spread by limiting ECM degradation [55]. Similarly, the uPA system, frequently dysregulated in multiple kinds of cancer, has been targeted with serine protease inhibitors to suppress invasion and migration [19]. In prostate cancer, protease inhibitors targeting cathepsins and MMPs have demonstrated the ability to reduce tumor growth and metastasis in preclinical models [56]. Cathepsin B and L inhibitors have been explored for their role in mitigating invasive potential in aggressive forms of prostate cancer and melanoma [57,58]. In colorectal cancer, MMP inhibitors have been tested for their ability to prevent angiogenesis and invasion [59]. Although the results have been promising in animal models, challenges with toxicity and efficacy have limited their broader application in human trials. Despite the challenges faced by broad-spectrum MMP inhibitors, certain protease inhibitors have successfully reached clinical use. Notably, proteasome inhibitors such as bortezomib (Velcade) and carfilzomib (Kyprolis) have been approved for the treatment of multiple myeloma, demonstrating significant survival benefits [60,61]. These inhibitors selectively target the 26S proteasome, leading to the accumulation of misfolded proteins and induction of apoptosis in rapidly dividing cancer cells [62]. Additionally, nelfinavir, originally an HIV protease inhibitor, has shown promise in combination therapies for pancreatic and cervical cancers by inhibiting stress response pathways [63]. The success of these inhibitors underscores the importance of target specificity and mechanism-based design in overcoming toxicity and resistance challenges in clinical settings.
Despite the mixed clinical success of protease inhibitors, several cancer types and molecular subtypes have shown promising responses to these agents. Multiple myeloma and hematologic malignancies have benefited from proteasome inhibitors such as bortezomib and carfilzomib, which selectively block the degradation of pro-apoptotic proteins, leading to tumor cell death [64]. In triple-negative breast cancer (TNBC), protease inhibitors targeting cathepsin B and uPA have demonstrated preclinical efficacy in reducing metastasis [65,66]. Pancreatic cancer has been another area of active research, with uPA inhibitors in combination with chemotherapy showing potential in reducing tumor invasion [67]. These findings suggest that protease inhibitors may be most effective in cancers where proteolytic remodeling is a dominant driver of disease progression.

Table 1.
Protease inhibitors clinical status in cancer research.
Table 1.
Protease inhibitors clinical status in cancer research.
| Name | Cancer Type | Clinical Trial Status (ClinicalTrials.gov ID) | Type of Protease Inhibited | Ref. |
|---|---|---|---|---|
| Bortezomib (Velcade) | Multiple Myeloma | FDA approved (NCT00006362) | Proteasome | [60,68] |
| Carfilzomib (Kyprolis) | Multiple Myeloma | FDA approved (NCT00461045) | Proteasome | [69] |
| Ixazomib (Ninlaro) | Multiple Myeloma | FDA approved (NCT00963820) | Proteasome | [70] |
| Andecaliximab (GS-5745) | Colorectal Carcinoma | Not FDA-Approved. (NCT01803282) | MMP-9 | [71] |
| CA-074 | Breast Cancer | Preclinical | Cathepsin B | [72] |
| CA-030 | Breast Cancer | Preclinical | Cathepsin B | [73] |
| CLIK-148 | Breast Cancer | Preclinical | Cathepsin L | [74,75] |
| CLIK-195 | Breast Cancer | Preclinical | Cathepsin L | [76] |
| L-235 | Breast Cancer Bone Metastasis | Preclinical | Cathepsin K | [77] |
| Fsn0503 (anti-CtsS antibody) | Colorectal Carcinoma | Preclinical | Cathepsin S | [78] |
| Marimastat | Various Cancers | Not FDA-Approved. (NCT00002911) | MMPs | [79] |
| Prinomastat | Various Cancers | Not FDA-Approved. (NCT00004199) | MMPs | [80] |
| Neovastat | Various Cancers | Not FDA-Approved. (NCT00026117) | MMPs | [81] |
| Rebimastat | Various Cancers | Not FDA-Approved. (NCT00040755) | MMPs | [82] |
| COL-3 | Sarcomas | Not FDA-Approved. (NCT00020683) | MMP-2 and MMP-9 | [83] |
| WX-671 | Pancreatic cancer | Phase II (NCT00499265) | uPA | [84] |
| AE-941 | NSCLC | Not FDA-Approved. (NCT00005838) | MMP-2 and MMP-9 | [85] |
| Bowman–Birk inhibitor | Various Cancers | Phase II (NCT00330382) | Serine protease | [86] |
| AG3340 | NSCLC | Not FDA-Approved. (NCT00004199) | MMPs | [87] |
| BMS-275291 | NSCLC | Not FDA-Approved. (NCT00006229) | MMPs | [88] |
| Nelfinavir | Pancreatic cancer | Phase II (NCT02024009) | HIV protease | [89] |
4.1. Clinical Trials
Protease inhibitors have shown promise in oncology by targeting key enzymes involved in tumor growth, invasion, and survival. In oncology, WX-UK1, a uPA inhibitor, is being developed for its role in reducing tumor invasion and metastasis. Its oral prodrug, upamostat (WX-671), is under clinical evaluation for cancers such as pancreatic cancer [84,90]. This serine protease inhibitor disrupts tumor metastasis by inhibiting the uPA system, crucial for extracellular matrix degradation. Despite preclinical efficacy and early clinical evaluations, no data on regulatory approval or large-scale success are currently available.
Proteasome inhibitors are well-established in oncology, and bortezomib, approved for multiple myeloma and mantle cell lymphoma, targets the 26S proteasome, inducing apoptosis in rapidly dividing cancer cells [91]. Clinical trials demonstrated improved progression-free survival, but common side effects include peripheral neuropathy, fatigue, and hematological toxicities [92,93]. Other threonine protease inhibitors, such as ER-807446 [94] and MLN-519 [95], showed preclinical promise in reducing tumor growth, but further development has been limited due to toxicity or lack of efficacy in advanced trials. TMC-95A, a natural proteasome inhibitor with a unique non-covalent binding mechanism, remains a research tool, without clinical translation [96,97].
MMP inhibitors specifically targeting cancer have had mixed outcomes. Marimastat, a broad-spectrum MMP inhibitor, reached Phase III for various cancers but was discontinued due to dose-limiting musculoskeletal toxicity [98]. Similarly, Rebimastat failed to secure approval after Phase III trials. These failures highlight the challenge of balancing therapeutic efficacy with minimizing side effects such as joint pain and systemic toxicity which arise due to the broad physiological roles of MMPs [99]. Prinomastat is a synthetic, hydroxamic acid-based MMP inhibitor designed to target MMPs like MMP-2, MMP-9, and MMP-14, which are involved in tumor invasion and metastasis. It was evaluated in Phase III clinical trials for non-small cell lung cancer (NSCLC) and despite promising preclinical data, the trials demonstrated limited efficacy in improving survival or disease progression and revealed significant side effects, including musculoskeletal pain, due to the off-target inhibition of MMPs involved in normal tissue remodeling [100]. S-3304 is an orally administered MMP inhibitor, developed by Shionogi & Co., Ltd., (Osaka, Japan), primarily targeting MMP-2 and MMP-9, and as of 2024, S-3304 has not received regulatory approval for cancer treatment. The lack of substantial efficacy in clinical trials, despite a favorable safety profile, has resulted in the discontinuation of its development for oncological indications. No ongoing clinical trials or further investigations involving S-3304 are currently registered [101,102]. SC-77964 is a small-molecule MMP inhibitor developed by Pfizer Inc. designed to target specific MMPs involved in cancer progression. Regardless of encouraging preclinical results, SC-77964 has not advanced beyond the preclinical stage. There are no registered clinical trials involving SC-77964 and it has not received regulatory approval for any indication [103]. Ro 28-2653 is a synthetic MMP inhibitor developed by Roche, exhibiting high selectivity for MMP-2, MMP-9, and MT1-MMP. Ro 28-2653 has not advanced to clinical trials and remains in the preclinical stage of development. The lack of clinical progression may be attributed to various factors, including challenges in demonstrating sufficient efficacy, potential safety concerns, or strategic decisions by the developing company [104,105].
