
Semisynthetic Flavonoids as GSK-3β Inhibitors: Computational Methods and Enzymatic Assay

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
2.2. Biological Testing
GSK-3β Enzymatic Assay
2.3. Computational Methods
Molecular Docking
2.4. Molecular Dynamics
2.5. Pharmacokinetic Prediction
3. Results
3.1. GSK-3β Inhibitory Activity of Flavonoid Derivatives
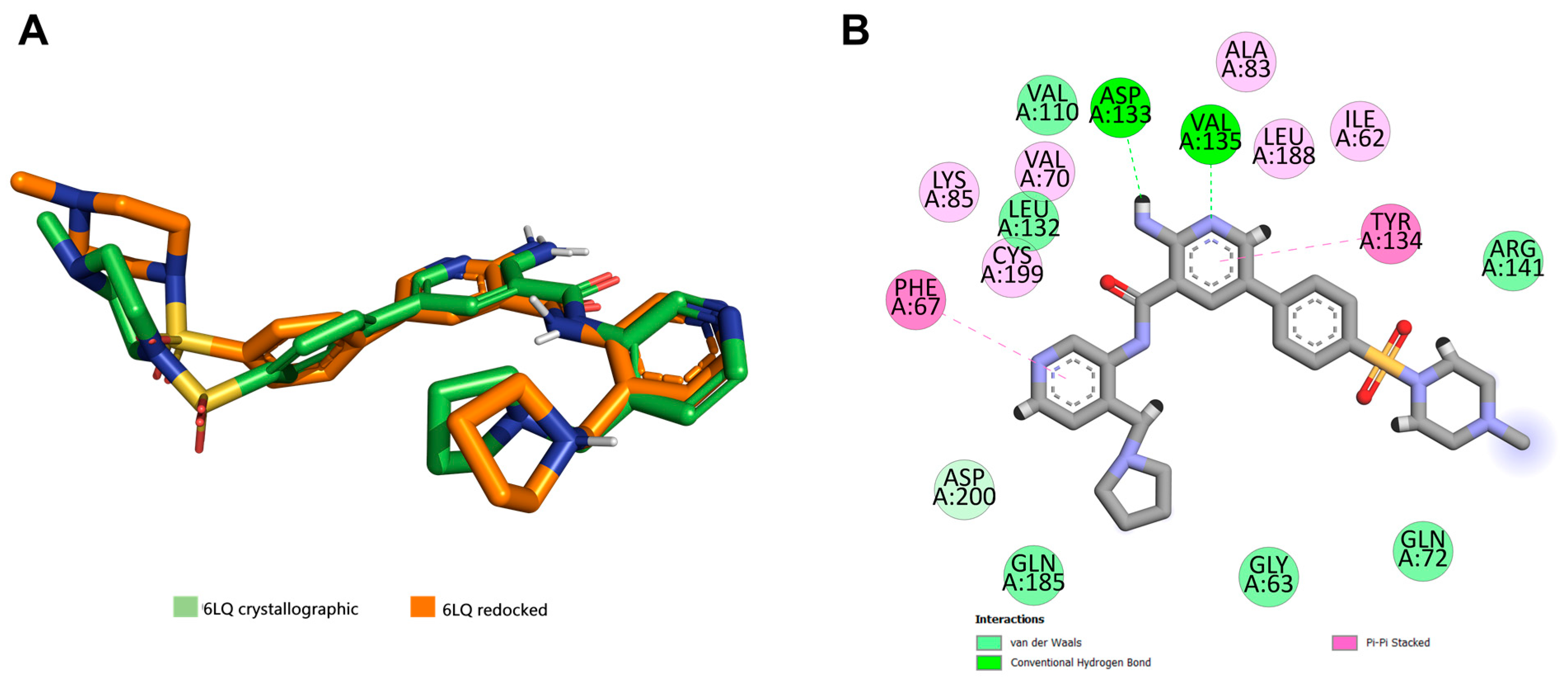
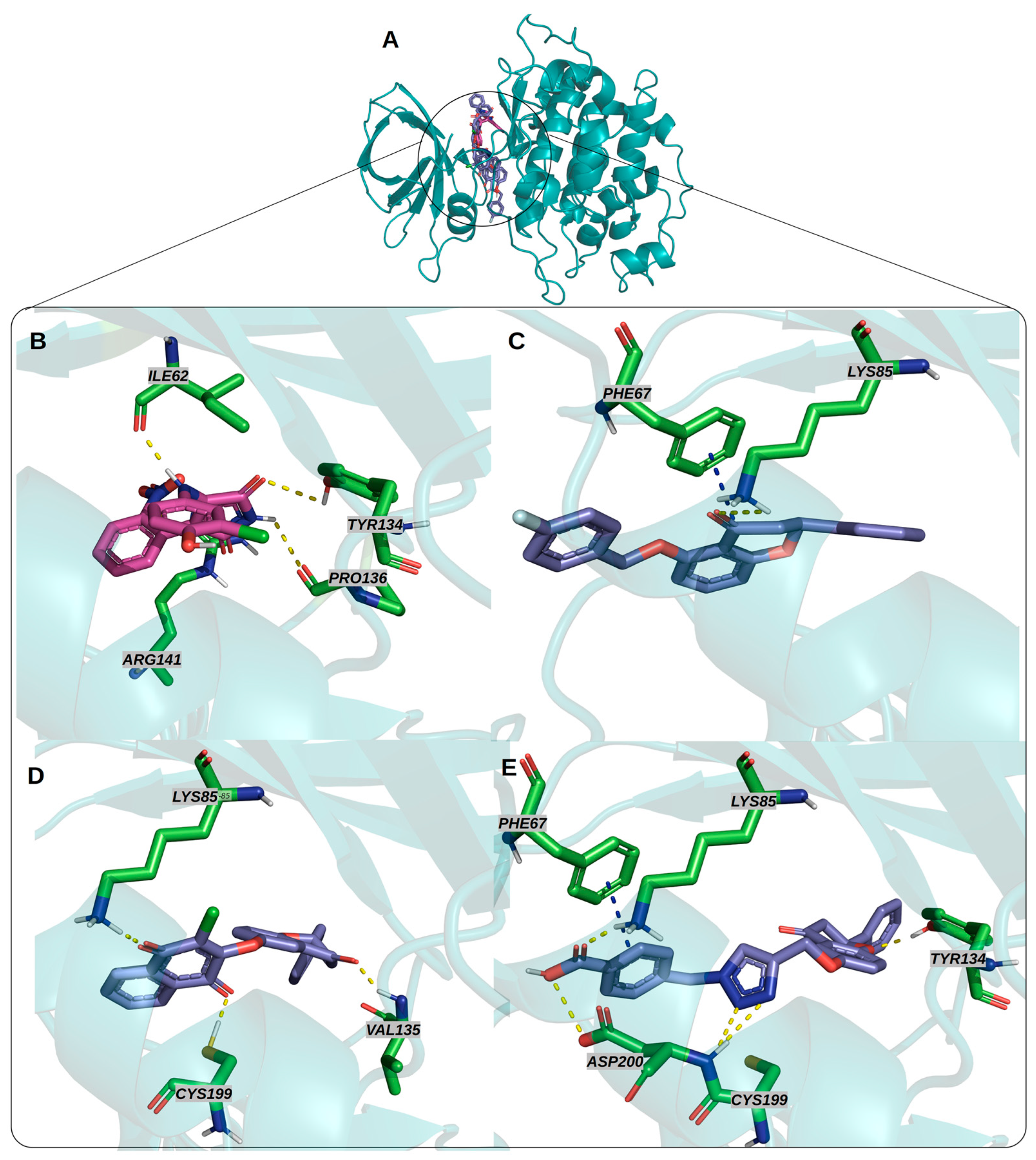
3.2. Docking Studies
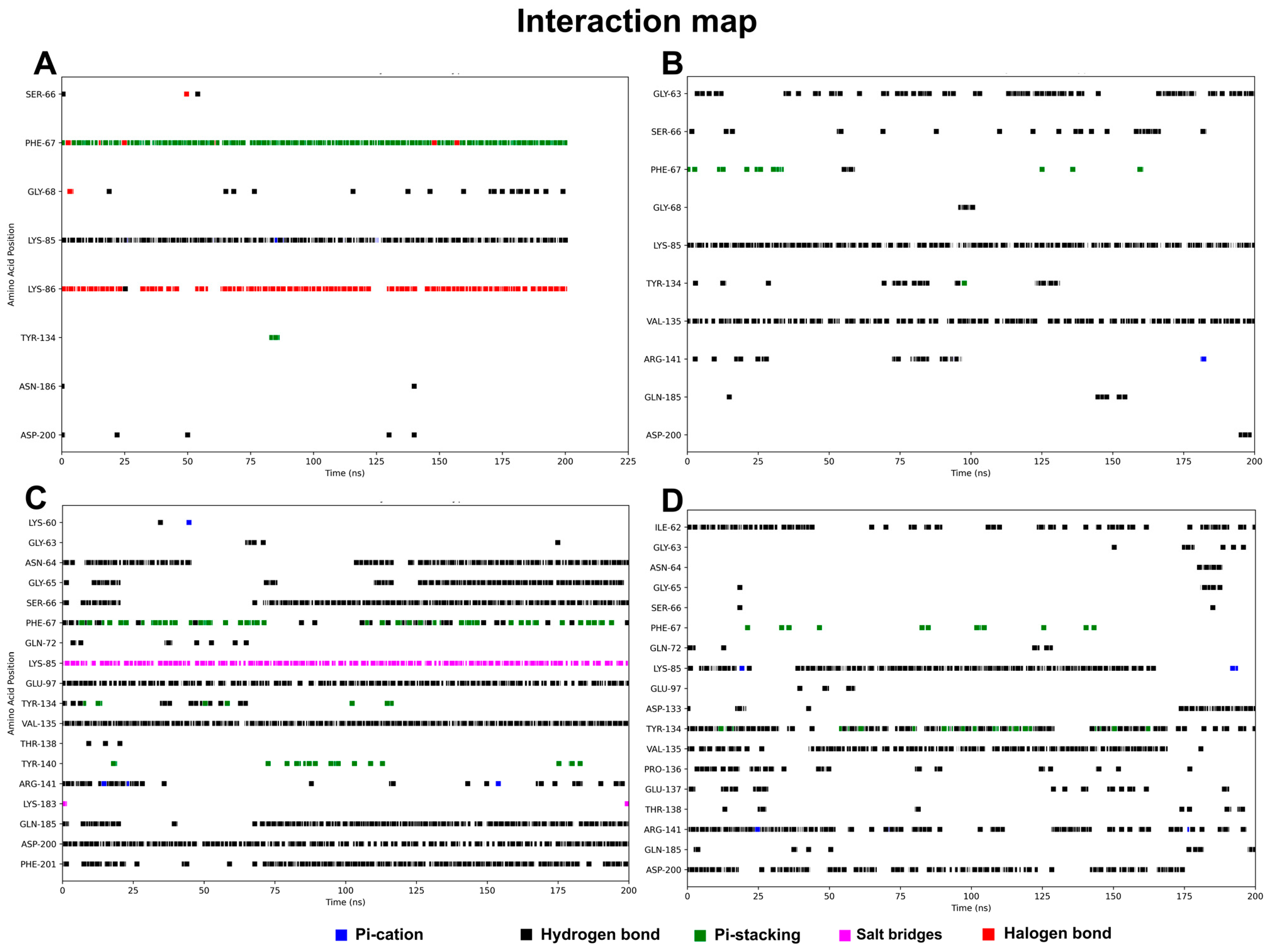
3.3. Molecular Dynamics Results
3.4. Free Energy Calculations
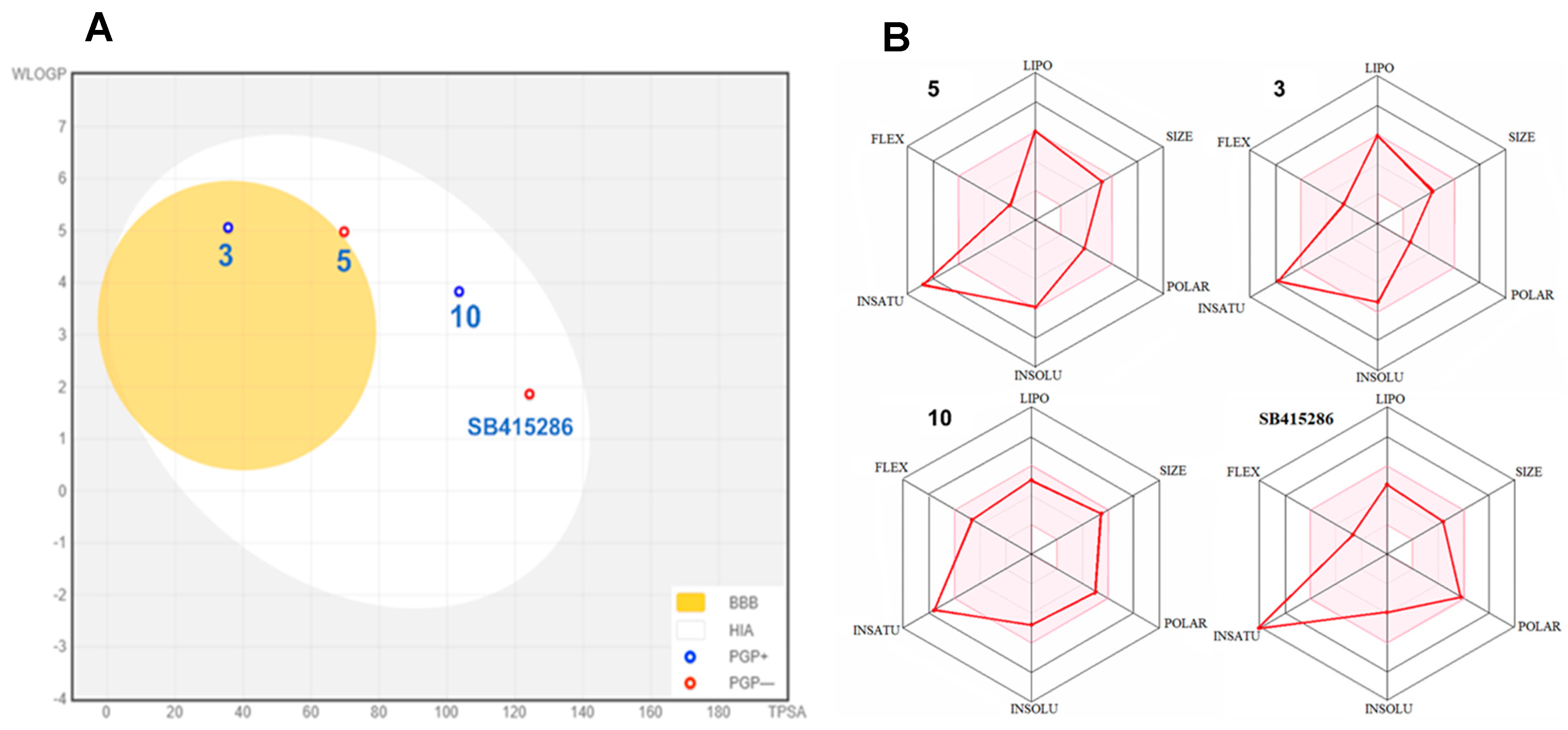
3.5. Pharmacokinetic Prediction of Selected Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kockeritz, L.; Doble, B.; Patel, S.; Woodgett, J.R. Glycogen synthase kinase-3-an overview of an over-achieving protein kinase. Curr. Drug Targets 2006, 7, 1377–1388. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef] [PubMed]
