
Geometry Change of 1,3-Dicyanobenzene upon Electronic Excitation from a Franck–Condon Fit of Several Fluorescence Emission Spectra †


Abstract
1. Introduction
2. Computational Methods
2.1. Quantum Chemical Calculations
2.2. Franck–Condon Fits
3. Experimental Methods
4. Results and Discussion
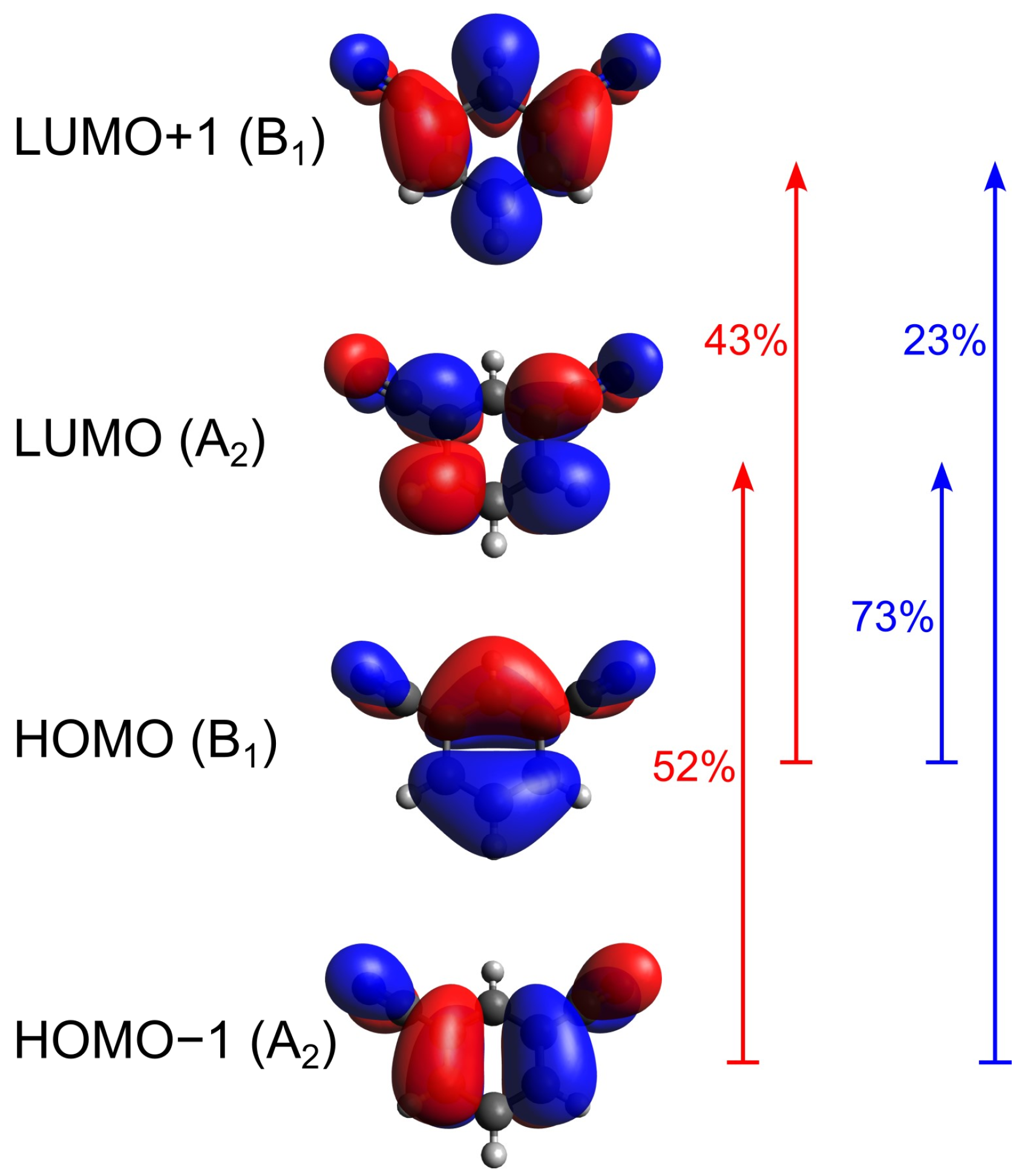
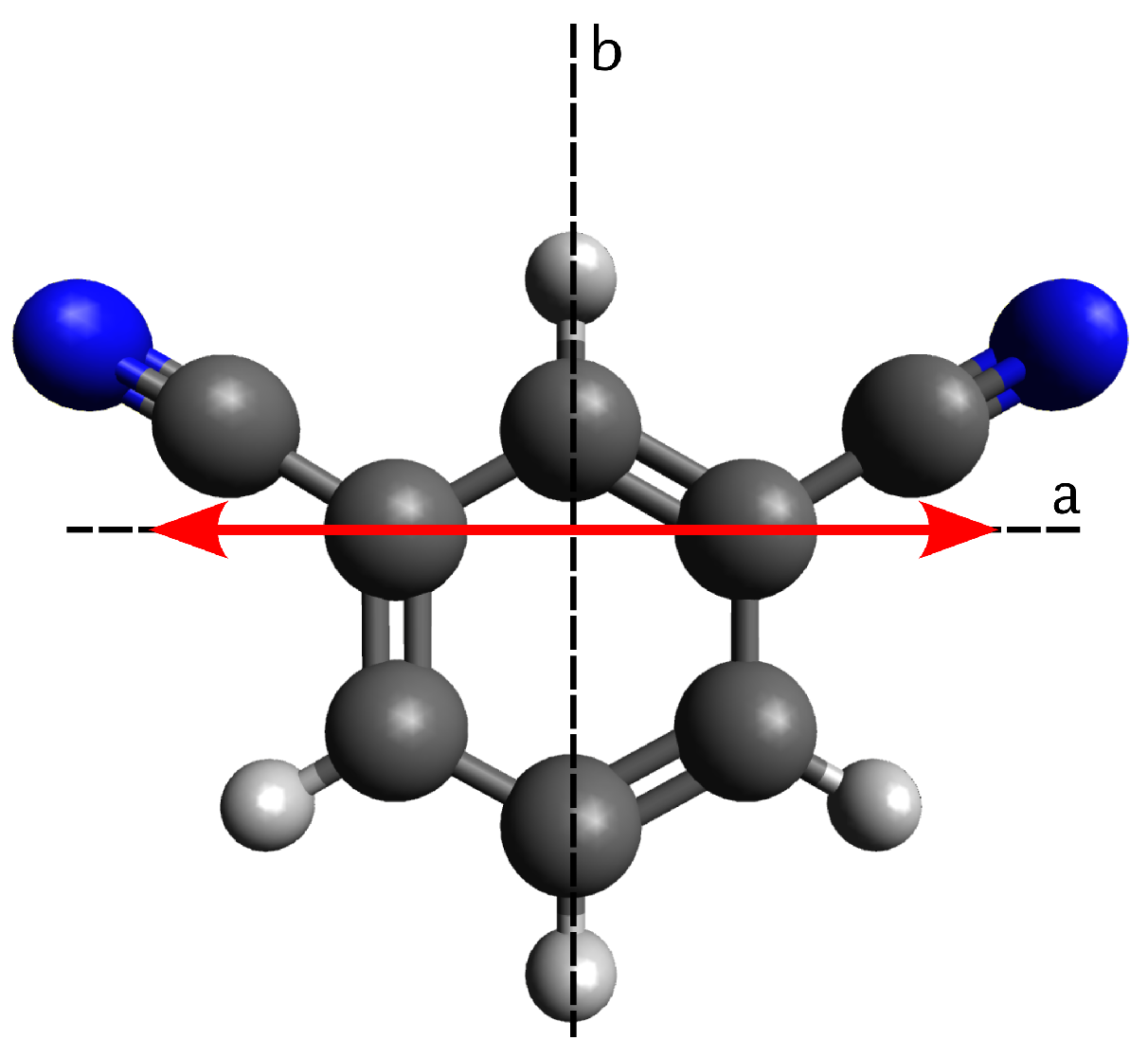
4.1. Computational Results
4.2. Experimental Results
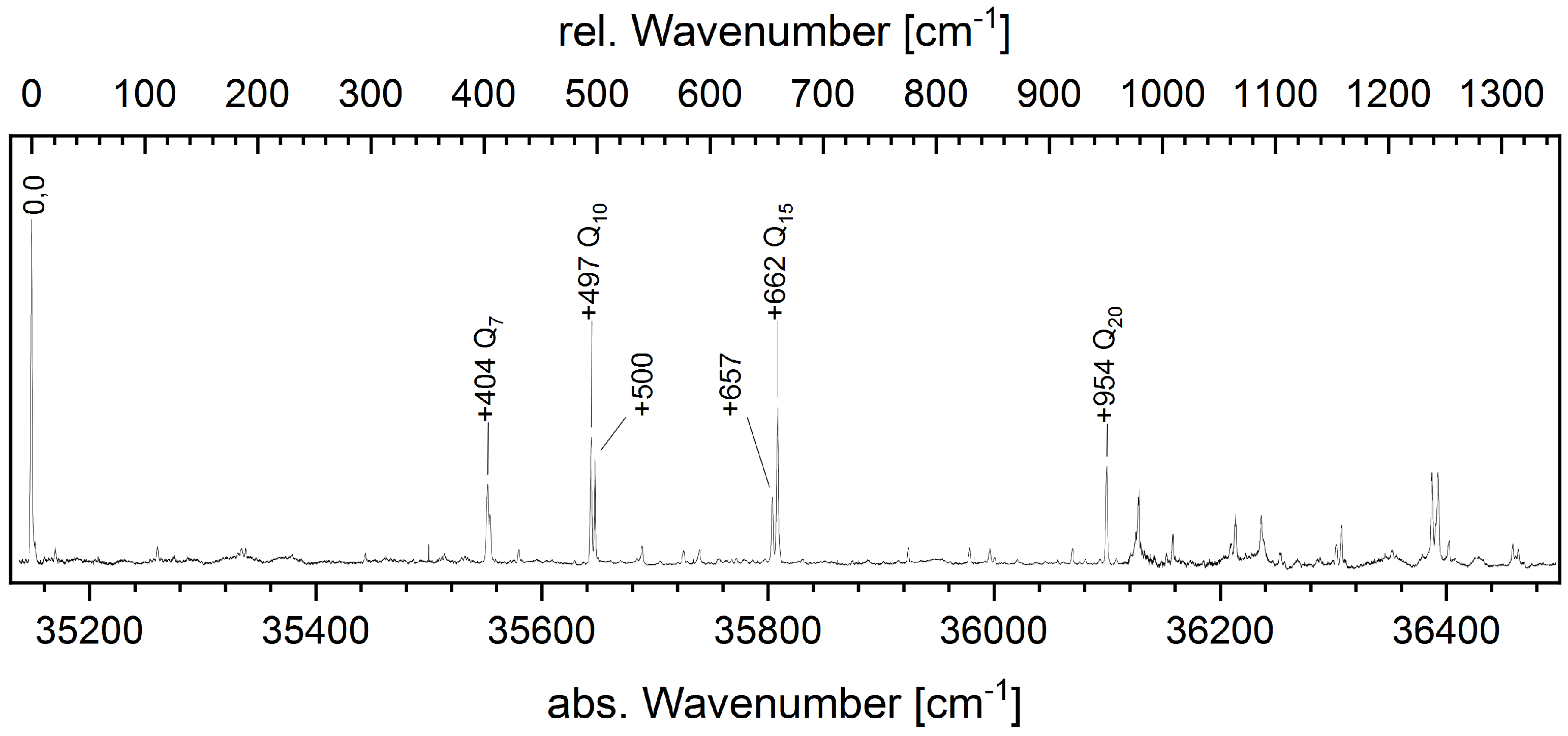
4.2.1. The LIF Spectrum of 1,3-Dicyanobenzene
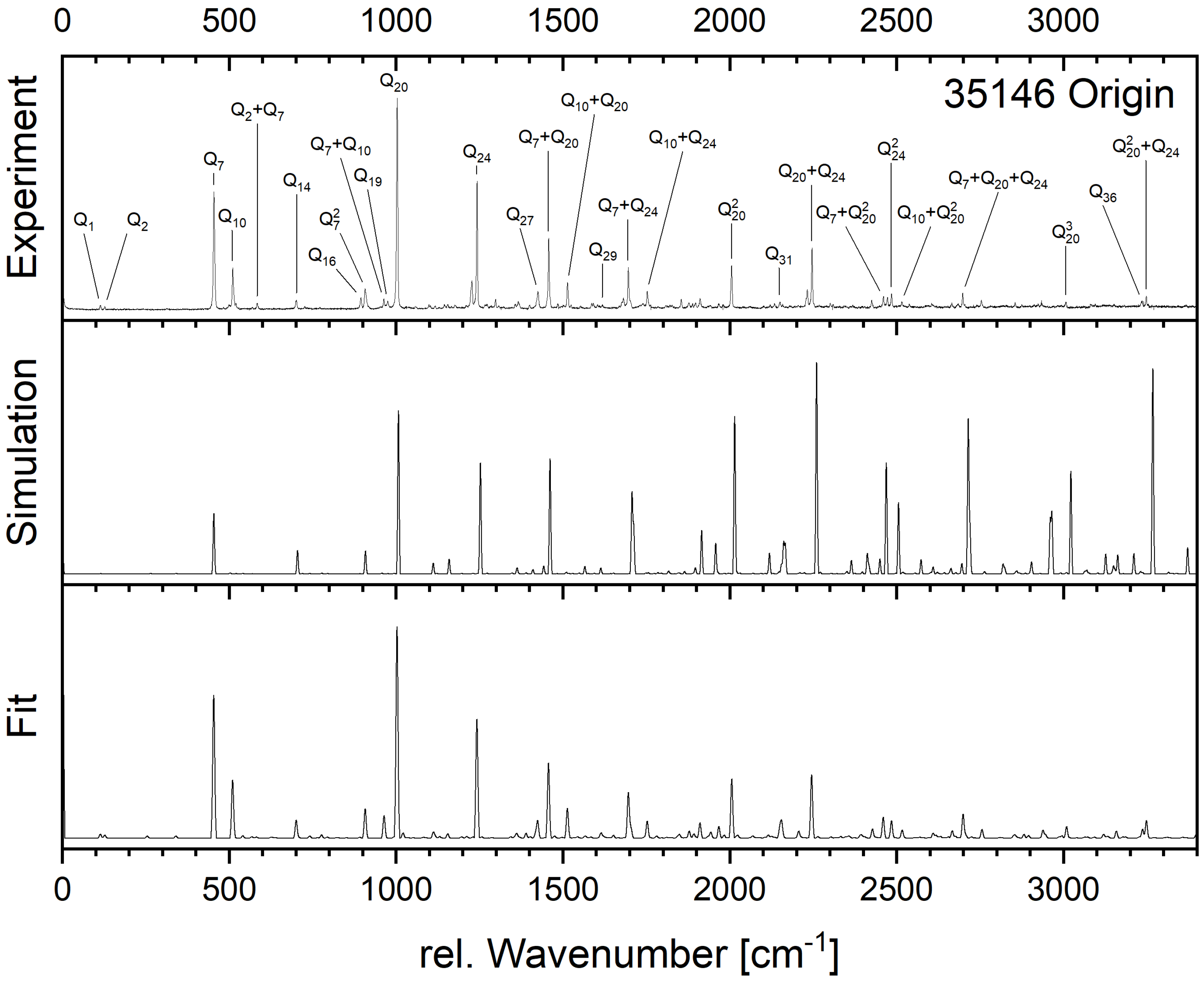
4.2.2. Fluorescence Emission
4.3. Franck–Condon Fit of Geometry Changes upon Electronic Excitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karle, J.; Mark, H.; Hargittai, I. Stereochemical Applications of Gas-Phase Electron Diffraction; Hargittai, I., Hargittai, M., Eds.; VCH: Deerfield Beach, FL, USA, 1988. [Google Scholar]
- Küpper, J.; Stern, S.; Holmegaard, L.; Filsinger, F.; Rouzée, A.; Rudenko, A.; Johnsson, P.; Martin, A.V.; Adolph, M. X-ray Diffraction from Isolated and Strongly Aligned Gas-Phase Molecules with a Free-Electron Laser. Phys. Rev. Lett. 2014, 112, 083002. [Google Scholar] [CrossRef]