One of the key reasons for the failure of MMP inhibitors in clinical trials is the ability of tumors to compensate for MMP blockade by activating alternative proteolytic pathways. Studies have shown that in response to MMP inhibition, tumors can upregulate ADAM proteases, which facilitate invasion through alternative extracellular matrix degradation mechanisms [2]. Additionally, certain tumors rely on plasminogen activator (uPA) systems or serine proteases to maintain metastatic potential [36]. Recent preclinical investigations have explored dual-targeting strategies where MMP inhibitors are co-administered with urokinase inhibitors or cathepsin inhibitors to prevent tumor escape mechanisms. For example, a combination of batimastat (MMP inhibitor) with serine protease inhibitor (aprotinin) has effectively inhibited the degradation of collagen IV and casein by tumor cells [106].
Among these, bortezomib, as a proteasome inhibitor, remains the only cancer protease inhibitor fully approved for clinical use. It has transformed the treatment landscape for multiple myeloma and mantle cell lymphoma. Other inhibitors, including WX-UK1 and upamostat, are still in various stages of clinical development, while compounds like marimastat and rebimastat have been discontinued due to adverse effects. The field continues to evolve, with ongoing research focused on improving specificity, minimizing side effects, and expanding therapeutic applications.
4.2. Combination Therapies
To address the limitations of protease inhibitors as monotherapies, combination strategies have emerged as a promising approach [107]. Combining protease inhibitors with chemotherapy has shown synergistic effects, with inhibitors reducing ECM remodeling and enhancing drug penetration into tumors [8]. For example, MMP inhibitors have been paired with cytotoxic agents to target both the tumor microenvironment and cancer cells directly [108]. Similarly, the combination of protease inhibitors with immunotherapy has gained traction, particularly in enhancing the efficacy of immune checkpoint inhibitors [109]. While still in early stages, combining protease inhibitors with immunotherapy agents is an area of active research. Studies are exploring the potential of combining proteasome inhibitors with immune checkpoint inhibitors to enhance anti-tumor immune responses [110]. The synergistic effects observed in these combination therapies can be attributed to several mechanisms. Protease inhibitors can sensitize cancer cells to apoptosis induced by chemotherapy agents [111]. Some protease inhibitors, like nelfinavir, can inhibit multidrug efflux pumps, potentially overcoming drug resistance [112]. Protease inhibitors can induce endoplasmic reticulum (ER) stress, which may sensitize cancer cells to other therapies [113]. Combining different inhibitors can target multiple oncogenic pathways simultaneously, leading to more robust anti-tumor effects [114]. Several clinical trials have explored the potential of protease inhibitor combinations in cancer therapy. A phase II trial demonstrated that indinavir combined with chemotherapy improved outcomes in elderly patients with Kaposi sarcoma, without additional toxicity [115]. Ongoing trials are investigating the combination of nelfinavir with various chemotherapy regimens in different cancer types [116].
Several studies have demonstrated the potential of combining protease inhibitors with chemotherapy or immunotherapy to improve therapeutic efficacy and overcome resistance. For example, the uPA inhibitor WX-UK1 has shown promise in combination with gemcitabine for pancreatic cancer, with early clinical trials suggesting a reduction in tumor metastasis [84]. Similarly, proteasome inhibitors like bortezomib have been successfully paired with immune checkpoint inhibitors in multiple myeloma, enhancing T-cell responses and improving survival rates [117]. The combination of MMP inhibitors such as marimastat with cytotoxic agents has also been explored, with mixed results due to toxicity challenges but promising synergistic effects in specific cancer types [118,119]. These studies highlight the importance of combinatorial approaches in enhancing the effectiveness of protease inhibitors. Protease inhibitors have been increasingly recognized for their ability to enhance immune checkpoint therapy by modulating the tumor immune microenvironment [120,121]. MMP inhibitors can improve T-cell infiltration by reducing the physical barriers imposed by excessive extracellular matrix deposition, making tumors more susceptible to anti-PD-1 and anti-PD-L1 therapies [122]. Additionally, cathepsin S inhibition has been shown to enhance antigen presentation by dendritic cells, thereby improving immune surveillance [123]. These mechanisms highlight the potential for protease inhibitors to be used in combination with immune checkpoint blockade to enhance anti-tumor immunity.
HIV protease inhibitors have demonstrated the ability to enhance the effects of traditional chemotherapy agents. For example, ritonavir has been shown to potentiate the antiproliferative and proapoptotic effects of docetaxel in hormone-independent prostate cancer cells [124]. Indinavir combined with chemotherapy has shown promise in elderly patients with advanced progressive classic Kaposi sarcoma, potentially boosting and extending the duration of chemotherapy effects [115]. Proteasome inhibitors, particularly when combined with other targeted therapies, have shown significant potential. Carfilzomib combined with nelfinavir or lopinavir has demonstrated increased efficacy in triple-negative breast cancer models, potentially overcoming resistance mechanisms [112]. Bortezomib, when used in combination with DNA-damaging drugs like doxorubicin and melphalan, has shown promise in the treatment of multiple myeloma [125].
By modulating the tumor microenvironment, protease inhibitors can improve immune cell infiltration and activity. Furthermore, the integration of protease inhibitors with radiotherapy has been explored to enhance the effects of radiation by disrupting tumor stroma and improving oxygenation, thereby increasing tumor radiosensitivity [126]. Protease inhibitors continue to hold significant potential in cancer therapy, with advancements in understanding their mechanisms and refining their applications leading to promising results. The focus on combination therapies and innovative delivery systems offers hope for overcoming current limitations and realizing their full therapeutic potential.
5. Challenges and Limitations
While protease inhibitors hold great promise as therapeutic agents in cancer treatment, their clinical development and application have been met with several significant challenges. These limitations include issues related to target specificity, toxicity, and the development of resistance mechanisms, which have collectively hindered their broader adoption and effectiveness in oncology [127].
One of the primary challenges in the development of protease inhibitors is achieving target selectivity. Proteases are ubiquitous enzymes with diverse physiological functions, and many shares structural similarities within their catalytic domains. This overlap often results in inhibitors affecting multiple proteases, leading to unintended off-target effects [128]. For example, MMP inhibitors such as batimastat and marimastat were found to lack sufficient specificity, inadvertently inhibiting multiple MMPs and disrupting normal physiological processes. This lack of selectivity not only reduces therapeutic efficacy but also increases the risk of adverse effects, necessitating the development of inhibitors with enhanced specificity and refined targeting mechanisms [42].
