

Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2α′Cys336Ser as an Exquisite Crystallographic Tool



Abstract
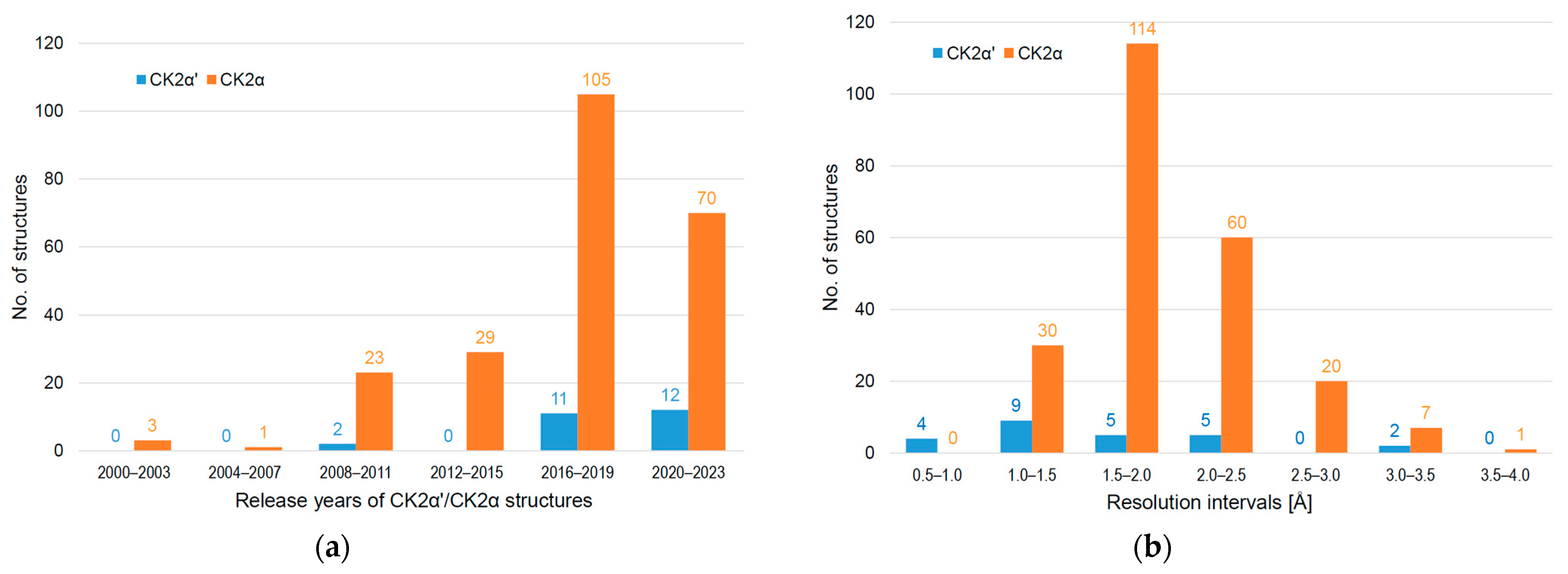
:1. Introduction
- For some members of a series of halogenated triazolo pyridines, the exact binding mode at the ATP site could only be clarified via high-resolution complex structures with CK2α′Cys336Ser [24];
- In the case of some 2-aminothiazole compounds supposed to occupy an allosteric binding site [25,26], CK2α′Cys336Ser complex structures revealed that in fact the ATP cavity harboured these inhibitory ligands [27]; with resolutions of 0.833 Å (PDB_ID 6TGU) and 0.922 Å (PDB_ID 6TE2), two of these structures are currently the best resolved among about 4500 X-ray diffraction entries in the PDB [1,2] with protein kinase chains;
- The power of the approach became particularly obvious when the αD pocket, an allosteric site originally discovered with CK2α [28,29], could be occupied via soaking of CK2α′Cys336Ser/MB002 crystals, although this required extensive local conformational rearrangements in the crystalline state of the protein [30].