- Majewski, W.A.; Meerts, W.L. Near UV spectra with fully resolved structure of naphthalene and perdeurated naphthalene. J. Mol. Spec. 1984, 104, 271–281. [Google Scholar] [CrossRef]
- Majewski, W.A.; Plusquellic, D.F.; Pratt, D.W. The rotationally resolved fluorescence excitation spectrum of 1-fluoronaphthalene. J. Chem. Phys. 1989, 90, 1362–1367. [Google Scholar] [CrossRef]
- Felker, P.M. Rotational coherence spectroscopy: Studies of the geometries of large gasphase species by picosecond time-domain methods. J. Phys. Chem. 1992, 96, 7844–7857. [Google Scholar] [CrossRef]
- Riehn, C. High-resolution pump-probe rotational coherence spectroscopy - rotational constants and structure of ground and electronically excited states of large molecular systems. Chem. Phys. 2002, 297, 283. [Google Scholar] [CrossRef]
- Williamson, J.; Dantus, M.; Kim, S.; Zewail, A. Ultrafast diffraction and molecular structure. Chem. Phys. Lett. 1992, 196, 529–534. [Google Scholar] [CrossRef]
- Bohm, M.; Tatchen, J.; Krügler, D.; Kleinermanns, K.; Nix, M.G.D.; LeGreve, T.A.; Zwier, T.S.; Schmitt, M. High-resolution and Dispersed Fluorescence Examination of Vibronic bands of Tryptamine: Spectroscopic signatures for La/Lb mixing near a conical intersection. J. Phys. Chem. A 2009, 113, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef] [PubMed]