Toxicity represents another significant hurdle in the clinical use of protease inhibitors. Off-target effects arising from poor selectivity can lead to adverse reactions, as seen in early clinical trials of MMP inhibitors. Musculoskeletal toxicity, characterized by joint pain and stiffness, was a prominent side effect that limited the tolerability and use of these agents in patients [129]. Additionally, the systemic inhibition of proteases that play critical roles in normal tissue homeostasis can result in unintended consequences, further complicating their clinical application. Efforts to mitigate toxicity have included optimizing dosing regimens, exploring localized delivery methods, and designing next-generation inhibitors with improved pharmacological profiles [130].
Tumor resistance to protease inhibitors poses another major challenge. Tumors are highly adaptive and capable of developing mechanisms to circumvent the effects of targeted therapies [131]. In the case of protease inhibitors, cancer cells may upregulate alternative proteases or signaling pathways to maintain tumor progression despite the inhibition of a specific target [132]. For instance, inhibiting one MMP may lead to compensatory activity by other MMPs or protease families, rendering the treatment less effective over time. Furthermore, changes in the tumor microenvironment, such as altered ECM composition or the increased expression of protease activators, can contribute to resistance. Addressing these resistance mechanisms requires a combination of approaches, including multi-target therapies, combination treatments with other agents, and strategies to anticipate and counteract tumor adaptation.
The dual role of proteases in both promoting and suppressing tumor progression presents a significant challenge in the development of protease inhibitors. While many proteases facilitate tumor invasion and metastasis, others contribute to immune surveillance and tumor suppression in specific contexts. For instance, MMP-8 has been shown to exert protective effects against cancer progression by modulating the extracellular matrix [133]. Consequently, recent efforts have shifted toward biomarker-driven approaches to selectively inhibit pathological protease activity while preserving physiological functions [134]. Advances in single-cell transcriptomics and proteomic profiling allow for the stratification of patients based on tumor-specific protease expression, thereby reducing unintended effects and improving therapeutic outcomes [134].
To overcome the selectivity challenges that contributed to the failure of early MMP inhibitors, recent advancements in computational drug design, proteomics, and structural biology have enabled the development of more precise inhibitors [135,136,137]. Structure-based drug design (SBDD) has been employed to identify inhibitors that selectively target disease-associated MMPs while sparing physiologically important ones [138]. Additionally, peptidomimetic inhibitors, which mimic natural protease substrates but resist degradation, have shown increased specificity [7]. Furthermore, advances in activity-based profiling using proteomics have facilitated the identification of cancer-specific protease signatures, guiding patient stratification for targeted therapy.
The challenges of selectivity, toxicity, and resistance underscore the complexity of translating protease inhibitors into effective cancer therapies. Continued research and innovation are essential to address these limitations and unlock the full therapeutic potential of protease inhibitors in oncology. The future of protease inhibitors in cancer therapy lies in addressing the challenges of specificity, toxicity, and resistance while leveraging innovative research and emerging technologies. Advancements in drug delivery, the identification of novel protease targets, and the integration of personalized medicine approaches are pivotal for optimizing the clinical potential of protease inhibitors.
6. Research Innovations
Despite these challenges, advances in drug design and delivery offer hope for overcoming these limitations. The development of highly selective small-molecule inhibitors, monoclonal antibodies, and peptide-based inhibitors has improved the precision of targeting specific proteases [139]. Novel drug delivery systems, such as nanoparticles and antibody–drug conjugates, have enabled the localized and controlled release of inhibitors, reducing systemic toxicity [140]. Additionally, combining protease inhibitors with other therapeutic modalities, such as chemotherapy, immunotherapy, or radiotherapy, has shown promise in enhancing efficacy and preventing resistance [141,142,143]. Nanoparticle-based delivery systems, for instance, allow for the encapsulation of protease inhibitors, enabling targeted delivery to tumor sites while sparing healthy tissues. These systems can be engineered to release their payload in response to specific stimuli within the tumor microenvironment, such as pH changes or enzymatic activity, further improving selectivity [144,145]. Bortezomib and carfilzomib (proteasome inhibitors) have been loaded into PEGylated lipid micelles, liposomes, gold nanoparticles, and mesoporous silica nanoparticles. These formulations demonstrated superior therapeutic effects, including higher efficacy, increased circulation time, and decreased systemic toxicity compared to free drug formulations in breast cancer, non-small cell lung cancer, and colon carcinoma models [146,147]. Doxorubicin and epoxomicin (a proteasome inhibitor) have been co-encapsulated in poly(lactic-co-glycolic acid) (PLGA) nanoparticles. This formulation showed enhanced anticancer efficiency, reduced drug resistance, and decreased toxicity to normal cells compared to free drugs [148].
Additionally, antibody–drug conjugates (ADCs) offer a promising approach by linking protease inhibitors to monoclonal antibodies that specifically bind to tumor-associated antigens, achieving both precision and efficacy [149]. ADCs offer several potential benefits for cancer therapy, including improved selectivity, enhanced efficacy, and reduced systemic toxicity [150]. By targeting tumor-associated antigens, ADCs can deliver cytotoxic agents specifically to cancer cells while sparing healthy tissues. The combination of antibody specificity and potent payloads allows for the delivery of highly cytotoxic agents that would be too toxic for systemic administration, potentially improving the therapeutic index [151].The concept of using protease inhibitors as ADC payloads aligns with the general principles of ADC design, including tumor microenvironment targeting, payload activation, and versatility. When developing protease inhibitor ADCs, several factors need to be considered, such as target selection, linker design, payload potency, and optimization, to achieve the desired balance of efficacy and safety [152].
Emerging technologies like CRISPR-Cas9 genome editing also hold promise for targeting proteases in cancer. By selectively knocking out or modulating the expression of genes encoding dysregulated proteases, CRISPR-based approaches can provide highly specific and durable therapeutic effects. These strategies may complement traditional protease inhibitors, particularly in addressing resistance mechanisms driven by compensatory upregulation of alternative proteases [153]. Despite its promise, the clinical application of CRISPR-Cas9 in cancer therapy faces several challenges which must be addressed before clinical implementation. One major concern is the off-target effects of CRISPR-Cas9, where unintended genetic modifications can lead to unpredictable consequences, raising safety and regulatory issues [154]. Additionally, the potential immunogenicity of Cas9 proteins poses a challenge for in vivo applications, necessitating further research into reducing immune responses against these components. Another significant challenge is the regulatory and ethical barriers associated with personalized medicine, as patient-specific protease inhibitor therapies require extensive validation, making clinical translation costly and time-consuming. Addressing these challenges will be essential for advancing the integration of these cutting-edge technologies into clinical practice [155].
The ongoing identification of novel proteases implicated in cancer progression opens new avenues for therapeutic intervention. While traditional efforts have focused on well-studied proteases such as MMPs, cathepsins, and uPA, recent research has uncovered additional protease families with significant roles in tumor biology. For example, the ADAM (a disintegrin and metalloprotease) family has been implicated in cell signaling and adhesion processes critical for tumor growth and metastasis [156]. Similarly, proteases involved in the unfolded protein response (UPR), such as those in the calpain family, are emerging as potential targets due to their role in regulating cancer cell survival under stress conditions [157]. Targeting these novel proteases may provide opportunities to disrupt cancer-specific pathways while avoiding the toxicity associated with inhibiting ubiquitously expressed proteases.
