Abstract
While originally intending to explore the venom gland microbiome of the desert hairy scorpion Hadrurus arizonensis Ewing, 1928, nanopore sequencing serendipitously recovered complete mitochondrial genomes for this iconic arachnid. Phylogenetic analysis of these high-quality genomes places Hadrurus as sister to Uroctonus, in agreement with some phylogenomic hypotheses. Additionally, we reveal significant genetic variation among individuals from the same population, highlighting the potential of mitogenomics for population genetics and phylogeography. This study showcases the effectiveness and affordability of nanopore sequencing for research with non-model organisms, opening new avenues for investigating arachnid biodiversity, evolution, and biogeography.
1. Introduction
Mitochondrial genomes, or mitogenomes, of non-model organisms offer a powerful window into evolutionary relationships and are useful across timescales, from ancient divergences to nuanced intraspecific processes. Advancements in DNA sequencing technology, coupled with accessible bioinformatics tools, have led to an increase in mitogenomic studies, fueling phylogenetic analyses across diverse taxonomic levels. Recent examples range from elucidating relationships within an orthopteran species complex [1] to clarifying the phylogeny at the family-level for an infraorder of deep-sea crustaceans [2]. Yet, the potential of these tools often extends beyond their initial targets as unanticipated discoveries in the bycatch of sequencing data [3,4]. Here, we present a fascinating case in point, presenting the first mitogenomes of the iconic desert hairy scorpion, Hadrurus arizonensis Ewing, 1928 (Figure 1), which were uncovered not while seeking them but as bycatch during an investigation into the microbiome of its venom-containing telson.
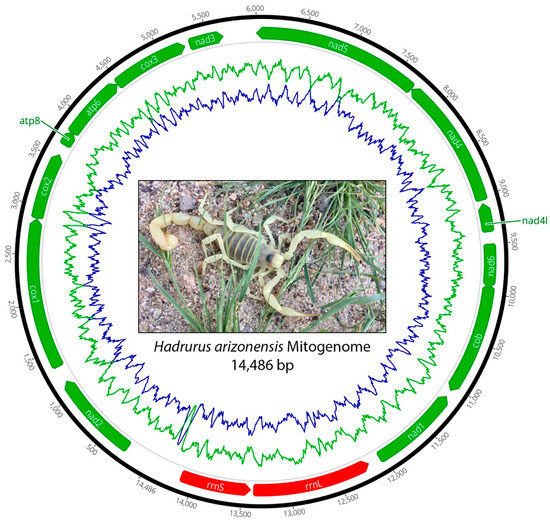
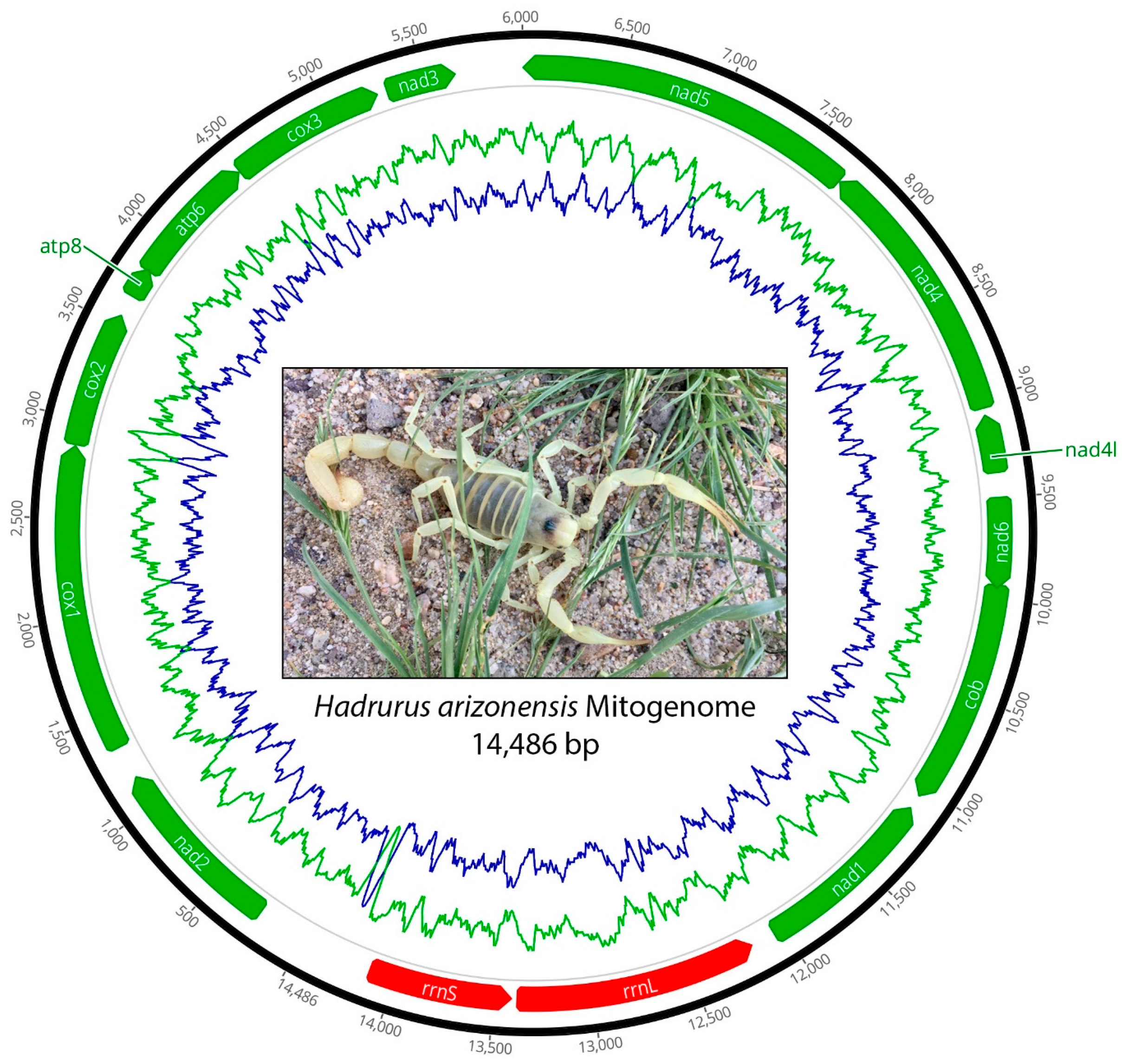
Figure 1.