- Geon Hyeong Lee, D.Y.K.; Kim, Y.S. High-Efficiency Dicyanobenzene-Based Organic Light-Emitting Diodes Exhibiting Thermally Activated Delayed Fluorescence. Mol. Cryst. Liq. Cryst. 2015, 621, 1–7. [Google Scholar]
- McGuire, B.A.; Loomis, R.A.; Burkhardt, A.M.; Lee, K.L.K.; Shingledecker, C.N.; Charnley, S.B.; Cooke, I.R.; Cordiner, M.A.; Herbst, E.; Kalenskii, S.; et al. Detection of two interstellar polycyclic aromatic hydrocarbons via spectral matched filtering. Science 2021, 371, 1265–1269. [Google Scholar] [CrossRef]
- McGuire, B.A.; Burkhardt, A.M.; Kalenskii, S.; Shingledecker, C.N.; Remijan, A.J.; Herbst, E.; McCarthy, M.C. Detection of the aromatic molecule benzonitrile (C6H5CN) in the interstellar medium. Science 2018, 359, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Chitarra, O.; Lee, K.L.K.; Buchanan, Z.; Melosso, M.; McGuire, B.A.; Goubet, M.; Pirali, O.; Martin-Drumel, M.-A. Hunting the relatives of benzonitrile: Rotational spectroscopy of dicyanobenzenes. Astron. Astrophys 2021, 652, A163. [Google Scholar] [CrossRef]