- Ugolkov, A.; Gaisina, I.; Zhang, J.-S.; Billadeau, D.D.; White, K.; Kozikowski, A.; Jain, S.; Cristofanilli, M.; Giles, F.; O’Halloran, T. GSK-3 inhibition overcomes chemoresistance in human breast cancer. Cancer Lett. 2016, 380, 384–392. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, P.; Larsson, S.; Holmgren, S.; Olsson, S.; Minthon, L.; Zetterberg, H.; Blennow, K.; Nilsson, S.; Sjölander, A. Genetic variants of GSK3B are associated with biomarkers for Alzheimer’s disease and cognitive function. J. Alzheimer’s Dis. 2015, 44, 1313–1322. [Google Scholar] [CrossRef]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef]
- De Groot, R.; Auwerx, J.; Bourouis, M.; Sassone-Corsi, P. Negative regulation of Jun/AP-1: Conserved function of glycogen synthase kinase 3 and the Drosophila kinase shaggy. Oncogene 1993, 8, 841–847. [Google Scholar] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef]
- Edderkaoui, M.; Chheda, C.; Soufi, B.; Zayou, F.; Hu, R.W.; Ramanujan, V.K.; Pan, X.; Boros, L.G.; Tajbakhsh, J.; Madhav, A. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology 2018, 155, 1985–1998.e5. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International (ADI): London, UK, 2015. [Google Scholar]
- Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Evans, D.A. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord. 2001, 15, 169–173. [Google Scholar] [CrossRef]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, Å.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels ofβ-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef]
- Sayas, C.L.; Ávila, J. GSK-3 and tau: A key duet in alzheimer’s disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Kumar, S.; Ma, B.; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999, 8, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Shatsky, M.; Wolfson, H.J.; Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Dolenc, J.; Mark, A.E. Molecular simulation as an aid to experimentalists. Curr. Opin. Struct. Biol. 2008, 18, 149–153. [Google Scholar] [CrossRef]
- LaConte, L.E.; Voelz, V.; Nelson, W.; Enz, M.; Thomas, D.D. Molecular dynamics simulation of site-directed spin labeling: Experimental validation in muscle fibers. Biophys. J. 2002, 83, 1854–1866. [Google Scholar] [CrossRef]
- Hole, K.L.; Williams, R.J. Flavonoids as an Intervention for Alzheimer’s disease: Progress and hurdles towards defining a mechanism of action. Brain Plast. 2020, 6, 167–192. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.-P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Mandelkow, E.-M.; Biernat, J.; Piwnica-Worms, H.; Mandelkow, E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993, 336, 417–424. [Google Scholar] [CrossRef]
- Drewes, G.; Lichtenberg-Kraag, B.; Döring, F.; Mandelkow, E.; Biernat, J.; Goris, J.; Doree, M.; Mandelkow, E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992, 11, 2131–2138. [Google Scholar] [CrossRef]
- Fernandez, J.W.; Rezai-Zadeh, K.; Obregon, D.; Tan, J. EGCG functions through estrogen receptor-mediated activation of ADAM10 in the promotion of non-amyloidogenic processing of APP. FEBS Lett. 2010, 584, 4259–4267. [Google Scholar] [CrossRef]
- Ribeiro, R.; Eloy, M.A.; Francisco, C.S.; Javarini, C.L.; Ayusso, G.M.; Da Rocha Fonseca, V.; Romão, W.; Regasini, L.O.; Araujo, S.C.; Almeida, M.O. Flavonoid derivatives targeting BCR-ABL kinase: Semisynthesis, Molecular dynamic simulations and Enzymatic inhibition. Curr. Top. Med. Chem. 2021, 21, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.; Hess, B.; Lindahl, E. GROMACS 2022.3 Source Code. 2022. Available online: https://zenodo.org/records/7037338 (accessed on 27 February 2025).