Circular map of the mitochondrial genome of Hadrurus arizonensis showing the arrangement of PCGs (green) and rRNA genes (red). The proportion of guanine and cytosine (GC) nucleotides is depicted by the green line, and the proportion of adenine and thymine (AT) nucleotides is shown by the blue line plotted along the mitochondrial genome.
The phylogenetic position of Hadrurus spp. and its sister genus Hoffmannihadrurus, together forming the family Hadruridae, has remained enigmatic within the scorpion tree of life. Initially classified within Caraboctonidae, subsequent phylogenomic analyses elevated the subfamily Hadrurinae to the family Hadruridae of the superfamily Hadruroidea [5,6]. However, the precise placement of Hadruroidea has remained elusive, forming an unstable relationship with Uroctonus (currently incertae sedis), Scorpionoidea, and Vaejovoidea [6,7,8].
Our investigation, initially focused on characterizing venom gland microbiomes using the MinION sequencing platform by Oxford Nanopore Technologies (ONT: Oxford, UK), serendipitously yielded high-quality complete mitogenomes for seven Hadrurus arizonensis specimens. These unexpected genetic data presented a unique opportunity to investigate intraspecific genetic diversity and explore the evolutionary history of Hadrurus. We focused on achieving four key objectives: (1) assemble and annotate the first complete mitogenomes of H. arizonensis, (2) clarify the phylogenetic position of Hadruroidea within the scorpion tree of life, (3) characterize intraspecific genetic diversity across genes, and (4) showcase the potential of nanopore sequencing for studies of non-model organisms like scorpions.
The fulfillment of these objectives will not only unveil novel insights into the genetics of Hadrurus but also underscore the potential for important discoveries from the bycatch of sequencing data. Our work highlights the often serendipitous nature of scientific discovery, as valuable revelations can be hidden beneath the surface of seemingly focused inquiries.
2. Materials and Methods
Seven Hadrurus arizonenesis were collected from just east of Cattail Cove State Park in Arizona, USA (34.356159°, −114.147638°), for ongoing investigations of scorpion tissue and venom microbiomes. Specimens were identified based on morphology, habitat, geographical location, and by submitting COI sequences to the NCBI Basic Local Alignment Search Tool (BLAST). A modified Epicentre Master Pure DNA Purification (Lucigen, Middleton, WI 53562, USA) protocol was used to extract DNA from frozen scorpion telsons, as seen in Shimwell et al. [9]. Telson tissue was cut into small pieces, mixed with 300 μL Tissue and Cell Lysis Buffer and 50 μg Proteinase K, prior to being homogenized with a sterile homogenizer to form a liquid suspension. Samples were incubated overnight in a 56 °C shaking water bath. Samples were iced and mixed with 175 μL of MPC Protein Precipitation Reagent and then centrifuged at 10,000× g for 10 min at 4 °C. The DNA was precipitated at −20 °C for 30 min or overnight by adding 500 μL of cold (−20 °C) isopropanol. Samples were warmed to 4 °C and centrifuged at 10,000× g for 10 min at 4 °C to pellet the DNA. Supernatants were decanted, and the pellets were washed twice with 70% ethanol. The pellets were air-dried for 30 min and resuspended in 50 μL of TE buffer (10 mM Tris, 1 mM EDTA, pH 8). Final concentrations of DNA ranged from 216 to 1676 ng/μL.
DNA from telson tissues was used to generate genomic and metagenomic libraries using a Native Barcoding Kit 24 V14 for ligation sequencing of gDNA (SQK-NBD114.24). DNA repair and end-prep was conducted using NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs Inc., Ipswich, MA, USA) with a 5 min incubation at 20 °C for 5 min followed by 65 °C for 5 min. Samples were purified by incubation with AMPure XP beads (Beckman Coulter, Indianapolis, IN, USA) on a hula mixer, followed by separation on a magnetic rack. Sample barcodes were ligated to DNA fragments using Blunt/TA Ligase Master Mix (New England Biolabs Inc., Ipswich, MA, USA) with a 20 min incubation at room temperature, followed by bead purification. Samples were then pooled, and sequencing adapters were ligated using NEBNext Quick Ligation Reaction Buffer and Quick T4 DNA Ligase (New England Biolabs Inc., Ipswich, MA, USA) with a 20 min incubation at room temperature, followed by bead purification. DNA quantification was conducted on a Qubit 4 fluorometer (Invitrogen, Carlsbad, CA, USA) throughout the protocol.
The pooled library was combined with Library Beads (LIBs) and Sequencing Buffer (SB) and loaded into a new and primed FLO-MIN114 flow cell. Sequencing of the flow cell was conducted using a MinION Mk1C and MinKNOW v.3.6.5. Sequencing ran for 72 h. After sequencing, the raw reads were basecalled, demultiplexed, and converted from fast5 to fasta files in Guppy v.6.4.2 using high-accuracy (HAC) basecalling.