The integration of protease inhibitors into personalized medicine paradigms has the potential to transform cancer treatment. By leveraging proteomic profiling technologies, it is now possible to identify the specific protease expression patterns within individual tumors and this information can guide the selection of the most appropriate protease inhibitor for each patient, optimizing therapeutic outcomes and minimizing side effects [158]. For example, tumors with high expression of MMP-9 may benefit from targeted MMP inhibitors, while those with elevated cathepsin activity could respond better to cysteine protease inhibitors. Personalized approaches may also involve combining protease inhibitors with other targeted therapies based on the molecular characteristics of the tumor microenvironment.
The implementation of precision medicine in protease inhibitor development hinges on the identification of predictive biomarkers that determine patient responses. Technique such as mass spectrometry-based proteomics has been employed to analyze protease expression in tumor tissues [159]. For example, high MMP-9 expression in colorectal cancer has been correlated with responsiveness to MMP-targeted therapies [160], whereas elevated cathepsin B levels have been linked to aggressive breast cancer subtypes [161]. Furthermore, liquid biopsy-based protease activity assays are emerging as a non-invasive approach to monitor tumor responses to therapy in real time [162]. These advancements are paving the way for more stratified clinical trials that match protease inhibitors to patients most likely to benefit from them.
Additionally, advances in bioinformatics and artificial intelligence (AI) are facilitating the development of predictive models for treatment response. These tools can integrate data from genomic, transcriptomic, and proteomic analyses to predict which patients are most likely to benefit from specific protease inhibitors, enabling a more tailored and effective approach to cancer therapy [163,164,165].
The continuous exploration of protease inhibitors’ mechanisms and therapeutic applications remains crucial for advancing cancer treatment. By integrating novel drug delivery systems, personalized medicine approaches, and AI-driven predictive models, future therapies may achieve enhanced specificity and minimized toxicity. Ongoing clinical trials and combination strategies will play a key role in determining the success of protease inhibitors in oncology. Further interdisciplinary research is essential to fully realize their therapeutic potential.
In conclusion, the future of protease inhibitors in oncology is poised for transformation through innovative research and emerging technologies. Advanced drug delivery systems, novel protease targets, and personalized medicine approaches offer the potential to overcome current limitations and unlock new therapeutic possibilities. As these strategies continue to evolve, they hold the promise of improving the specificity, efficacy, and safety of protease inhibitors, ultimately enhancing their impact on cancer treatment. Protease inhibitors remain a promising yet challenging frontier in cancer treatment. Continued research and collaboration will be essential to unlock their potential and improve outcomes for cancer patients worldwide.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol. Cancer Res. 2023, 21, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S. Extracellular proteolysis in cancer: Proteases, substrates, and mechanisms in tumor progression and metastasis. J. Biol. Chem. 2024, 300, 107347. [Google Scholar] [CrossRef] [PubMed]
- Popova, N.V.; Jucker, M. The Functional Role of Extracellular Matrix Proteins in Cancer. Cancers 2022, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef]
- Park, K.C.; Dharmasivam, M.; Richardson, D.R. The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators that Inhibit their Activity. Int. J. Mol. Sci. 2020, 21, 6805. [Google Scholar] [CrossRef]
- Ashja Ardalan, A.; Khalili-Tanha, G.; Shoari, A. Shaping the Landscape of Lung Cancer: The Role and Therapeutic Potential of Matrix Metalloproteinases. Int. J. Transl. Med. 2024, 4, 661–679. [Google Scholar] [CrossRef]
- Fear, G.; Komarnytsky, S.; Raskin, I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol. Ther. 2007, 113, 354–368. [Google Scholar] [CrossRef]
- Rudzinska, M.; Daglioglu, C.; Savvateeva, L.V.; Kaci, F.N.; Antoine, R.; Zamyatnin, A.A., Jr. Current Status and Perspectives of Protease Inhibitors and Their Combination with Nanosized Drug Delivery Systems for Targeted Cancer Therapy. Drug Des. Dev. Ther. 2021, 15, 9–20. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef]
- DeClerck, Y.A.; Imren, S. Protease inhibitors: Role and potential therapeutic use in human cancer. Eur. J. Cancer 1994, 30, 2170–2180. [Google Scholar] [CrossRef]
- Almutairi, S.; Kalloush, H.M.; Manoon, N.A.; Bardaweel, S.K. Matrix Metalloproteinases Inhibitors in Cancer Treatment: An Updated Review (2013–2023). Molecules 2023, 28, 5567. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Borges, P.H.O.; Ferreira, S.B.; Silva, F.P., Jr. Recent Advances on Targeting Proteases for Antiviral Development. Viruses 2024, 16, 366. [Google Scholar] [CrossRef] [PubMed]
- Motyan, J.A.; Toth, F.; Tozser, J. Research applications of proteolytic enzymes in molecular biology. Biomolecules 2013, 3, 923–942. [Google Scholar] [CrossRef]
- Vizovisek, M.; Ristanovic, D.; Menghini, S.; Christiansen, M.G.; Schuerle, S. The Tumor Proteolytic Landscape: A Challenging Frontier in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 2514. [Google Scholar] [CrossRef]
- Tagirasa, R.; Yoo, E. Role of Serine Proteases at the Tumor-Stroma Interface. Front. Immunol. 2022, 13, 832418. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Di Carluccio, G.; Motti, M.L.; Carriero, M.V. Therapeutic Strategies Targeting Urokinase and Its Receptor in Cancer. Cancers 2022, 14, 498. [Google Scholar] [CrossRef]
- Kryza, T.; Silva, M.L.; Loessner, D.; Heuze-Vourc’h, N.; Clements, J.A. The kallikrein-related peptidase family: Dysregulation and functions during cancer progression. Biochimie 2016, 122, 283–299. [Google Scholar] [CrossRef]
- Rudzinska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Pranjol, M.Z.; Gutowski, N.; Hannemann, M.; Whatmore, J. The Potential Role of the Proteases Cathepsin D and Cathepsin L in the Progression and Metastasis of Epithelial Ovarian Cancer. Biomolecules 2015, 5, 3260–3279. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef]