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bahrami, H.; Salehabadi, H.; Nazari, Z.; Amanlou, M. Combined Virtual Screening, DFT Calculations and Molecular Dynamics Simulations to Discovery of Potent MMP-9 Inhibitors. Lett. Drug Des. Discov. 2018, 16, 892–903. [Google Scholar] [CrossRef]
- Bernetti, M.; Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Britto, K.B.; Francisco, C.S.; Ferreira, D.; Borges, B.J.P.; Conti, R.; Profeti, D.; Rodrigues, L.R.; Lacerda, V.; Morais, P.A.B.; Borges, W.S. Identifying new isatin derivatives with gsk-3β inhibition capacity through molecular docking and bioassays. J. Braz. Chem. Soc. 2020, 31, 476–487. [Google Scholar] [CrossRef]
- Atkins, R.J.; Stylli, S.S.; Luwor, R.B.; Kaye, A.H.; Hovens, C.M. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wu, Y.; Xu, L.; Jin, J. Theoretical Studies on the Selectivity Mechanisms of Glycogen Synthase Kinase 3β (GSK3β) with Pyrazine ATP-competitive Inhibitors by 3DQSAR, Molecular Docking, Molecular Dynamics Simulation and Free Energy Calculations. Curr. Comput.-Aided Drug Des. 2019, 16, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Arfeen, M.; Bhagat, S.; Patel, R.; Prasad, S.; Roy, I.; Chakraborti, A.K.; Bharatam, P.V. Design, synthesis and biological evaluation of 5-benzylidene-2-iminothiazolidin-4-ones as selective GSK-3β inhibitors. Eur. J. Med. Chem. 2016, 121, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Ma, Z.; Meng, X.; Li, W.; Liang, S.; Chen, X. Exploration of the selective binding mechanism of GSK3β via molecular modeling and molecular dynamics simulation studies. Med. Chem. Res. 2020, 29, 690–698. [Google Scholar] [CrossRef]
- de Paula, H.; Bolsoni, C.S.; de Souza, F.F.; Fonseca, V.D.R.; Romão, W.; de Sairre, M.I.; Honorio, K.M.; Lacerda Jr, V.; Morais, P.A.B. Semisynthetic p-Coumaric Acid Derivatives as Lead Dual Inhibitors Against DPP-IV and GSK-3β for Antidiabetic Therapy. Chem. Biol. Drug Des. 2024, 104, e70016. [Google Scholar] [CrossRef]
- Hua, L.; Anjum, F.; Shafie, A.; Ashour, A.A.; Almalki, A.A.; Alqarni, A.A.; Banjer, H.J.; Almaghrabi, S.A.; He, S.; Xu, N. Identifying promising GSK3β inhibitors for cancer management: A computational pipeline combining virtual screening and molecular dynamics simulations. Front. Chem. 2023, 11, 1200490. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | Compound |
|---|---|
| 29.08% [a] | 1 |
| 1.211 | 2 |
| 0.467 | 3 |
| 5.941 | 4 |
| 0.428 | 5 |
| 61.89 | 6 |
| 1.589 | 7 |
| 7.476 | 8 |
| 1.426 | 9 |
| 0.376 | 10 |
| 0.655 | 11 |
| 2.027 | 12 |
| 4.52% [a] | 13 |
| 0.959 | 14 |
| 0.607 | 15 |
| 2.752 | 16 |
| 0.096 | SB415286 |
| SEM * | ΔGMM/PBSA | Complex |
|---|---|---|
| ±0.12 | −31.08 | GSK3β-3 |
| ±0.44 | −28.43 | GSK3β-5 |
| ±0.83 | −50.10 | GSK3β-10 |
| ±0.69 | −19.88 | GSK3β-SB415286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Paula, H.; Souza, F.; Ferreira, L.; Silva, J.A.B.; Ribeiro, R.; Vilachã, J.; Emery, F.S.; Lacerda, V., Jr.; Morais, P.A.B. Semisynthetic Flavonoids as GSK-3β Inhibitors: Computational Methods and Enzymatic Assay. Targets 2025, 3, 13. https://doi.org/10.3390/targets3020013
de Paula H, Souza F, Ferreira L, Silva JAB, Ribeiro R, Vilachã J, Emery FS, Lacerda V Jr., Morais PAB. Semisynthetic Flavonoids as GSK-3β Inhibitors: Computational Methods and Enzymatic Assay. Targets. 2025; 3(2):13. https://doi.org/10.3390/targets3020013
Chicago/Turabian Stylede Paula, Heberth, Fernanda Souza, Lara Ferreira, Jéssica A. B. Silva, Rayssa Ribeiro, Juliana Vilachã, Flávio S. Emery, Valdemar Lacerda, Jr., and Pedro A. B. Morais. 2025. "Semisynthetic Flavonoids as GSK-3β Inhibitors: Computational Methods and Enzymatic Assay" Targets 3, no. 2: 13. https://doi.org/10.3390/targets3020013
APA Stylede Paula, H., Souza, F., Ferreira, L., Silva, J. A. B., Ribeiro, R., Vilachã, J., Emery, F. S., Lacerda, V., Jr., & Morais, P. A. B. (2025). Semisynthetic Flavonoids as GSK-3β Inhibitors: Computational Methods and Enzymatic Assay. Targets, 3(2), 13. https://doi.org/10.3390/targets3020013