2. Results and Discussion
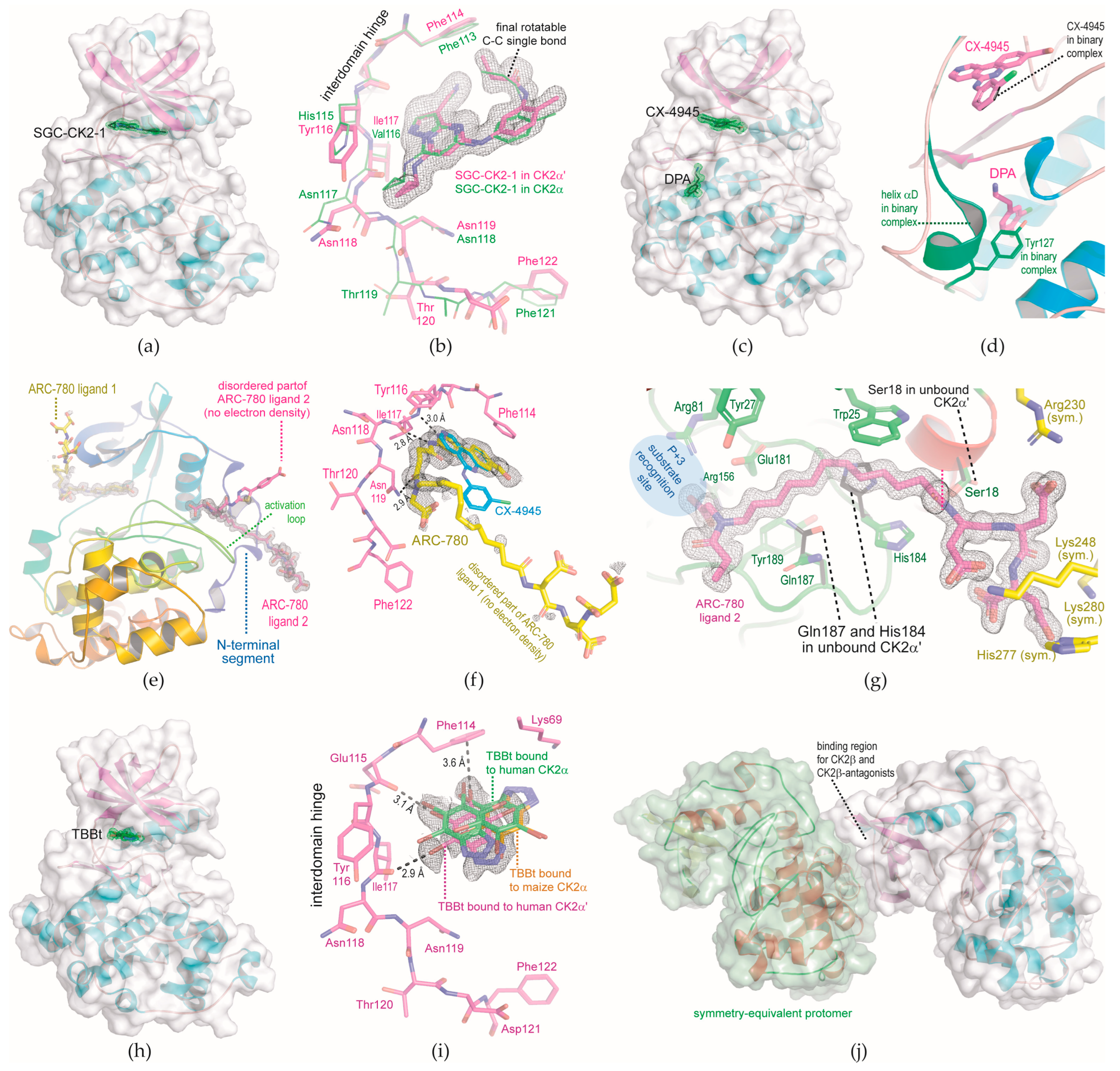
2.1. Complex Structures Determined with Triclinic CK2α′Cys336Ser Crystals
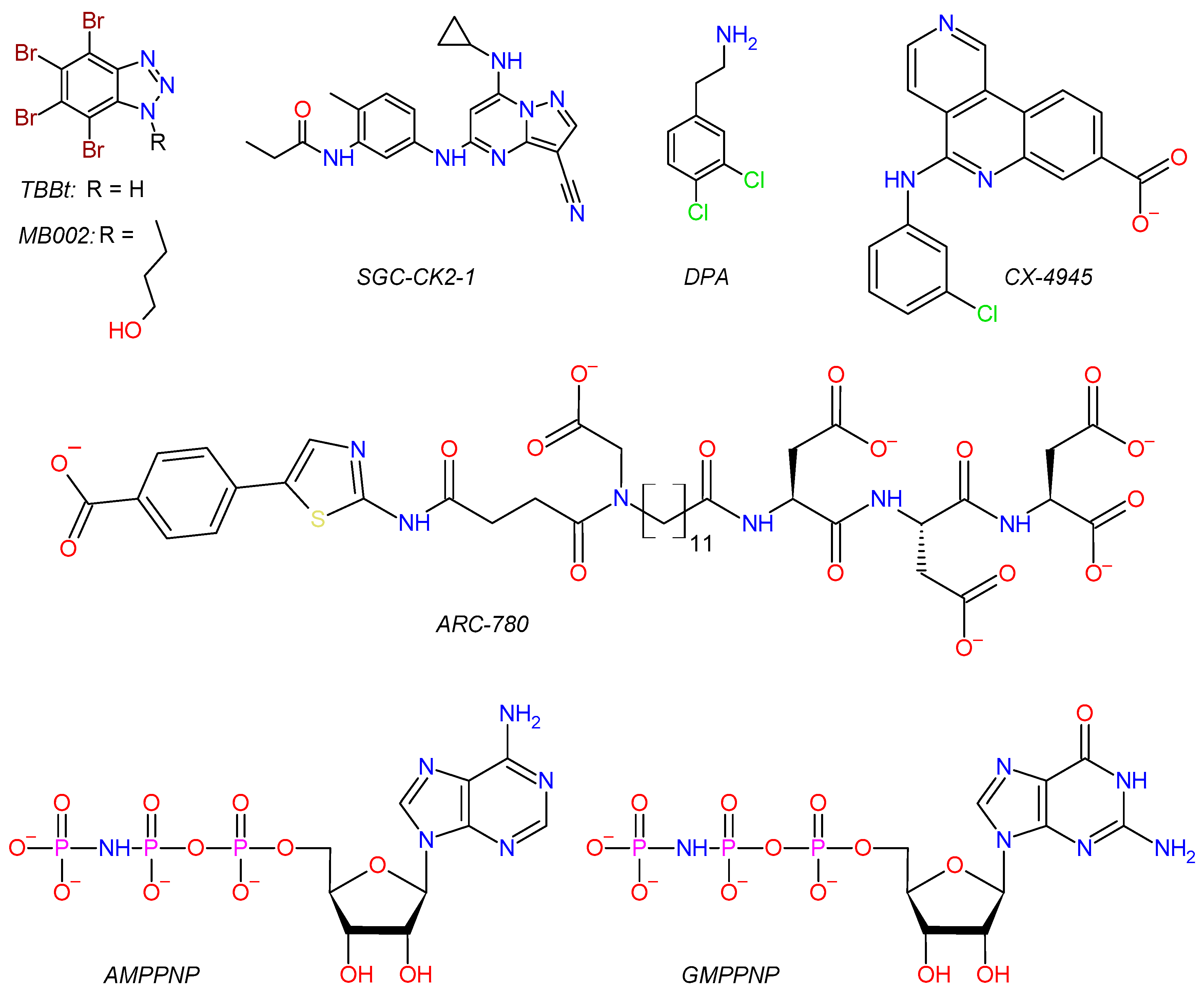
2.1.1. CK2α′Cys336Ser Structure in Complex with the ATP-Site Ligands SGC-CK2-1
2.1.2. Ternary Complex Structure of CK2α′Cys336Ser, CX-4945, and DPA
2.1.3. N-Terminal Segment Site: A Novel Ligand Binding Site Discovered in a CK2α′Cys336Ser Structure in Complex with the Bisubstrate Inhibitor ARC-780
2.1.4. CK2α′Cys336Ser Structure in Complex with TBBt
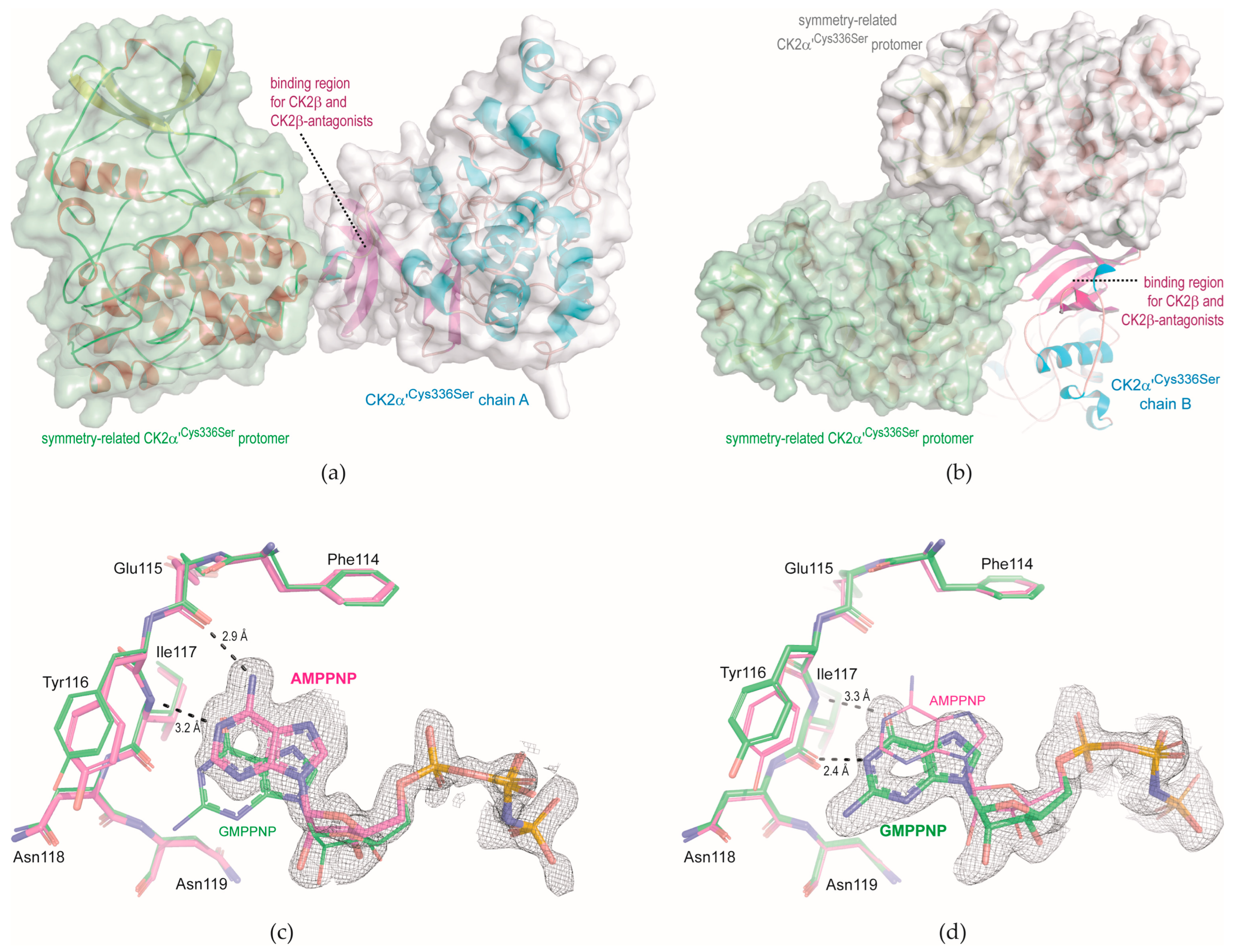
2.2. Complex Structures Determined with Monoclinic CK2α′Cys336Ser Crystals
3. Materials and Methods
3.1. Protein Expression and Purification
3.2. Preparation of ARC-780
3.3. Protein Crystallization
3.3.1. General Procedure
3.3.2. Preparation of CK2α′Cys336Ser/SGC-CK2-1 Binary Complex Crystals and of CK2α′Cys336Ser/CX-4945/DPA Ternary Complex Crystals
3.3.3. Preparation of CK2α′Cys336Ser/ARC-780 Complex Crystals
3.3.4. Co-Crystallization of CK2α′Cys336Ser and TBBt
3.3.5. Growth of CK2α′Cys336Ser Crystals in Complex with AMPPNP or GMPPNP
3.4. X-ray Diffraction Data Collection and Processing followed by Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- wwPDB consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolska, G.; Boldyreff, B.; Issinger, O.G. Cloning and sequencing of the casein kinase 2 α subunit from Zea mays. Biochim. Biophys. Acta 1991, 1129, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Boldyreff, B.; Meggio, F.; Dobrowolska, G.; Pinna, L.A.; Issinger, O.G. Expression and characterization of a recombinant maize CK-2 α subunit. Biochim. Biophys. Acta 1993, 1173, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Pinna, L.A.; Issinger, O.G.; Schomburg, D. Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 Å resolution. EMBO J. 1998, 17, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef]
- Battistutta, R. Protein kinase CK2 in health and disease: Structural bases of protein kinase CK2 inhibition. Cell. Mol. Life Sci. 2009, 66, 1868–1889. [Google Scholar] [CrossRef]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: A patent review. Expert Opin. Ther. Pat. 2012, 22, 1081–1097. [Google Scholar] [CrossRef]
- Atkinson, E.L.; Iegre, J.; Brear, P.D.; Zhabina, E.A.; Hyvönen, M.; Spring, D.R. Downfalls of Chemical Probes Acting at the Kinase ATP-Site: CK2 as a Case Study. Molecules 2021, 26, 1977. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. [Google Scholar] [CrossRef]
- Olsen, B.; Boldyreff, B.; Niefind, K.; Issinger, O. Purification and characterization of the CK2α’-based holoenzyme, an isozyme of CK2α: A comparative analysis. Protein Expr. Purif. 2006, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Rasmussen, T.; Niefind, K.; Issinger, O. Biochemical characterization of CK2 α and α’ paralogues and their derived holoenzymes: Evidence for the existence of a heterotrimeric CK2 α’-holoenzyme forming trimeric complexes. Mol. Cell. Biochem. 2008, 316, 37–47. [Google Scholar] [CrossRef]