- van Hinsbergh, V.W.; Engelse, M.A.; Quax, P.H. Pericellular proteases in angiogenesis and vasculogenesis. Arter. Thromb. Vasc. Biol. 2006, 26, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Sukharev, S.A.; Pleshakova, O.V.; Sadovnikov, V.B. Role of proteases in activation of apoptosis. Cell Death Differ. 1997, 4, 457–462. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Mukherjee, T.; Das, K. Coagulation Protease-Driven Cancer Immune Evasion: Potential Targets for Cancer Immunotherapy. Cancers 2024, 16, 1568. [Google Scholar] [CrossRef]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar]
- Xie, Z.; Zhao, M.Y.; Yan, C.X.; Kong, W.; Lan, F.; Zhao, S.X.; Yang, Q.H.; Bai, Z.T.; Qing, H.; Ni, J.J. Cathepsin B in programmed cell death machinery: Mechanisms of execution and regulatory pathways. Cell Death Dis. 2023, 14, 255. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Savvateeva, L.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis. Int. J. Mol. Sci. 2021, 22, 4669. [Google Scholar] [CrossRef]
- Shoari, A. Potential of MMP-2 and MMP-9 Gelatinase Blockade as a Therapeutic Strategy in Fibrosarcoma Treatment: A Decadal Review. Targets 2024, 2, 104–125. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Rabaglino, M.B.; Wood, C.E.; Siemann, D.W. Cathepsin L in tumor angiogenesis and its therapeutic intervention by the small molecule inhibitor KGP94. Clin. Exp. Metastasis 2016, 33, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, J.; Papiewska-Pajak, I.; Kowalska, M.A.; Boncela, J. Cathepsin B Is Upregulated and Mediates ECM Degradation in Colon Adenocarcinoma HT29 Cells Overexpressing Snail. Cells 2019, 8, 203. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Platet, N.; Liaudet, E.; Laurent, V.; Derocq, D.; Brouillet, J.P.; Rochefort, H. Biological and clinical significance of cathepsin D in breast cancer metastasis. Stem Cells 1996, 14, 642–650. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Flores-Soto, E.; Aquino-Galvez, A. Hypoxic Effects on Matrix Metalloproteinases’ Expression in the Tumor Microenvironment and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 16887. [Google Scholar] [CrossRef]
- Qu, P.; Yan, C.; Du, H. Matrix metalloproteinase 12 overexpression in myeloid lineage cells plays a key role in modulating myelopoiesis, immune suppression, and lung tumorigenesis. Blood 2011, 117, 4476–4489. [Google Scholar] [CrossRef]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: A versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev. 2024, 43, 1095–1116. [Google Scholar] [CrossRef]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Shoari, A.; Khalili-Tanha, G.; Coban, M.A.; Radisky, E.S. Structure and computation-guided yeast surface display for the evolution of TIMP-based matrix metalloproteinase inhibitors. Front. Mol. Biosci. 2023, 10, 1321956. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Myung, J.; Sin, N.; Crews, C.M. Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: Insights into specificity and potency. Bioorganic Med. Chem. Lett. 1999, 9, 3335–3340. [Google Scholar] [CrossRef]
- Lund, I.K.; Rasch, M.G.; Ingvarsen, S.; Pass, J.; Madsen, D.H.; Engelholm, L.H.; Behrendt, N.; Hoyer-Hansen, G. Inhibitory monoclonal antibodies against mouse proteases raised in gene-deficient mice block proteolytic functions. Front. Pharmacol. 2012, 3, 26921. [Google Scholar] [CrossRef]
- Kwok, H.F.; Buick, R.J.; Kuehn, D.; Gormley, J.A.; Doherty, D.; Jaquin, T.J.; McClurg, A.; Ward, C.; Byrne, T.; Jaworski, J.; et al. Antibody targeting of Cathepsin S induces antibody-dependent cellular cytotoxicity. Mol. Cancer 2011, 10, 147. [Google Scholar] [CrossRef]
- Dana, D.; Pathak, S.K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef]
- Wei, Y.; Huang, M.D.; Jiang, L.G. Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications. Catalysts 2024, 14, 787. [Google Scholar] [CrossRef]
- Farady, C.J.; Craik, C.S. Mechanisms of macromolecular protease inhibitors. Chembiochem 2010, 11, 2341–2346. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Allosteric modulators: An emerging concept in drug discovery. ACS Med. Chem. Lett. 2015, 6, 104–107. [Google Scholar] [CrossRef]
- Cheng, S.S.; Yang, G.J.; Wang, W.; Leung, C.H.; Ma, D.L. The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 2020, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef]
- Botkjaer, K.A.; Deryugina, E.I.; Dupont, D.M.; Gardsvoll, H.; Bekes, E.M.; Thuesen, C.K.; Chen, Z.; Ploug, M.; Quigley, J.P.; Andreasen, P.A. Targeting tumor cell invasion and dissemination in vivo by an aptamer that inhibits urokinase-type plasminogen activator through a novel multifunctional mechanism. Mol. Cancer Res. 2012, 10, 1532–1543. [Google Scholar] [CrossRef]
- Cwilichowska, N.; Swiderska, K.W.; Dobrzyn, A.; Drag, M.; Poreba, M. Diagnostic and therapeutic potential of protease inhibition. Mol. Asp. Med. 2022, 88, 101144. [Google Scholar] [CrossRef]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef]
- Nalla, A.K.; Gorantla, B.; Gondi, C.S.; Lakka, S.S.; Rao, J.S. Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer Gene Ther. 2010, 17, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Sudhan, D.R.; Pampo, C.; Rice, L.; Siemann, D.W. Cathepsin L inactivation leads to multimodal inhibition of prostate cancer cell dissemination in a preclinical bone metastasis model. Int. J. Cancer 2016, 138, 2665–2677. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Ascione, B.; Ciarlo, L.; Vona, R.; Leonetti, C.; Scarsella, M.; Mileo, A.M.; Catricalà, C.; Paggi, M.G.; Malorni, W. Cathepsin B inhibition interferes with metastatic potential of human melanoma: An study. Mol. Cancer 2010, 9, 207. [Google Scholar] [CrossRef]
- Shoari, A.; Ashja Ardalan, A.; Dimesa, A.M.; Coban, M.A. Targeting Invasion: The Role of MMP-2 and MMP-9 Inhibition in Colorectal Cancer Therapy. Biomolecules 2024, 15, 35. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Raedler, L.A. Kyprolis (Carfilzomib) Received New Indications as Combination Therapy for Use in Relapsed and/or Refractory Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 93–96. [Google Scholar] [PubMed]
- Lee, M.S.; Lim, S.H.; Yu, A.R.; Hwang, C.Y.; Kang, I.; Yeo, E.J. Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells. Biology 2021, 10, 153. [Google Scholar] [CrossRef]
- Davis, M.A.; Delaney, J.R.; Patel, C.B.; Storgard, R.; Stupack, D.G. Nelfinavir is effective against human cervical cancer cells in vivo: A potential treatment modality in resource-limited settings. Drug Des. Dev. Ther. 2016, 10, 1837–1846. [Google Scholar] [CrossRef][Green Version]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Victor, B.C.; Anbalagan, A.; Mohamed, M.M.; Sloane, B.F.; Cavallo-Medved, D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 2011, 13, R115. [Google Scholar] [CrossRef]
- Bevan, P.; Mala, C. The Role of uPA and uPA Inhibitors in Breast Cancer. Breast Care 2008, 3, 1–2. [Google Scholar] [CrossRef]
- Kumar, A.A.; Buckley, B.J.; Ranson, M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules 2022, 12, 152. [Google Scholar] [CrossRef]