- Zajonz, M.; Oberkirch, T.; Hebestreit, M.-L.; Lindic, M.M.; Hättig, C.; Schmitt, M. Excited state dipole moments of two dicyanobenzene isomers from thermochromic shifts and ab initio calculations. J. Photochem. Photobiol. A Chem. 2024, 452, 115589. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations onWorkstation Computers: The Program System TURBOMOLE. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Hättig, C.; Kohn, A. Transition moments and excited-state first-order properties in the coupled cluster model CC2 using the resolution-of-the-identity approximation. J. Chem. Phys. 2002, 117, 6939–6951. [Google Scholar] [CrossRef]
- Hättig, C. Geometry optimizations with the coupled-cluster model CC2 using the resolution-of-the-identity approximation. J. Chem. Phys. 2002, 118, 7751–7761. [Google Scholar] [CrossRef]
- Hellweg, A.; Grün, S.; Hättig, C. Benchmarking the performance of spin-component scaled cc2 in ground and electronically excited states. Phys. Chem. Chem. Phys. 2008, 10, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Deglmann, P.; Furche, F.; Ahlrichs, R. An efficient implementation of second analytical derivatives for density functional methods. Chem. Phys. Lett. 2002, 362, 511–518. [Google Scholar] [CrossRef]
- Spangenberg, D.; Imhof, P.; Kleinermanns, K. The S1 state geometry of phenol determined by simultaneous Franck–Condon and rotational constants fits. Phys. Chem. Chem. Phys. 2003, 5, 2505–2514. [Google Scholar] [CrossRef]
- Brause, R.; Schmitt, M.; Spangenberg, D.; Kleinermanns, K. Determination of the excited state structure of 7-azaindole using a Franck–Condon analysis. Mol. Phys. 2004, 102, 1615–1623. [Google Scholar] [CrossRef]
- Doktorov, E.V.; Malkin, I.A.; Man’ko, V.I. Dynamical symmetry of vibronic transitions in polyatomic molecules and the Franck-Condon principle. J. Mol. Spec. 1975, 56, 1–20. [Google Scholar] [CrossRef]
- Doktorov, E.V.; Malkin, I.A.; Man’ko, V.I. Dynamical symmetry of vibronic transitions in polyatomic molecules and the Franck-Condon principle. J. Mol. Spec. 1977, 64, 302–326. [Google Scholar] [CrossRef]
- Levenberg, K. A method for the solution of certain non-linear problems in least squares. Q. Appl. Math. Meth. 1944, 2, 164–168. [Google Scholar] [CrossRef]