Demultiplexed nanopore sequencing reads were imported into Geneious Prime v2023.0.4 (Biomatters Ltd., New Zealand). All ends were trimmed to a quality score of 10 or greater with BBDuk and reads with minimum lengths between 50 and 500 bp were discarded per sample. Reads from the H. arizonensis sample with the most data were mapped to the complete mitogenome of Uroctonus mordax Thorell, 1876 (NC_010782) as a reference using the Highest Sensitivity option and five fine-tuning iterations. A consensus sequence was generated and aligned to 17 available scorpion mitogenomes. The H. arizonensis consensus sequence clearly aligned more closely with those of parvorder Iurida, so samples belonging to parvorder Buthida were removed, and samples were realigned. The resulting alignment was used to manually inspect the consensus sequence for indels, which are the most common error in ONT data [10]. Geneious Prime was used to determine open reading frames to make sure divergent nuclear mitochondrial segments (NUMTs) did not influence the result. No evidence of NUMTs was found. The resulting H. arizonensis mitogenome was then used as a reference to assemble reads into contigs and generate consensus sequences for the remaining six samples. Mitogenomes were annotated using the MITOS web server [11]. Gene boundaries were checked manually in Artemis 17.0 [12], as well as in Geneious Prime via manual inspection and comparison to published and annotated mitogenomes of Iurida scorpions. The seven annotated mitogenomes were deposited in GenBank under accession numbers OR995570–OR995576.
For each rRNA gene and protein-coding gene (PCG), we used DnaSP v.6.2.01 [13] to determine the number of variable sites, number of haplotypes, haplotype diversity, and nucleotide diversity. We visually assessed the diversity of these genes by constructing simple (ε = 0) median-joining networks using POPART v.1.7.2 [14]. This approach prioritizes short connections by merging minimum spanning trees into a single network, making it well suited for revealing patterns of intraspecific haplotype diversity.
The usefulness of mitogenomic data in phylogenetic reconstruction has been tested in other chelicerates [15,16]. To address the phylogenetic position of Hadruridae (or Hadruroidea) using mitochondrial genomic data, we leveraged 11 scorpion mitogenomes available (see below), plus the seven mitogenomes generated here, and constructed matrices using the amino acid (m1) and nucleotide (m2) sequences of the 13 protein-coding genes in scorpion mitochondria. Species used in this study included two buthids (Centruroides vittatus (Say, 1821) (NC_037222) and Olivierus martensii (Karsch, 1879) (NC_009738)) and nine iurids. From these iurids, our sample included Scorpiops tibetanus Hirst, 1911 (Scorpiopidae, NC_053569), Heterometrus longimanus (Herbst 1800) (Scorpionidae, KR190462), U. mordax (insertae sedis, NC_010782), and six species of family Vaejovidae (Chihuahuanus coahuilae (Williams, 1968) (BK061405), Konetontli acapulco (Armas & Martin-Frias, 2001) (BK061399), Kuarapu purhepecha Francke & Ponce-Saavedra, 2010 (BK061401), Paravaejovis spinigerus (Wood, 1863) (BK061400), Vaejovis smithi Pocock, 1902 (KX520650), Vaejovis mexicanus C. L. Koch, 1836 (BK061398)). The amino acid and nucleotide sequences from each of 13 PCGs were aligned using MAFFT v.7.4 (--auto –anysymbol –quiet) [17] and trimmed using trimAl v.1.2 (-fasta -gappyout) [18]. Model selection, maximum likelihood phylogenetic analysis, and nodal support of each locus were performed using IQTREE v. 2.0.6 [19] implementing ModelFinder Plus [20] and ultrafast bootstraptraping [21] with the following command: iqtree2 -s LOCUS.aligned.fasta -m MFP -B 1000. Then, these loci (amino acids and nucleotides) were concatenated and analyzed using IQTREE implementing the model selected in each partition and 1000 ultrafast bootstrap replicates (-p Partition_file.txt -B 1000).
We calculated the length of the gene and intergenic regions in the 12 scorpion species and plotted them next to the phylogeny using the R packages phytools [22], ggplot2 [23], and ggtree [24].
3. Results
Nanopore sequencing of the seven samples yielded 5.9 million reads that were basecalled and passed initial filtering in Guppy (Table 1). Additional quality filtering and the removal of short reads with BBDuk reduced the number of reads per sample to 236,000 to 1,760,000, with an average of 842,857. Of these, 709 to 7838 mapped to reference sequences, with an average of 2537 per sample. Coverage ranged from 14 to 227 across the seven assemblies.

Table 1.
Nanopore read data used for assembly of Hadrurus arizonensis mitogenomes. Passed reads indicate the number of reads after filtering with Guppy. Reads were then trimmed in Geneious Prime using BBDuk and mapped to reference mitogenomes (see Methods). Coverage is the number of reads that align to each base pair in the reference sequences.
The mitochondrial genome of H. arizonensis is a circular double-stranded DNA molecule with a length of 14,486 bp (Figure 1). The mitogenome comprises 13 PCGs (ATP6, ATP8, COI-II, NAD1-6, and NAD4L), 24 tRNA genes, and 2 rRNA genes (16S and 18S). Of these 38 genes, 23 are encoded on the leading strand and 15 are on the lagging strand. The AT content ranges from 69.64% to 69.86%, with a mean of 69.69%.