- Bischoff, N.; Olsen, B.; Raaf, J.; Bretner, M.; Issinger, O.G.; Niefind, K. Structure of the human protein kinase CK2 catalytic subunit CK2α’ and interaction thermodynamics with the regulatory subunit CK2β. J. Mol. Biol. 2011, 407, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nakaniwa, T.; Kinoshita, T.; Sekiguchi, Y.; Tada, T.; Nakanishi, I.; Kitaura, K.; Suzuki, Y.; Ohno, H.; Hirasawa, A.; Tsujimoto, G. Structure of human protein kinase CK2α2 with a potent indazole-derivative inhibitor. Acta Crystallogr. F Struct. Biol. Commun. 2009, 65, 75–79. [Google Scholar] [CrossRef]
- Tsuyuguchi, M.; Nakaniwa, T.; Kinoshita, T. Crystal structures of human CK2α2 in new crystal forms arising from a subtle difference in salt concentration. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Bretner, M.; Najda-Bernatowicz, A.; Łebska, M.; Muszyńska, G.; Kilanowicz, A.; Sapota, A. New inhibitors of protein kinase CK2, analogues of benzimidazole and benzotriazole. Mol. Cell. Biochem. 2008, 316, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Hochscherf, J.; Lindenblatt, D.; Witulski, B.; Birus, R.; Aichele, D.; Marminon, C.; Bouaziz, Z.; Le Borgne, M.; Jose, J.; Niefind, K. Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2α and Its Paralog CK2α’. Pharmaceuticals 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Bischoff, N.; Golub, A.G.; Bdzhola, V.G.; Balanda, A.O.; Prykhod’ko, A.O.; Yarmoluk, S.M. Structural Hypervariability of the Two Human Protein Kinase CK2 Catalytic Subunit Paralogs Revealed by Complex Structures with a Flavonol- and a Thieno[2,3-d]pyrimidine-Based Inhibitor. Pharmaceuticals 2017, 10, 9. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Nickelsen, A.; Applegate, V.M.; Hochscherf, J.; Witulski, B.; Bouaziz, Z.; Marminon, C.; Bretner, M.; Le Borgne, M.; Jose, J.; et al. Diacritic binding of an indenoindole inhibitor by CK2α paralogs explored by a reliable path to atomic resolution CK2α′ structures. ACS Omega 2019, 4, 5471–5478. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic Acid (CX-4945), the First Clinical Stage Inhibitor of Protein Kinase CK2 for the Treatment of Cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, K.; Lindenblatt, D.; Wińska, P.; Wielechowska, M.; Toelzer, C.; Niefind, K.; Bretner, M. Synthesis, biological properties and structural study of new halogenated azolo[4,5-b]pyridines as inhibitors of CK2 kinase. Bioorganic Chem. 2021, 106, 104502. [Google Scholar] [CrossRef]