- Raedler, L. Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma. Am. Health Drug Benefits 2015, 8, 135–140. [Google Scholar]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. U.S. Food and Drug Administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef]
- Shah, M.A.; Starodub, A.; Sharma, S.; Berlin, J.; Patel, M.; Wainberg, Z.A.; Chaves, J.; Gordon, M.; Windsor, K.; Brachmann, C.B.; et al. Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.C.; Christy, M.P.; Phan, V.V.; Gerwick, W.H.; Hook, G.; O’Donoghue, A.J.; Hook, V. Molecular Features of CA-074 pH-Dependent Inhibition of Cathepsin B. Biochemistry 2022, 61, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Turk, D.; Podobnik, M.; Popovic, T.; Katunuma, N.; Bode, W.; Huber, R.; Turk, V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. [Google Scholar] [CrossRef] [PubMed]
- Jakos, T.; Pislar, A.; Pecar Fonovic, U.; Svajger, U.; Kos, J. Cysteine cathepsins L and X differentially modulate interactions between myeloid-derived suppressor cells and tumor cells. Cancer Immunol. Immunother. 2020, 69, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, H.; Nishimura, T.; Tada, Y.; Asao, T.; Turk, D.; Turk, V.; Katunuma, N. Inhibition mechanism of cathepsin L-specific inhibitors based on the crystal structure of papain-CLIK148 complex. Biochem. Biophys. Res. Commun. 1999, 266, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Katunuma, N.; Murata, E.; Kakegawa, H.; Matsui, A.; Tsuzuki, H.; Tsuge, H.; Turk, D.; Turk, V.; Fukushima, M.; Tada, Y.; et al. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999, 458, 6–10. [Google Scholar] [CrossRef]
- Bennacef, I.; Rubins, D.; Riffel, K.; Williams, M.; Posavec, D.J.; Holahan, M.A.; Purcell, M.L.; Haley, H.D.; Wolf, M.; Stachel, S.J.; et al. Preclinical evaluation of [(11) C]L-235 as a radioligand for Positron Emission Tomography cathepsin K imaging in bone. J. Label. Comp. Radiopharm. 2021, 64, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, R.; Astorgues-Xerri, L.; Bekradda, M.; Gormley, J.; Buick, R.; Kerr, P.; Cvitkovic, E.; Raymond, E.; D’Incalci, M.; Frapolli, R.; et al. Fsn0503h antibody-mediated blockade of cathepsin S as a potential therapeutic strategy for the treatment of solid tumors. Biochimie 2015, 108, 101–107. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.E.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef]
- Scatena, R. Prinomastat, a hydroxamate-based matrix metalloproteinase inhibitor. A novel pharmacological approach for tissue remodelling-related diseases. Expert Opin. Investig. Drugs 2000, 9, 2159–2165. [Google Scholar] [CrossRef]
- Falardeau, P.; Champagne, P.; Poyet, P.; Hariton, C.; Dupont, E. Neovastat, a naturally occurring multifunctional antiangiogenic drug, in phase III clinical trials. Semin. Oncol. 2001, 28, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Carpinteiro, I.; Portugal, J.; Azul, A.; Polido, M.; Petrova, K.T.; Salema-Oom, M.; Caldeira, J. Challenges in Matrix Metalloproteinases Inhibition. Biomolecules 2020, 10, 717. [Google Scholar] [CrossRef]
- Dezube, B.J.; Krown, S.E.; Lee, J.Y.; Bauer, K.S.; Aboulafia, D.M. Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi’s sarcoma: An AIDS Malignancy Consortium Study. J. Clin. Oncol. 2006, 24, 1389–1394. [Google Scholar] [CrossRef]
- Heinemann, V.; Ebert, M.P.; Laubender, R.P.; Bevan, P.; Mala, C.; Boeck, S. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br. J. Cancer 2013, 108, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Lee, J.J.; Komaki, R.; Herbst, R.S.; Feng, L.; Evans, W.K.; Choy, H.; Desjardins, P.; Esparaz, B.T.; Truong, M.T.; et al. Chemoradiotherapy with or without AE-941 in stage III non-small cell lung cancer: A randomized phase III trial. J. Natl. Cancer Inst. 2010, 102, 859–865. [Google Scholar] [CrossRef]
- Armstrong, W.B.; Taylor, T.H.; Kennedy, A.R.; Melrose, R.J.; Messadi, D.V.; Gu, M.; Le, A.D.; Perloff, M.; Civantos, F.; Goodwin, W.J.; et al. Bowman birk inhibitor concentrate and oral leukoplakia: A randomized phase IIb trial. Cancer Prev. Res. 2013, 6, 410–418. [Google Scholar] [CrossRef]
- Griffioen, A.W. AG-3340 (Agouron Pharmaceuticals Inc.). IDrugs 2000, 3, 336–345. [Google Scholar]
- Leighl, N.B.; Paz-Ares, L.; Douillard, J.Y.; Peschel, C.; Arnold, A.; Depierre, A.; Santoro, A.; Betticher, D.C.; Gatzemeier, U.; Jassem, J.; et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J. Clin. Oncol. 2005, 23, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Qi, C.; Shaw, R.; Jones, C.M.; Bridgewater, J.A.; Radhakrishna, G.; Patel, N.; Holmes, J.; Virdee, P.S.; Tranter, B.; et al. Standard or high dose chemoradiotherapy, with or without the protease inhibitor nelfinavir, in patients with locally advanced pancreatic cancer: The phase 1/randomised phase 2 SCALOP-2 trial. Eur. J. Cancer 2024, 209, 114236. [Google Scholar] [CrossRef]
- Lai, X.; Cheng, D.; Xu, H.; Wang, J.; Lv, X.; Yao, H.; Li, L.; Wu, J.; Ye, S.; Li, Z. Phase I Trial of Upamostat Combined With Gemcitabine in Locally Unresectable or Metastatic Pancreatic Cancer: Safety and Preliminary Efficacy Assessment. Cancer Med. 2025, 14, e70550. [Google Scholar] [CrossRef]
- Shah, J.J.; Orlowski, R.Z. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia 2009, 23, 1964–1979. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Flinn, I.; Richardson, P.G.; Hari, P.; Callander, N.; Noga, S.J.; Stewart, A.K.; Turturro, F.; Rifkin, R.; Wolf, J.; et al. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood 2012, 119, 4375–4382. [Google Scholar] [CrossRef] [PubMed]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, K.; Akasaka, K.; Wu, J.; Murphy, E.; Jiang, Y.; Li, X.-Y.; Tendyke, K.; Schiller, S.; Reardon, C.; Decosta, B.; et al. Novel proteasome inhibitors show potent anti-tumor efficacy. Cancer Res. 2004, 64, 925. [Google Scholar]
- Ostrowska, H. The ubiquitin-proteasome system: A novel target for anticancer and anti-inflammatory drug research. Cell Mol. Biol. Lett. 2008, 13, 353–365. [Google Scholar] [CrossRef]
- Yang, Z.Q.; Kwok, B.H.; Lin, S.; Koldobskiy, M.A.; Crews, C.M.; Danishefsky, S.J. Simplified synthetic TMC-95A/B analogues retain the potency of proteasome inhibitory activity. Chembiochem 2003, 4, 508–513. [Google Scholar] [CrossRef]
- Guedes, R.A.; Grilo, J.H.; Carvalho, A.N.; Fernandes, P.M.P.; Ressurreiçao, A.S.; Brito, V.; Santos, A.O.; Silvestre, S.; Gallerani, E.; Gama, M.J.; et al. New Scaffolds of Proteasome Inhibitors: Boosting Anticancer Potential by Exploiting the Synergy of In Silico and In Vitro Methodologies. Pharmaceuticals 2023, 16, 1096. [Google Scholar] [CrossRef]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J. Clin. Oncol. 2004, 22, 4683–4690. [Google Scholar] [CrossRef]
- Park, S.; Ko, Y.H.; Lee, B.; Shin, B.; Beck, B.R. Abstract 35: Molecular optimization of phase III trial failed anticancer drugs using target affinity and toxicity-centered multiple properties reinforcement learning. Clin. Cancer Res. 2020, 26, 35. [Google Scholar] [CrossRef]