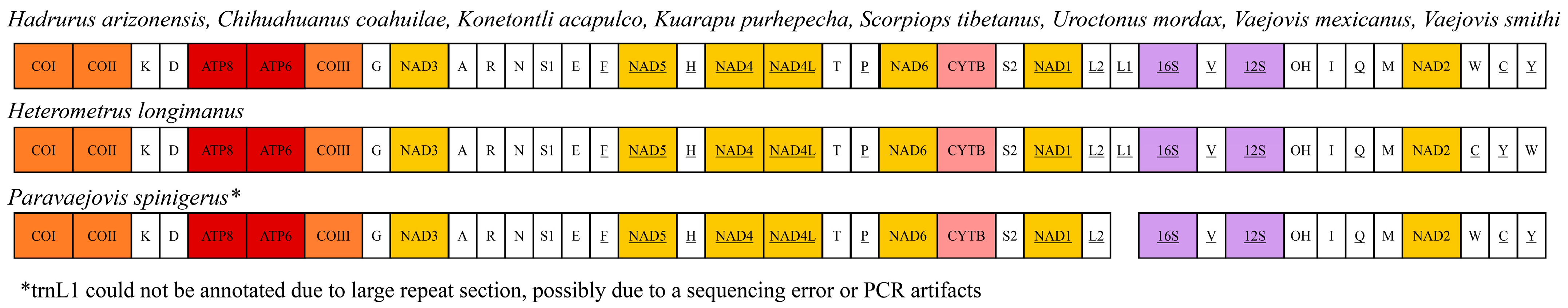
The gene order of the H. arizonensis mitogenome is identical to that of scorpions in the family Vaejovidae (C. choahuilae, K. acapulco, K. purhepecha, V. mexicanus, and V. smithi), Scorpiopidae (S. tebetanus) and U. mordax (Figure 2). The gene order in the mitogenome of H. arizonensis was largely identical to that of both P. spinigerus (whose mitogenome is partially sequenced) and H. longimanus. However, the last three transfer RNA (tRNA) genes differed from H. longimanus in their arrangement. In H. arizonensis, these tRNAs were ordered tryptophan (W), cysteine (C), and tyrosine (Y), while in H. longimanus, they were ordered cytosine (C), tyrosine (Y), and tryptophan (W).
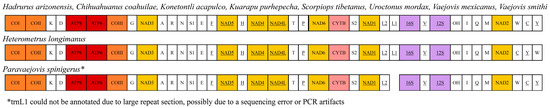
Figure 2.
Comparison of mitochondrial gene arrangement across Hadrurus arizonensis and other Iurida scorpions with sequenced mitogenomes. Full gene names are provided in Table 2.
Analysis of the seven mitogenomes revealed moderate levels of polymorphism and diversity across 15 protein-coding and ribosomal genes (Table 2). The 12S and 16S rRNA genes exhibit the lowest variation, with 1.11% and 1.92% of sites polymorphic, respectively. Protein-coding genes displayed higher levels, ranging from 2.24% in COII to 4.02% in NAD3. Haplotype diversity followed a similar pattern, with rRNAs showing lower values (0.714 and 0.952) compared to that of most protein-coding genes (0.810 to 1.000). Nucleotide diversity also trended higher in protein-coding genes, reaching 0.01286 in NAD3 and ranging from 0.00733 to 0.01092 in other genes.
Table 2.
Diversity metrics for two rRNA genes and 13 protein-coding genes from seven Hadrurus arizonensis mitogenomes.
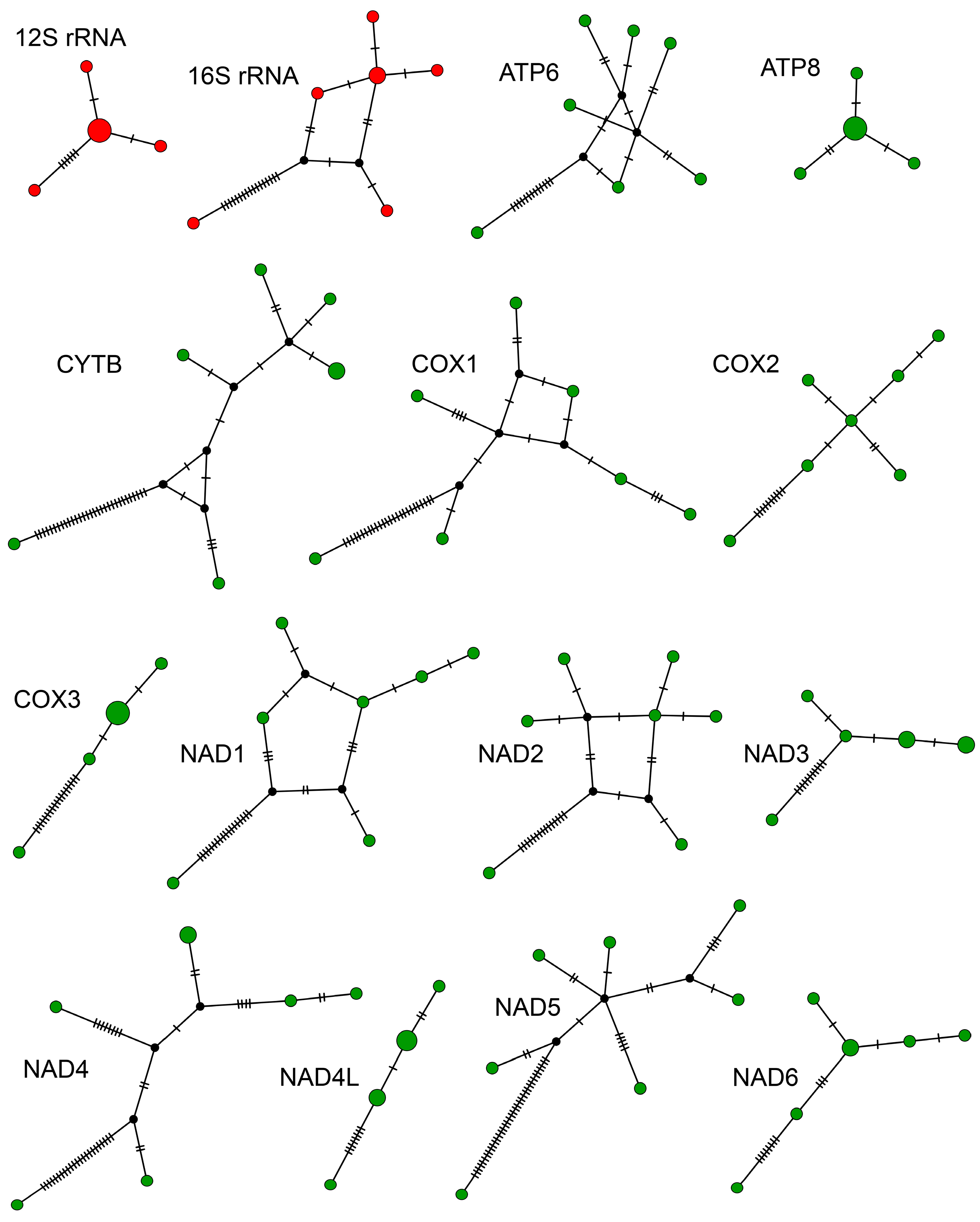
Haplotype networks for rRNA and protein-coding genes reveal a genetic structure (Figure 3) indicative of a complex phylogeographical history. The simplest networks consisted of four haplotypes arranged linearly (COIII and NAD4L) or with a central haplotype with remaining haplotypes attached (12S and ATP8). Six genes produced reticulated haplotype networks, connecting six to seven haplotypes, and contained two to five hypothetical haplotypes (16S, ATP6, CYTB, COI, NAD1, and NAD2). The remaining genes (COI, NAD3, NAD4, NAD5, and NAD6) produced non-reticulated networks with five to seven haplotypes and zero to three hypothetical haplotypes. In all networks, a single haplotype differed conspicuously from the other six haplotypes by a number of unique mutations, indicative of two different mitochondrial clades occurring at the same site.