- Bestgen, B.; Krimm, I.; Kufareva, I.; Kamal, A.A.M.; Seetoh, W.G.; Abell, C.; Hartmann, R.W.; Abagyan, R.; Cochet, C.; Le Borgne, M.; et al. 2-Aminothiazole Derivatives as Selective Allosteric Modulators of the Protein Kinase CK2. 1. Identification of an Allosteric Binding Site. J. Med. Chem. 2019, 62, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Bestgen, B.; Kufareva, I.; Seetoh, W.; Abell, C.; Hartmann, R.W.; Abagyan, R.; Le Borgne, M.; Filhol, O.; Cochet, C.; Lomberget, T.; et al. 2-Aminothiazole Derivatives as Selective Allosteric Modulators of the Protein Kinase CK2. 2. Structure-Based Optimization and Investigation of Effects Specific to the Allosteric Mode of Action. J. Med. Chem. 2019, 62, 1817–1836. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Nickelsen, A.; Applegate, V.M.; Jose, J.; Niefind, K. Structural and Mechanistic Basis of the Inhibitory Potency of Selected 2-Aminothiazole Compounds on Protein Kinase CK2. J. Med. Chem. 2020, 63, 7766–7772. [Google Scholar] [CrossRef]
- Brear, P.; De Fusco, C.; Georgiou, K.H.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvönen, M. Specific inhibition of CK2α from an anchor outside the active site. Chem. Sci. 2016, 7, 6839–6845. [Google Scholar] [CrossRef] [PubMed]
- De Fusco, C.; Brear, P.; Iegre, J.; Georgiou, K.H.; Sore, H.F.; Hyvönen, M.; Spring, D.R. A fragment-based approach leading to the discovery of a novel binding site and the selective CK2 inhibitor CAM4066. Bioorganic Med. Chem. 2017, 25, 3471–3482. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Applegate, V.; Nickelsen, A.; Klußmann, M.; Neundorf, I.; Götz, C.; Jose, J.; Niefind, K. Molecular Plasticity of Crystalline CK2α’ Leads to KN2, a Bivalent Inhibitor of Protein Kinase CK2 with Extraordinary Selectivity. J. Med. Chem. 2022, 65, 1302–1312. [Google Scholar] [CrossRef]
- Wells, C.I.; Drewry, D.H.; Pickett, J.E.; Tjaden, A.; Krämer, A.; Müller, S.; Gyenis, L.; Menyhart, D.; Litchfield, D.W.; Knapp, S.; et al. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem. Biol. 2021, 28, 546–558.e10. [Google Scholar] [CrossRef] [PubMed]
- Davis-Gilbert, Z.W.; Krämer, A.; Dunford, J.E.; Howell, S.; Senbabaoglu, F.; Wells, C.I.; Bashore, F.M.; Havener, T.M.; Smith, J.L.; Hossain, M.A.; et al. Discovery of a Potent and Selective Naphthyridine-Based Chemical Probe for Casein Kinase 2. ACS Med. Chem. Lett. 2023, 14, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Pack, M.; Götz, C.; Wrublewsky, S.; Montenarh, M. SGC-CK2-1 Is an Efficient Inducer of Insulin Production and Secretion in Pancreatic β-Cells. Pharmaceutics 2021, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kinoshita, C.; Axtman, A.D.; Young, J.E. Evaluation of a Selective Chemical Probe Validates That CK2 Mediates Neuroinflammation in a Human Induced Pluripotent Stem Cell-Derived Mircroglial Model. Front. Mol. Neurosci. 2022, 15, 824956. [Google Scholar] [CrossRef] [PubMed]
- Boewe, A.S.; Wemmert, S.; Kulas, P.; Schick, B.; Götz, C.; Wrublewsky, S.; Montenarh, M.; Menger, M.D.; Laschke, M.W.; Ampofo, E. Inhibition of CK2 Reduces NG2 Expression in Juvenile Angiofibroma. Biomedicines 2022, 10, 966. [Google Scholar] [CrossRef] [PubMed]