- Bissett, D.; O’Byrne, K.J.; von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 842–849. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Eckhardt, S.G.; Bukowski, R.; Sullivan, D.M.; Ikeda, M.; Yano, Y.; Yamada-Sawada, T.; Kambayashi, Y.; Tanaka, K.; Javle, M.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of s-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2007, 13, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- van Marle, S.; van Vliet, A.; Sollie, F.; Kambayashi, Y.; Yamada-Sawada, T. Safety, tolerability and pharmacokinetics of oral S-3304, a novel matrix metalloproteinase inhibitor, in single and multiple dose escalation studies in healthy volunteers. Int. J. Clin. Pharmacol. Ther. 2005, 43, 282–293. [Google Scholar] [CrossRef]
- Lutz, M.R., Jr.; Flieger, S.; Colorina, A.; Wozny, J.; Hosmane, N.S.; Becker, D.P. Carborane-Containing Matrix Metalloprotease (MMP) Ligands as Candidates for Boron Neutron-Capture Therapy (BNCT). ChemMedChem 2020, 15, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Heinemann, P.V.; Heiland, S.; Koziol, J.; Gardner, H.; Wagner, S. Selective MMP-inhibition with Ro 28-2653 in acute experimental stroke--a magnetic resonance imaging efficacy study. Brain Res. 2011, 1368, 264–270. [Google Scholar] [CrossRef]
- Lein, M.; Jung, K.; Ortel, B.; Stephan, C.; Rothaug, W.; Juchem, R.; Johannsen, M.; Deger, S.; Schnorr, D.; Loening, S.; et al. The new synthetic matrix metalloproteinase inhibitor (Roche 28-2653) reduces tumor growth and prolongs survival in a prostate cancer standard rat model. Oncogene 2002, 21, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Della Porta, P.; Soeltl, R.; Krell, H.W.; Collins, K.; O’Donoghue, M.; Schmitt, M.; Kruger, A. Combined treatment with serine protease inhibitor aprotinin and matrix metalloproteinase inhibitor Batimastat (BB-94) does not prevent invasion of human esophageal and ovarian carcinoma cells in vivo. Anticancer Res. 1999, 19, 3809–3816. [Google Scholar]
- Lin, Z.; Wang, L.; Xing, Z.; Wang, F.; Cheng, X. Update on Combination Strategies of PARP Inhibitors. Cancer Control 2024, 31, 10732748241298329. [Google Scholar] [CrossRef]
- Zhong, S.W.; Jeong, J.H.; Chen, Z.K.; Chen, Z.H.; Luo, J.L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef]
- Mempel, T.R.; Krappmann, D. Combining precision oncology and immunotherapy by targeting the MALT1 protease. J. Immunother. Cancer 2022, 10, e005442. [Google Scholar] [CrossRef]
- Li, W.K.; Wei, J.; Cheng, M.; Liu, M. Unveiling promising targets in gastric cancer therapy: A comprehensive review. Mol. Ther. Oncol. 2024, 32, 200857. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Sedlarikova, L.; Buechler, L.; Kraus, M.; Yang, C.H.; Strakova, N.; Soucek, K.; Navratil, J.; Svoboda, M.; Welm, A.L.; et al. HIV-protease inhibitors potentiate the activity of carfilzomib in triple-negative breast cancer. Br. J. Cancer 2024, 131, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Subeha, M.R.; Telleria, C.M. The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir. Cancers 2020, 12, 3437. [Google Scholar] [CrossRef]
- Wen, W.; Han, E.S.; Dellinger, T.H.; Lu, L.X.; Wu, J.; Jove, R.; Yim, J.H. Synergistic Anti-Tumor Activity by Targeting Multiple Signaling Pathways in Ovarian Cancer. Cancers 2020, 12, 2586. [Google Scholar] [CrossRef]
- Sgadari, C.; Scoppio, B.; Picconi, O.; Tripiciano, A.; Gaiani, F.M.; Francavilla, V.; Arancio, A.; Campagna, M.; Palladino, C.; Moretti, S.; et al. Clinical Efficacy of the HIV Protease Inhibitor Indinavir in Combination with Chemotherapy for Advanced Classic Kaposi Sarcoma Treatment: A Single-Arm, Phase II Trial in the Elderly. Cancer Res. Commun. 2024, 4, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Soto, A.E.; McKenzie, N.D.; Whicker, M.E.; Pearson, J.M.; Jimenez, E.A.; Portelance, L.; Hu, J.J.; Lucci, J.A., III; Qureshi, R.; Kossenkov, A.; et al. Phase 1 trial of nelfinavir added to standard cisplatin chemotherapy with concurrent pelvic radiation for locally advanced cervical cancer. Cancer 2021, 127, 2279–2293. [Google Scholar] [CrossRef]
- Paul, B.; Kang, S.; Zheng, Z.; Kang, Y. The challenges of checkpoint inhibition in the treatment of multiple myeloma. Cell Immunol. 2018, 334, 87–98. [Google Scholar] [CrossRef]
- Goffin, J.R.; Anderson, I.C.; Supko, J.G.; Eder, J.P., Jr.; Shapiro, G.I.; Lynch, T.J.; Shipp, M.; Johnson, B.E.; Skarin, A.T. Phase I trial of the matrix metalloproteinase inhibitor marimastat combined with carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 3417–3424. [Google Scholar] [CrossRef]
- Watson, S.A.; Morris, T.M.; Collins, H.M.; Bawden, L.J.; Hawkins, K.; Bone, E.A. Inhibition of tumour growth by marimastat in a human xenograft model of gastric cancer: Relationship with levels of circulating CEA. Br. J. Cancer 1999, 81, 19–23. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, L. New insight into the agonism of protease-activated receptors as an immunotherapeutic strategy. J. Biol. Chem. 2024, 300, 105614. [Google Scholar] [CrossRef]
- de Magalhaes, M.T.Q.; Mambelli, F.S.; Santos, B.P.O.; Morais, S.B.; Oliveira, S.C. Serine protease inhibitors containing a Kunitz domain: Their role in modulation of host inflammatory responses and parasite survival. Microbes Infect. 2018, 20, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef] [PubMed]
- Dheilly, E.; Battistello, E.; Katanayeva, N.; Sungalee, S.; Michaux, J.; Duns, G.; Wehrle, S.; Sordet-Dessimoz, J.; Mina, M.; Racle, J.; et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell 2020, 37, 674–689. [Google Scholar] [CrossRef]
- Ikezoe, T.; Hisatake, Y.; Takeuchi, T.; Ohtsuki, Y.; Yang, Y.; Said, J.W.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitor, ritonavir: A potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004, 64, 7426–7431. [Google Scholar] [CrossRef]
- Bernstein, W.B.; Dennis, P.A. Repositioning HIV protease inhibitors as cancer therapeutics. Curr. Opin. HIV AIDS 2008, 3, 666–675. [Google Scholar] [CrossRef]
- Yoder, A.K.; Lakomy, D.S.; Dong, Y.Q.; Raychaudhury, S.; Royse, K.; Hartman, C.; Richardson, P.; White, D.L.; Kramer, J.R.; Lin, L.L.L.; et al. The association between protease inhibitors and anal cancer outcomes in veterans living with HIV treated with definitive chemoradiation: A retrospective study. BMC Cancer 2021, 21, 776. [Google Scholar] [CrossRef]
- Song, R.; Qiao, W.; He, J.; Huang, J.; Luo, Y.; Yang, T. Proteases and Their Modulators in Cancer Therapy: Challenges and Opportunities. J. Med. Chem. 2021, 64, 2851–2877. [Google Scholar] [CrossRef]
- Deu, E.; Verdoes, M.; Bogyo, M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat. Struct. Mol. Biol. 2012, 19, 9–16. [Google Scholar] [CrossRef]