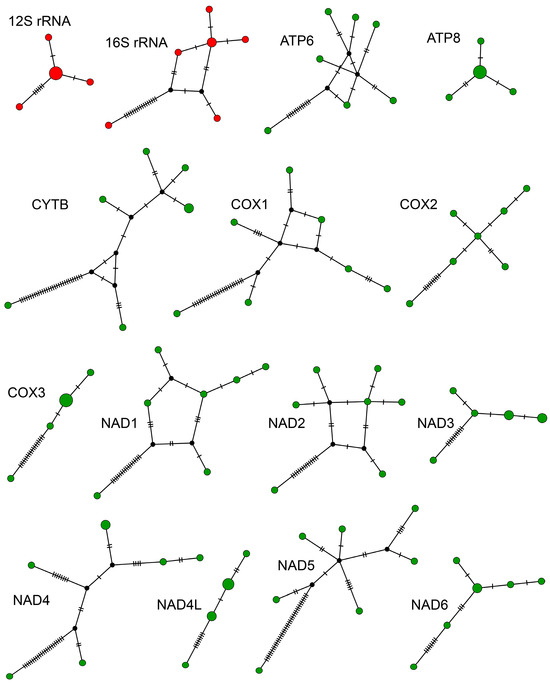
Figure 3.
Haplotype networks showing amount of genetic diversity among genes coding for proteins (green dots) and ribosomes (red dots) from seven Hadrurus arizonensis mitogenomes. Black dots represent hypothetical haplotypes.
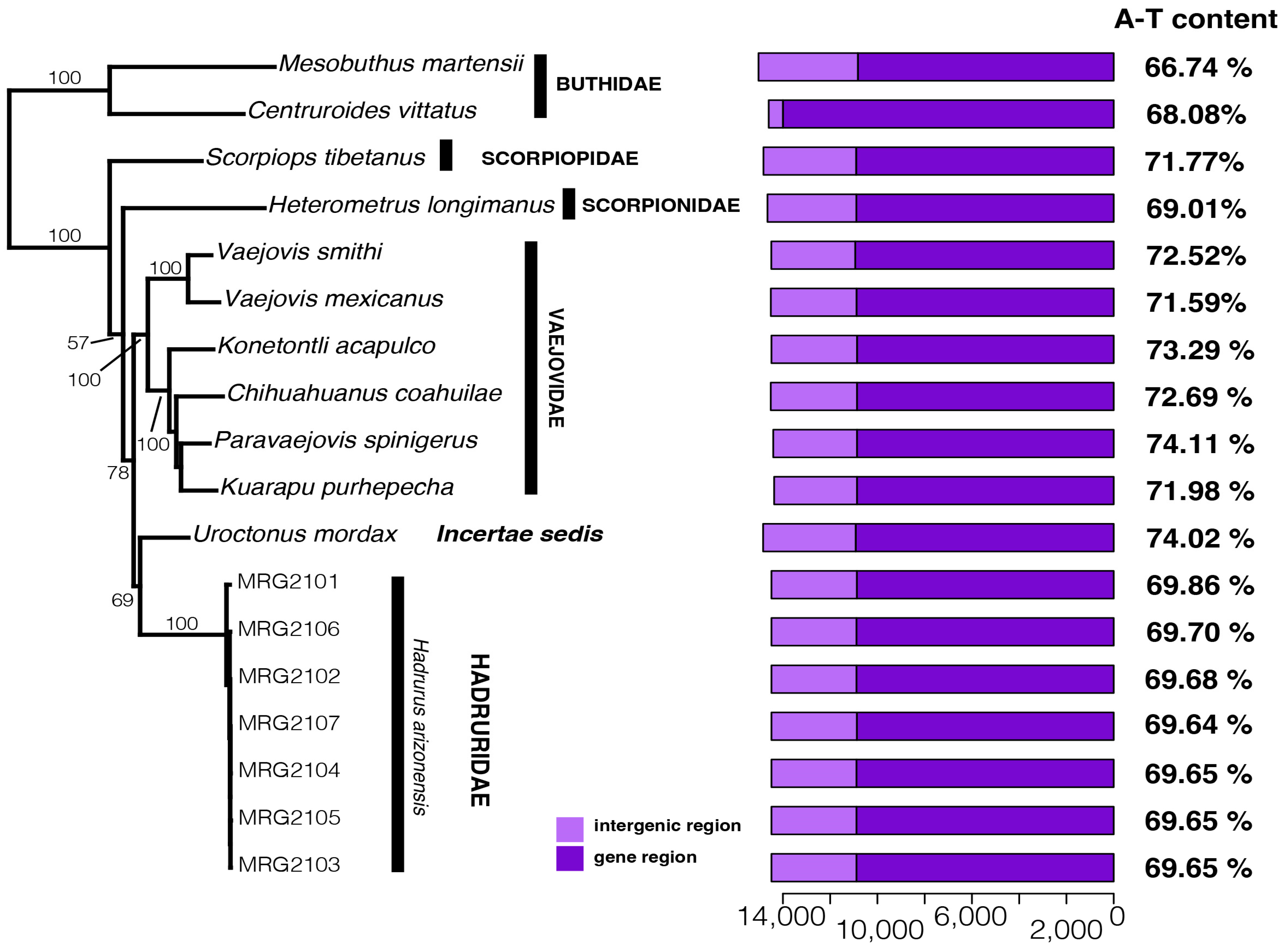
Maximum likelihood phylogenetic analysis of Iurida mitogenomes, rooted with two Buthida mitogenomes, produced a phylogeny with strong bootstrap support for most nodes (Figure 4). The seven H. arizonensis individuals were grouped together as a monophyletic group that recently shared a common ancestor. These samples formed a divergent lineage that is sister to U. mordax with low support. The H. arizonensis and U. mordax clade was sister to a clade containing members of the family Vaejovidae: V. smithi, V. mexicanus, K. acapulco, C. coahuilae, P. spinigerus, and K. purhepecha. Within the vaejovid clade, V. smithi and V. mexicanus formed a clade sister to a clade containing K. acapulco, C. coahuilae, P. spinigerus, and K. purhepecha. Scorpiops tibetanus and H. longimanus were recovered as sister taxa to the clade containing the superfamilies Vaejovoidea, Hadruroidea, and genus Uroctonus.