- PyMOL. The PyMOL Molecular Graphics System, version 1.7; Schrödinger, LLC: New York, NY, USA, 2013. [Google Scholar]
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef]
- Winiewska-Szajewska, M.; Czapinska, H.; Kaus-Drobek, M.; Fricke, A.; Mieczkowska, K.; Dadlez, M.; Bochtler, M.; Poznański, J. Competition between electrostatic interactions and halogen bonding in the protein-ligand system: Structural and thermodynamic studies of 5,6-dibromobenzotriazole-hCK2α complexes. Sci. Rep. 2022, 12, 18964. [Google Scholar] [CrossRef]
- Tickle, I.J.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. STARANISO; Global Phasing Ltd.: Cambridge, UK, 2018. [Google Scholar]
- Janeczko, M.; Orzeszko, A.; Kazimierczuk, Z.; Szyszka, R.; Baier, A. CK2α and CK2α’ subunits differ in their sensitivity to 4,5,6,7-tetrabromo- and 4,5,6,7-tetraiodo-1H-benzimidazole derivatives. Eur. J. Med. Chem. 2012, 47, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Tsuyuguchi, M.; Kitagawa, D.; Sawa, M.; Nakamura, S.; Nakanishi, I.; Kinoshita, T. Bivalent binding mode of an amino-pyrazole inhibitor indicates the potentials for CK2α1-selective inhibitors. Biochem. Biophys. Res. Commun. 2022, 630, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Weinkam, P.; Rettenmaier, T.J.; Bichmann, L.; Keedy, D.A.; Woldeyes, R.A.; Schneidman-Duhovny, D.; Demerdash, O.N.; Mitchell, J.C.; Wells, J.A.; et al. CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites. J. Mol. Biol. 2016, 428, 709–719. [Google Scholar] [CrossRef]
- Pietsch, M.; Viht, K.; Schnitzler, A.; Ekambaram, R.; Steinkrüger, M.; Enkvist, E.; Nienberg, C.; Nickelsen, A.; Lauwers, M.; Jose, J.; et al. Unexpected CK2β-antagonistic functionality of bisubstrate inhibitors targeting protein kinase CK2. Bioorganic Chem. 2020, 96, 103608. [Google Scholar] [CrossRef] [PubMed]
- Czapinska, H.; Winiewska-Szajewska, M.; Szymaniec-Rutkowska, A.; Piasecka, A.; Bochtler, M.; Poznański, J. Halogen Atoms in the Protein-Ligand System. Structural and Thermodynamic Studies of the Binding of Bromobenzotriazoles by the Catalytic Subunit of Human Protein Kinase CK2. J. Phys. Chem. B 2021, 125, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Wąsik, R.; Wińska, P.; Poznański, J.; Shugar, D. Synthesis and physico-chemical properties in aqueous medium of all possible isomeric bromo analogues of benzo-1H-triazole, potential inhibitors of protein kinases. J. Phys. Chem. B 2012, 116, 7259–7268. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Bortolato, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The ATP-binding site of protein kinase CK2 holds a positive electrostatic area and conserved water molecules. Chembiochem 2007, 8, 1804–1809. [Google Scholar] [CrossRef]