- Krzeski, P.; Buckland-Wright, C.; Balint, G.; Cline, G.A.; Stoner, K.; Lyon, R.; Beary, J.; Aronstein, W.S.; Spector, T.D. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: A randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res. Ther. 2007, 9, R109. [Google Scholar] [CrossRef]
- Abbenante, G.; Fairlie, D.P. Protease inhibitors in the clinic. Med. Chem. 2005, 1, 71–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Lai, Y.; Xu, W.; Lei, S.; Chen, G.; Wang, Z. A computer-aided, heterodimer-based “triadic” carrier-free drug delivery platform to mitigate multidrug resistance in lung cancer and enhance efficiency. J. Colloid Interface Sci. 2025, 677, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef]
- Decock, J.; Hendrickx, W.; Thirkettle, S.; Gutierrez-Fernandez, A.; Robinson, S.D.; Edwards, D.R. Pleiotropic functions of the tumor- and metastasis-suppressing matrix metalloproteinase-8 in mammary cancer in MMTV-PyMT transgenic mice. Breast Cancer Res. 2015, 17, 38. [Google Scholar] [CrossRef]
- Mishra, P.; Laha, D.; Grant, R.; Nilubol, N. Advances in Biomarker-Driven Targeted Therapies in Thyroid Cancer. Cancers 2021, 13, 6194. [Google Scholar] [CrossRef]
- Singh, T.; Jayaram, B.; Adekoya, O.A. Computational Approaches to Matrix Metalloprotease Drug Design. Methods Mol. Biol. 2017, 1579, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.J.; Butler, G.S.; Rodriguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Amin, S.A.; Banerjee, P.; Gayen, S.; Jha, T. A review of MMP-2 structures and binding mode analysis of its inhibitors to strategize structure-based drug design. Bioorg Med. Chem. 2022, 74, 117044. [Google Scholar] [CrossRef]
- Xie, X.; Yu, T.; Li, X.; Zhang, N.; Foster, L.J.; Peng, C.; Huang, W.; He, G. Recent advances in targeting the “undruggable” proteins: From drug discovery to clinical trials. Signal Transduct. Target. Ther. 2023, 8, 335. [Google Scholar] [CrossRef]
- Ding, X.Y.; Ma, J.T.; Fan, T.; Issa, R.; Li, Y.X.; Weng, D.; Zhang, D.G.; Chen, Y.X. Inorganic nanoparticles-based strategies for the microbial detection in infectious diseases. Interdiscip. Med. 2024, 2, e20230045. [Google Scholar] [CrossRef]
- Ueki, N.; Lee, S.; Sampson, N.S.; Hayman, M.J. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat. Commun. 2013, 4, 2735. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, Z.; Liu, H.; Man, J.; Oladejo, A.O.; Ibrahim, S.; Wang, S.; Hao, B. Novel Drug Delivery Systems: An Important Direction for Drug Innovation Research and Development. Pharmaceutics 2024, 16, 674. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, C.; Wang, J.; Zhao, L.; Li, G. Research progress in nano-drug delivery systems based on the characteristics of the liver cancer microenvironment. Biomed. Pharmacother. 2024, 170, 116059. [Google Scholar] [CrossRef]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 293. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Allela, O.Q.B.; Pecho, R.D.C.; Jayasankar, N.; Rao, D.P.; Thamaraikani, T.; Vasanthan, M.; Viktor, P.; Lakshmaiya, N.; et al. Progressing nanotechnology to improve targeted cancer treatment: Overcoming hurdles in its clinical implementation. Mol. Cancer 2023, 22, 169. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Du, X.J.; Liu, J.; Sun, R.; Zhu, Y.H.; Wang, J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J. Control. Release 2015, 208, 14–24. [Google Scholar] [CrossRef]
- Park, J.E.; Park, J.; Jun, Y.; Oh, Y.; Ryoo, G.; Jeong, Y.S.; Gadalla, H.H.; Min, J.S.; Jo, J.H.; Song, M.G.; et al. Expanding therapeutic utility of carfilzomib for breast cancer therapy by novel albumin-coated nanocrystal formulation. J. Control. Release 2019, 302, 148–159. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Bozkurt, F.; Yilmaz, M.T.; Sircan-Kucuksayan, A.; Hanikoglu, A.; Ozben, T. A new combination strategy to enhance apoptosis in cancer cells by using nanoparticles as biocompatible drug delivery carriers. Sci. Rep. 2021, 11, 13027. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef]
- Esapa, B.; Jiang, J.; Cheung, A.; Chenoweth, A.; Thurston, D.E.; Karagiannis, S.N. Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers 2023, 15, 1845. [Google Scholar] [CrossRef]
- Theocharopoulos, C.; Lialios, P.P.; Samarkos, M.; Gogas, H.; Ziogas, D.C. Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines 2021, 9, 1111. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Lang, N.; Tarantelli, C.; Spriano, F.; Barraja, P.; Bertoni, F. Antibody-drug conjugates for lymphoma patients: Preclinical and clinical evidences. Explor. Target. Antitumor Ther. 2022, 3, 763–794. [Google Scholar] [CrossRef]
- Meng, H.; Nan, M.; Li, Y.; Ding, Y.; Yin, Y.; Zhang, M. Application of CRISPR-Cas9 gene editing technology in basic research, diagnosis and treatment of colon cancer. Front. Endocrinol. 2023, 14, 1148412. [Google Scholar] [CrossRef] [PubMed]
- Zischewski, J.; Fischer, R.; Bortesi, L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017, 35, 95–104. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, J.; Ge, S.; Lai, L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021, 8, 649896. [Google Scholar] [CrossRef]
- Mochizuki, S.; Okada, Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007, 98, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef]
- Koomen, J.M.; Haura, E.B.; Bepler, G.; Sutphen, R.; Remily-Wood, E.R.; Benson, K.; Hussein, M.; Hazlehurst, L.A.; Yeatman, T.J.; Hildreth, L.T.; et al. Proteomic contributions to personalized cancer care. Mol. Cell. Proteom. 2008, 7, 1780–1794. [Google Scholar] [CrossRef]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef]
- Regalado, C.R.; Balogh, M. MMP9: Link between neuropathy and colorectal cancer? Front. Mol. Biosci. 2024, 11, 1451611. [Google Scholar] [CrossRef]
- Sun, T.; Jiang, D.Q.; Zhang, L.; Su, Q.L.; Ma, W.L.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Dathathri, E.; Isebia, K.T.; Abali, F.; Lolkema, M.P.; Martens, J.W.M.; Terstappen, L.; Bansal, R. Liquid Biopsy Based Circulating Biomarkers in Metastatic Prostate Cancer. Front. Oncol. 2022, 12, 863472. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Slikker, W. Integrating artificial intelligence with bioinformatics promotes public health. Exp. Biol. Med. 2023, 248, 1905–1907. [Google Scholar] [CrossRef]
- Hu, F.; Wang, L.; Hu, Y.; Wang, D.; Wang, W.; Jiang, J.; Li, N.; Yin, P. A novel framework integrating AI model and enzymological experiments promotes identification of SARS-CoV-2 3CL protease inhibitors and activity-based probe. Briefings Bioinform. 2021, 22, bbab301. [Google Scholar] [CrossRef] [PubMed]
- Elend, L.; Jacobsen, L.; Cofala, T.; Prellberg, J.; Teusch, T.; Kramer, O.; Solov’yov, I.A. Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations. Molecules 2022, 27, 4020. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).