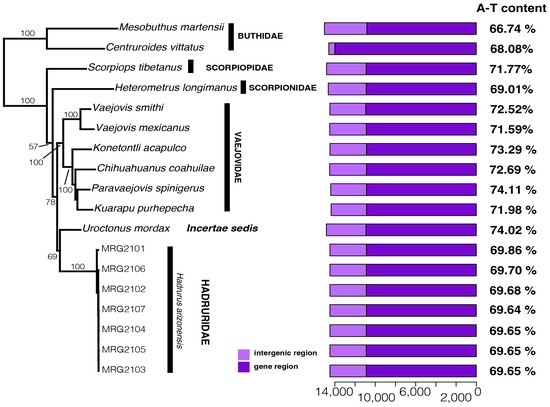
Figure 4.
Left: Maximum likelihood tree (log-likelihood score of −81,800.5705) based on the concatenated nucleotide sequences of 13 mitochondrial orthologs (11,114 bp) of 12 scorpion species (Hadrurus arizonensis is represented by seven specimens). Numbers on/near the nodes represent ultrafast bootstrap support values. Right: bar plots indicating the length (bp) of the gene and intergenic regions.
4. Discussion
The present study describes the first complete mitogenomes for the iconic desert hairy scorpion, H. arizonensis, using nanopore sequencing. Our high-quality mitogenomes reveal not only substantial intraspecific genetic diversity in mitochondrial genes in a population of scorpions, but also hint at an interesting phylogenetic position for Hadruridae within Iurida. These results highlight the potential of nanopore sequencing for fast and relatively inexpensive mitogenomic sequencing of non-model organisms like scorpions.
Mitochondrial Gene Diversity: As far as we know, this is the first study analyzing intraspecific variation across all complete ribosomal RNA (rRNA) genes and protein-coding genes (PCGs) from scorpion mitochondria. As expected, rRNA genes showed less diversity than PCGs. Traditionally, scorpion phylogenetics has relied on the slower evolving 16S rRNA gene and the faster changing COI gene [25,26,27]. While COI is a popular DNA barcoding tool and is often used for intraspecific scorpion studies, our results reveal that other mitochondrial genes are even more variable. This is evident in the highly structured and diverse haplotype networks generated from our mitogenome data (Figure 3). Among these, genes like CYTB, NAD4, and NAD5 stand out due to their higher percentage of variable sites and relatively larger size relative to most other mitochondrial genes (Table 2), making them potentially more informative markers for intraspecific studies. Despite their potential utility, however, COI remains the established choice for many intraspecific scorpion analyses.
Previous analysis of 256 COI sequences by Graham et al. [28] uncovered complex phylogeographic patterns in H. arizonensis, revealing six main clades linked to the Pleistocene. Interestingly, BLAST searching the COI gene sequences from our mitogenomes against the NCBI database revealed that our seven specimens belong to two of the six clades. Specifically, one sample aligns with a northern clade distributed along the Lower Colorado River Valley (Group III), while the remaining six belong to a larger, widespread clade found across the central part of the species’ range in the Mojave and Sonoran deserts (Group I). Notably, our sampling site was not included in the previous study, and our findings extend the southern distribution of mitochondrial Group III by roughly 40 km.
Phylogenetic Relationships: Our phylogenetic analysis using Iurida mitogenomes places H. arizonensis of the family Hadruridae as a sister lineage to U. mordax (incertae sedis; Figure 4). Together, Hadruridae and Uroctonus formed a clade sister to Vaejovidae. Recent studies that analyzed transcriptome and ultra-conserved element (UCE) data generally placed superfamilies Scorpionoidea and Vaejovoidea as sister taxa [6,7,8]. As such, we expected the six vaejovid species in our analysis (superfamily Vaejovoidea) to form a clade with Heterometrus of Scorpionidae (superfamily Scorpionoidea). Intriguingly, in our phylogeny, Heterometrus instead formed a lineage sister to a clade made up of six Vaejoviodea spp., U. mordax, and H. arizonensis. What could have caused this discrepancy?
Mitochondrial genes have a higher mutation rate than that of most nuclear genes, making them powerful markers to study recent diversification events, but sometimes they are too saturated with homoplasious mutations to resolve deeper relationships. Diversification among the Iurida scorpion families in this study has been estimated to have occurred in the Cretaceous period [29], perhaps a timeframe too old for mitogenome data. In insects, mitogenomes have been found to be useful for resolving low-level phylogenetic relationships but to be inadequate at the subfamily-level and above [30]. In addition, our phylogenetic analysis was admittedly limited due to the availability of mitogenomes for only nine Iurida scorpions other than our seven for H. arizonensis.