- Winiewska-Szajewska, M.; Maciejewska, A.M.; Speina, E.; Poznański, J.; Paprocki, D. Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2. Molecules 2021, 26, 3163. [Google Scholar] [CrossRef]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 2007, 408, 363–373. [Google Scholar] [CrossRef]
- Laudet, B.; Moucadel, V.; Prudent, R.; Filhol, O.; Wong, Y.S.; Royer, D.; Cochet, C. Identification of chemical inhibitors of protein-kinase CK2 subunit interaction. Mol. Cell. Biochem. 2008, 316, 63–69. [Google Scholar] [CrossRef]
- Brear, P.; North, A.; Iegre, J.; Hadje Georgiou, K.; Lubin, A.; Carro, L.; Green, W.; Sore, H.F.; Hyvönen, M.; Spring, D.R. Novel non-ATP competitive small molecules targeting the CK2 α/β interface. Bioorganic Med. Chem. 2018, 26, 3016–3020. [Google Scholar] [CrossRef]
- Kufareva, I.; Bestgen, B.; Brear, P.; Prudent, R.; Laudet, B.; Moucadel, V.; Ettaoussi, M.; Sautel, C.F.; Krimm, I.; Engel, M.; et al. Discovery of holoenzyme-disrupting chemicals as substrate-selective CK2 inhibitors. Sci. Rep. 2019, 9, 15893. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Horn, M.; Götz, C.; Niefind, K.; Neundorf, I.; Pietsch, M. Design of CK2β-Mimicking Peptides as Tools To Study the CK2α/CK2β Interaction in Cancer Cells. ChemMedChem 2019, 14, 833–841. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, N.; Zhou, Y.; Cortopassi, W.A.; Jacobson, M.P.; Zhao, L.J.; Zhong, R.G. Structure-based Discovery of Novel CK2α-Binding Cyclic Peptides with Anti-cancer Activity. Mol. Infor. 2019, 38, e1800089. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, E.L.; Iegre, J.; D’Amore, C.; Brear, P.; Salvi, M.; Hyvönen, M.; Spring, D.R. Development of small cyclic peptides targeting the CK2α/β interface. Chem. Commun. 2022, 58, 4791–4794. [Google Scholar] [CrossRef] [PubMed]
- Rodnight, R.; Lavin, B.E. Phosvitin kinase from brain: Activation by ions and subcellular distribution. Biochem. J. 1964, 93, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, G.M.; Traugh, J.A. Cyclic nucleotide-independent protein kinases from rabbit reticulocytes. Purification of casein kinases. J. Biol. Chem. 1979, 254, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Becher, I.; Savitski, M.M.; Savitski, M.F.; Hopf, C.; Bantscheff, M.; Drewes, G. Affinity profiling of the cellular kinome for the nucleotide cofactors ATP, ADP, and GTP. ACS Chem. Biol. 2013, 8, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Pütter, M.; Guerra, B.; Issinger, O.G.; Schomburg, D. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Struct. Biol. 1999, 6, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Rahnel, H.; Viht, K.; Lavogina, D.; Mazina, O.; Haljasorg, T.; Enkvist, E.; Uri, A. A Selective Biligand Inhibitor of CK2 Increases Caspase-3 Activity in Cancer Cells and Inhibits Platelet Aggregation. ChemMedChem 2017, 12, 1723–1736. [Google Scholar] [CrossRef]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Struct. Biol. 2011, 67, 293–302. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Struct. Biol. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Struct. Biol. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Struct. Biol. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Struct. Biol. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Struct. Biol. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Struct. Biol. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Struct. Biol. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. Electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. D Struct. Biol. 2009, 65, 1074–1080. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| CK2α′Cys336Ser ligands | SGC-CK2-1 | CX-4945 + DPA | ARC-780 | TBBt |
| PDB code | 8Q9S | 8QBU | 8Q77 | 8QCD |
| X-ray diffraction data quality | ||||
| Wavelength (Å) | 0.9762 | 0.8856 | 0.9677 | 0.8856 |
| Synchrotron (beamline) | P13, EMBL/DESY | P13, EMBL/DESY | MASSIF-3, ESRF | ID23-1, ESRF |
| Space group | P1 | P1 | P1 | P1 |
| Unit cell: a, b, c (Å) α, β, γ (°) | 46.217, 47.682, 50.563 66.827, 89.429, 88.930 | 46.173, 47.714, 50.586 66.053, 89.722, 88.974 | 46.603, 47.919, 51.031 66.591, 89.667, 88.163 | 46.388, 47.701, 50.342 113.481, 90.448, 90.171 |
| Protomers per asymmetric unit | 1 | 1 | 1 | 1 |
| Resolution (Å) (highest resolution shell) | 32.060–1.352 (1.400–1.352) | 46.17–1.095 (1.134–1.095) | 46.578–1.255 (1.364–1.255) | 23.34–1.033 (1.070–1.033) |
| Rsym (%) | 9.7 (148.2) * | 10.5 (136.8) * | 5.2 (55.9) * | 12.8 (55.2) * |
| CC1/2 | 0.997 (0.589) * | 0.997 (0.608) * | 0.998 (0.806) * | 0.979 (0.561) * |
| Signal-to-noise ratio (I/σI) | 7.8 (1.6) * | 6.5 (1.7) * | 12.2 (1.6) * | 5.7 (1.8) * |
| No. of unique refl. | 56,703 (2835) * | 118,499 (5925) * | 80,300 (4015) * | 149,296 (7465) * |
| Completeness/spherical (%) | 65.3 (11.1) * | 72.9 (18.1) * | 72.5 (16.4) * | 76.9 (21.5) * |
| Completeness/ellipsoidal (%) § | 90.2 (57.4) * | 89.7 (53.6) * | 82.1 (31.7) * | 84.9 (37.0) * |
| Multiplicity | 7.2 (7.1) * | 6.5 (6.6) * | 3.9 (3.9) * | 2.6 (2.6) * |
| Wilson B-factor (Å2) | 19.46 | 11.97 | 10.32 | 1.45 |
| Structure refinement and quality | ||||
| No. of reflections for Rwork/Rfree | 54,699/1989 | 116,384/1984 | 79,069/1214 | 147,251/2014 |
| Rwork/Rfree (%) | 13.54/16.88 | 15.93/17.93 | 11.99/15.73 | 14.37/15.62 |
| No. of non-H-atoms | 3111 | 3275 | 3260 | 3307 |
| Protein | 2822 | 2898 | 2793 | 2876 |
| Ligand/ion | 69 | 176 | 281 | 44 |
| Water | 253 | 293 | 314 | 406 |
| Average B-factor (Å2) | 26.58 | 18.80 | 16.47 | 7.66 |
| Protein | 25.44 | 17.29 | 14.16 | 6.23 |
| Ligand/ion | 26.84 | 24.59 | 30.78 | 11.66 |
| water | 39.24 | 32.14 | 30.06 | 17.57 |
| RMS deviations | ||||
| Bond lengths (Å) | 0.008 | 0.016 | 0.023 | 0.009 |
| Bond angles (°) | 0.86 | 1.42 | 1.67 | 1.04 |
| Ramachandran plot | ||||