The conflicting results from different studies emphasize the complexity of scorpion phylogenetics and the need for further investigation. Our mitogenomic data provide a valuable piece of the puzzle, but data from additional taxa and nuclear genomic regions may help resolve the position of Hadruridae within the scorpion tree of life.
Potential of Nanopore Sequencing: Results from this study demonstrate the effectiveness of nanopore sequencing for mitogenomic studies in non-model organisms like scorpions. Our original intention for ONT sequencing was to identify bacterial DNA from genomic scorpion DNA, isolated from venom glands, without the prior need for PCR amplification. This would minimize inherent biases associated with PCR and allow us to capture longer reads compared to those of methods like Illumina short-read sequencing. Despite our focus on bacterial DNA, we were able to obtain high-quality mitochondrial genomes for all seven Hadrurus arizonensis individuals that were sequenced together on a single ONT flow cell. Given the depth of coverage for each individual (Table 1), a few more samples could probably have been added to the sequencing run. Excitingly, we suspect that even more samples could be included on the same flow cell if nanopore adaptive sampling (NAS) is leveraged. The NAS approach allows users to selectively enrich target sequences by rejecting reads that are not a close enough match to reference sequences [31]. For example, we could have used all other published scorpion mitogenomes as reference sequences and had the sequencer reject reads that were less than a 70% match, resulting in more mitochondrial reads. Since our original intention was to sequence microbiome data (bacterial DNA) we did not use NAS but were still able to generate enough mitochondrial reads to assemble high-quality and complete mitochondrial genomes. This is particularly encouraging for studies of poorly characterized species where reference genomes are unavailable, as prior information, like conserved regions for primer design, are not even necessary.
Our nanopore sequencing approach, combined with NAS, will be particularly useful for generating phylogenetic data for low or moderately diverse taxa like the family Hadruridae, which contains only nine species. Most recent phylogenetic analyses of animal mitogenomes have used high-throughput sequencing, i.e., [32,33,34] or Sanger sequencing of PCR amplicons produced with a variety of primer pairs and techniques, i.e., [35,36,37]. We propose, however, that the phylogeny of a small taxon like Hadruridae could be resolved by running representatives of each species together in a single run on an ONT flow cell. This streamlined approach should yield complete mitochondrial genomes for all species simultaneously. The mitogenomes could then be used to estimate phylogenetic relationships among these closely related species, as we did with Iurida (Figure 4). Furthermore, H. arizonensis mitogenomes could now be used as NAS references for targeting and the enrichment of mitochondrial reads. This approach streamlines phylogenetic analyses for low-diversity groups by combining species in a single run, optimizing mitochondrial read capture and requiring minimal resources, all at a reduced cost.
5. Conclusions
This study unveils the first complete mitogenomes for an icon of the American Southwest: the desert hairy scorpion. Utilizing nanopore sequencing as an investigative tool targeting bacterial DNA from venom glands, our study successfully generated high-quality mitogenomes as bycatch. Phylogenetic analysis of these data allowed us to explore relationships among Iurida scorpions for which mitogenomic data are available. Furthermore, the exploration of genetic diversity yielded insights into the potential of alternative mitochondrial genes, such as CYTB, NAD4, and NAD5, whose high intraspecific variability suggests their possible utility as informative markers for future studies of closely related scorpions, such as population genetics and phylogeography. Looking ahead, the use of nanopore sequencing with adaptive sampling presents a powerful tool for phylogenetic analyses of taxonomically limited groups like the Hadruridae. This streamlined approach envisions a single sequencing run yielding complete mitogenomes for multiple species, accelerating our understanding of the tree of life for non-model organisms like scorpions.
Author Contributions
Specimen provision, M.R.G.; conceptualization, M.R.G. and B.M.; laboratory procedures, M.R.G., D.S.T. and B.M.; data analysis, M.R.G., C.E.S.-L. and J.R.Z.; resources, M.R.G. and B.M.; writing—original draft preparation, M.R.G.; writing—review and editing, C.E.S.-L., J.R.Z., D.S.T. and B.M.; All authors have read and agreed to the published version of the manuscript.
Funding
This project benefitted from an NSF grant DEB-1754030 awarded to MRG.
Data Availability Statement
Seven complete and annotated mitochondrial genomes for H. arizonensis were deposited in GenBank under accession numbers OR995570–OR995576.