| Favoured (%) | 96.00 | 96.93 | 97.24 | 97.85 |
| Allowed (%) | 4.00 | 2.76 | 2.76 | 2.15 |
| Outliers (%) | 0.00 | 0.31 | 0.00 | 0.00 |
| Structure No. | 5 | 6 |
|---|---|---|
| Ligands of CK2α′Cys336Ser | AMPPNP | GMPPNP |
| PDB code | 8QCG | 8QF1 |
| X-ray diffraction data quality | ||
| Wavelength (Å) | 0.8856 | 0.8856 |
| Synchrotron (beamline) | ID23-1, ESRF | ID23-1, ESRF |
| Space group | P21 | P21 |
| Unit cell: a, b, c (Å) α, β, γ (°) | 46.599, 71.852, 101.984 90.000, 92.423, 90.000 | 46.628, 71.590, 102.101 90.000, 92.094, 90.000 |
| Protomers per asymmetric unit | 2 | 2 |
| Resolution (Å) (highest resolution shell) | 101.893–1.045 (1.157–1.045) * | 102.033–1.318 (1.493–1.318) * |
| Rsym (%) | 8.5 (97.6) | 14.4 (47.2) * |
| CC1/2 | 0.997 (0.732) * | 0.973 (0.367) * |
| Signal-to-noise ratio (I/σI) | 9.5 (2.0) * | 4.1 (1.7) * |
| No. of unique refl. | 229,873 (11494) * | 91,444 (4572) * |
| Completeness/spherical (%) | 72.6 (13.8) * | 57.8 (9.3) * |
| Completeness/ellipsoidal (%) § | 94.1 (62.1) * | 90.9 (62.8) * |
| Multiplicity | 6.6 (5.9) * | 3.5 (2.8) * |
| Wilson B-factor (Å2) | 11.47 | 12.80 |
| Structure refinement and quality | ||
| No. of reflections for Rwork/Rfree | 227,816/2005 | 89,975/1370 |
| Rwork/Rfree (%) | 15.06/17.03 | 21.40/23.22 |
| No. of non-H-atoms | 6447 | 6251 |
| Protein | 5630 | 5597 |
| Ligand/ion | 91 | 48 |
| Water | 752 | 619 |
| Average B-factor (Å2) | 20.41 | 23.67 |
| Protein | 18.48 | 22.39 |
| Ligand/ion | 25.21 | 24.86 |
| Water | 34.48 | 35.26 |
| RMS deviations | ||
| Bond lengths (Å) | 0.007 | 0.004 |
| Bond angles (°) | 0.82 | 0.66 |
| Ramachandran plot | ||
| Favoured (%) | 96.91 | 95.82 |
| Allowed (%) | 2.94 | 4.02 |
| Outliers (%) | 0.15 | 0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werner, C.; Lindenblatt, D.; Viht, K.; Uri, A.; Niefind, K. Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2α′Cys336Ser as an Exquisite Crystallographic Tool. Kinases Phosphatases 2023, 1, 306-322. https://doi.org/10.3390/kinasesphosphatases1040018
Werner C, Lindenblatt D, Viht K, Uri A, Niefind K. Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2α′Cys336Ser as an Exquisite Crystallographic Tool. Kinases and Phosphatases. 2023; 1(4):306-322. https://doi.org/10.3390/kinasesphosphatases1040018
Chicago/Turabian StyleWerner, Christian, Dirk Lindenblatt, Kaido Viht, Asko Uri, and Karsten Niefind. 2023. "Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2α′Cys336Ser as an Exquisite Crystallographic Tool" Kinases and Phosphatases 1, no. 4: 306-322. https://doi.org/10.3390/kinasesphosphatases1040018
APA StyleWerner, C., Lindenblatt, D., Viht, K., Uri, A., & Niefind, K. (2023). Discovery and Exploration of Protein Kinase CK2 Binding Sites Using CK2α′Cys336Ser as an Exquisite Crystallographic Tool. Kinases and Phosphatases, 1(4), 306-322. https://doi.org/10.3390/kinasesphosphatases1040018