Acknowledgments
We thank George Graham and Paula Cushing for their assistance in the field. We thank the Biology Department, Eastern Connecticut State University, for their continued support. We are grateful to the five anonymous reviewers whose insightful comments significantly improved the quality of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ruiz-Mendoza, P.X.; Jasso-Martínez, J.M.; Gutiérrez-Rodríguez, J.; Samacá-Sáenz, E.; Zaldívar-Riverón, A. Mitochondrial genome characterization and mitogenome phylogenetics in the central Mexican Stenopelmatus talpa complex (Orthoptera: Stenopelmatidae: Stenopelmatini). Rev. Mex. Biodivers. 2023, 94, e945094. [Google Scholar] [CrossRef]
- Kong, D.; Gan, Z.; Li, X. Phylogenetic relationships and adaptation in deep-sea carideans revealed by mitogenomes. Gene 2023, 896, 148054. [Google Scholar] [CrossRef] [PubMed]
- Galen, S.C.; Borner, J.; Perkins, S.L.; Weckstein, J.D. Phylogenomics from transcriptomic “bycatch” clarify the origins and diversity of avian trypanosomes in North America. PLoS ONE 2020, 15, e0240062. [Google Scholar] [CrossRef] [PubMed]
- Ghanavi, H.R.; Twort, V.G.; Duplouy, A. Exploring bycatch diversity of organisms in whole genome sequencing of Erebidae moths (Lepidoptera). Sci. Rep. 2021, 11, 24499. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Graham, M.R.; Sharma, P.P.; Ortiz, E.; Possani, L.D. Hadrurid scorpion toxins: Evolutionary conservation and selective pressures. Toxins 2019, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ojanguren-Affilastro, A.A.; Sharma, P.P. Another one bites the dust: Taxonomic sampling of a key genus in phylogenomic datasets reveals more non-monophyletic groups in traditional scorpion classification. Invertebr. Syst. 2020, 34, 133–143. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; González-Santillán, E.; Monod, L.; Sharma, P.P. Phylogenomics facilitates stable scorpion systematics: Reassessing the relationships of Vaejovidae and a new higher-level classification of Scorpiones (Arachnida). Mol. Phylogenet. Evol. 2019, 135, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Ojanguren-Affilastro, A.A.; Graham, M.R.; Sharma, P.P. Congruence between ultraconserved element-based matrices and phylotranscriptomic datasets in the scorpion Tree of Life. Cladistics 2023, 39, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Shimwell, C.; Atkinson, L.; Graham, M.R.; Murdoch, B. A first molecular characterization of the scorpion telson microbiota of Hadrurus arizonensis and Smeringurus mesaensis. PLoS ONE 2023, 18, e0277303. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 474, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Ban, X.C.; Shao, Z.K.; Wu, L.J.; Sun, J.T.; Xue, X.F. Highly diversified mitochondrial genomes provide new evidence for interordinal relationships in the Arachnida. Cladistics 2022, 38, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, W.T.; Zhang, Q.L.; Liu, M.; Xing, C.W.; Cao, Y.; Luo, F.Z.; Yuan, M.L. Mitochondrial phylogenomics provides insights into the phylogeny and evolution of spiders (Arthropoda: Araneae). Zool. Res. 2022, 43, 566–584. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeeseler, A.; Lanfear, R. IQTREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Revell, L. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Fet, V.; Soleglad, M.E.; Barker, M.D. Phylogenetic analysis of the “hirsutus” group of the genus Hadrurus Thorell (Scorpiones: Iuridae) based on morphology and mitochondrial DNA. In Scorpions 2001. In Memoriam Gary A. Polis; Fet, V., Selden, P.A., Eds.; British Arachnological Society: Burnham Beeches, UK, 2001; pp. 139–160. [Google Scholar]
- Gantenbein, B.; Fet, V.; Largiader, C.; Scholl, A. First DNA phylogeny of the genus Euscorpius Thorell 1876 (Scorpiones, Euscorpiidae) and its bearing on the taxonomy and biogeography of this genus. Biogeographica 1999, 75, 59–72. [Google Scholar]
- Gantenbein, B.; Largiadèr, C.R. The phylogeographic importance of the Strait of Gibraltar as a gene flow barrier in terrestrial arthropods: A case study with the scorpion Buthus occitanus as model organism. Mol. Phylogenet. Evol. 2003, 28, 119–130. [Google Scholar] [CrossRef]
- Graham, M.R.; Jaeger, J.R.; Prendini, L.; Riddle, B.R. Phylogeography of the Arizona hairy scorpion (Hadrurus arizonensis) supports a model of biotic assembly in the Mojave Desert and adds a new Pleistocene refugium. J. Biogeogr. 2013, 40, 1298–1312. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Aharon, S.; Ballesteros, J.A.; Gainett, G.; Baker, C.M.; González-Santillán, E.; Harvey, M.S.; Hassan, M.K.; Almaaty, A.H.A.; Monod, L.; et al. Phylogenomics of scorpions reveal contemporaneous diversification of scorpion mammalian predators and mammal-active sodium channel toxins. Syst. Biol. 2022, 71, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Ghanavi, H.R.; Twort, V.; Hartman, T.J.; Zahiri, R.; Wahlberg, N. The (non) accuracy of mitochondrial genomes for family-level phylogenetics in Erebidae (Lepidoptera). Zool. Scr. 2022, 51, 695–707. [Google Scholar] [CrossRef]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore adaptive sampling: A tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef]
- Abramson, N.I.; Bodrov, S.Y.; Bondareva, O.V.; Genelt-Yanovskiy, E.A.; Petrova, T.V. A mitochondrial genome phylogeny of voles and lemmings (Rodentia: Arvicolinae): Evolutionary and taxonomic implications. PLoS ONE 2021, 16, e0248198. [Google Scholar] [CrossRef]
- Pan, D.; Shi, B.; Du, S.; Gu, T.; Wang, R.; Xing, Y.; Zhang, Z.; Chen, J.; Cumberlidge, N.; Sun, H. Mitogenome phylogeny reveals Indochina Peninsula origin and spatiotemporal diversification of freshwater crabs (Potamidae: Potamiscinae) in China. Cladistics 2022, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Dietrich, C.H.; Yu, X.; Jiao, M.; Dai, R.; Yang, M. Mitogenomic phylogeny of Typhlocybinae (Hemiptera: Cicadellidae) reveals homoplasy in tribal diagnostic morphological traits. Ecol. Evol. 2022, 12, e8982. [Google Scholar] [CrossRef]
- Ding, L.; Zhou, Q.; Sun, Y.; Feoktistova, N.Y.; Liao, J. Two novel cricetine mitogenomes: Insight into the mitogenomic characteristics and phylogeny in Cricetinae (Rodentia: Cricetidae). Genomics 2020, 112, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; Uribe, J.E.; Eernisse, D.J.; Zardoya, R. A mitogenomic phylogeny of chitons (Mollusca: Polyplacophora). BMC Evol. Biol. 2020, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Catanese, G.; Morey, G.; Verger, F.; Grau, A.M. The Nursehound Scyliorhinus stellaris mitochondrial genome—Phylogeny, relationships among Scyliorhinidae and variability in waters of the Balearic Islands. Int. J. Mol. Sci. 2022, 23, 10355. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).