- Marquardt, D.D. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Schmitt, M.; Henrichs, U.; Müller, H.; Kleinermanns, K. Intermolecular vibrations of the phenol dimer revealed by spectral hole burning and dispersed fluorescence spectroscopy. J. Chem. Phys. 1995, 103, 9918–9928. [Google Scholar] [CrossRef]
- Roth, W.; Jacoby, C.; Westphal, A.; Schmitt, M. A study of 2H- and 2D-benzotriazole in their lowest electronic states by UV-laser double-resonance spectroscopy. J. Phys. Chem. A 1998, 102, 3048–3059. [Google Scholar] [CrossRef]
- Palmer, M.H.; Moyes, W.; Spiers, M. The Electronic Structure of Substituted Benzenes: Ab Initio Calculations and Photoelectron Spectra for Benzonitrile, the Tolunitriles, Ffluorobenzonitriles, Dicyanobenzenes and Ethynybenzene. J. Mol. Struct. 1980, 62, 165–167. [Google Scholar] [CrossRef]
- Wilson, E.B. The Normal Modes and Frequencies of Vibration of the Regular Plane Hexagon Model of the Benzene Molecule. Phys. Rev. 1934, 45, 706. [Google Scholar] [CrossRef]
- Varsanyi, G. Assignments for Vibrational Spectra of 700 Benzene Derivatives; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Mulliken, R.S. Report on Notation for the Spectra of Polyatomic Molecules. J. Chem. Phys. 1955, 23, 1997–2011. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra and Molecular Structure, III. Electronic Spectra and Electronic Structure of Polyatomic Molecules; van Nostrand Reinhold Company: New York, NY, USA, 1966. [Google Scholar]
- Kemp, D.J.; Tuttle, W.D.; Jones, F.M.; Gardner, A.M.; Andrejeva, A.; Wakefield, J.C.; Wright, T.G. Consistent assignment of the vibrations of symmetric and asymmetric meta-disubstituted benzenes. J. Mol. Spec. 2018, 346, 46–59. [Google Scholar] [CrossRef]
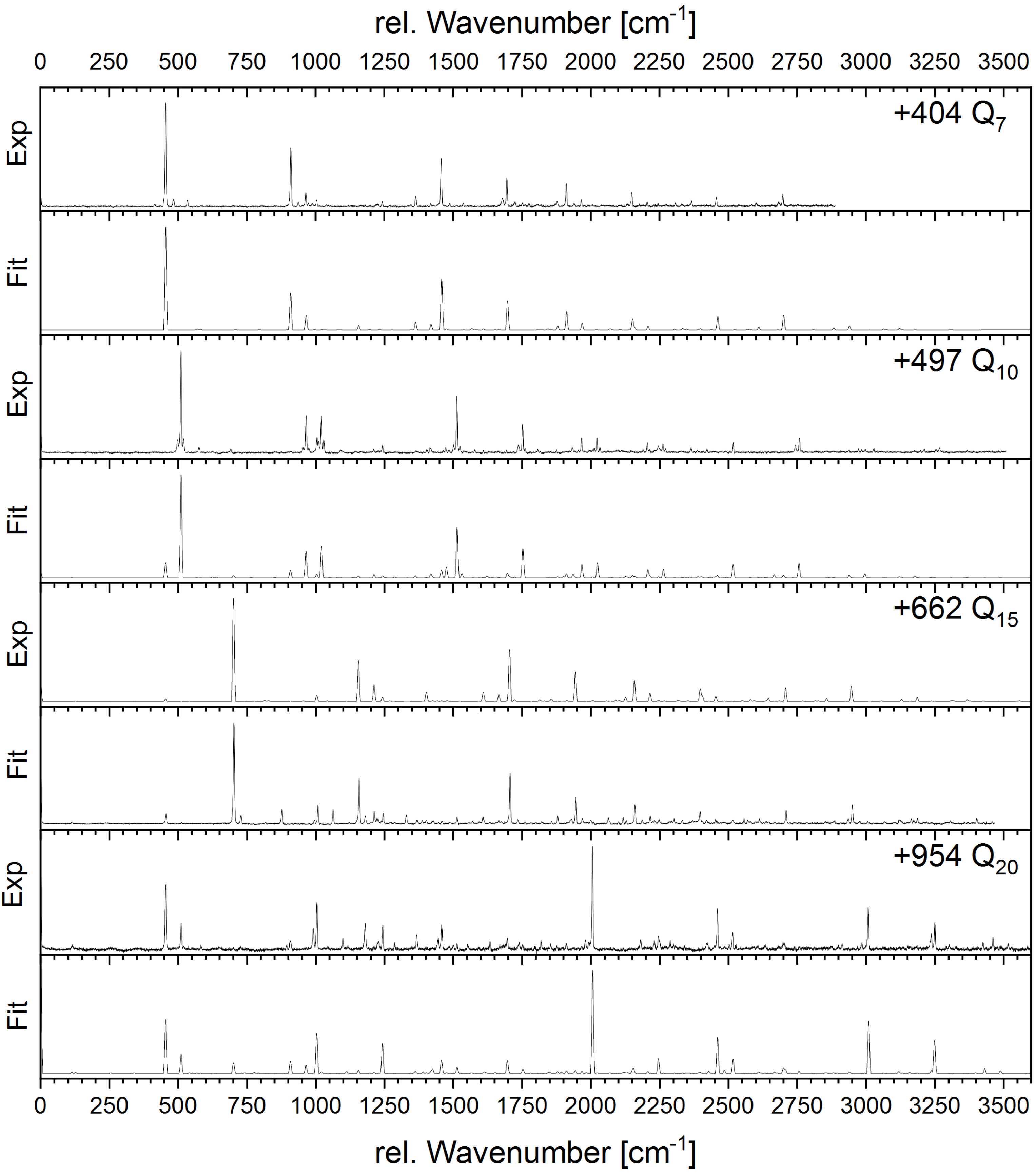

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCS-CC2/cc-pVTZ | Experiment | FCFit | |
|---|---|---|---|
| /MHz | 2706.7 | 2723.018609(46) 1 | 2706.7 2 |
| /MHz | 899.9 | 906.419893(21) 1 | 899.9 2 |
| /MHz | 675.4 | 679.859840(15) 1 | 675.4 2 |
| /amu Å2 | 0.0 | 0.207 | 0.0 |
| ()/MHz | 2632.9 | - | 2618.3 |
| ()/MHz | 885.9 | - | 884.3 |
| ()/MHz | 662.9 | - | 661.1 |
| () /amu Å2 | 0.0 | - | −0.014 |
| A(-)/MHz | −73.8 | - | −88.4 |
| B(-)/MHz | −14.0 | - | −15.6 |
| C(-)/MHz | −12.5 | - | −14.3 |
| () /∘ | 0.0 | - | - |
| () / | 35,958.0 | 35,146 3 | - |
| State | S0 | S1 | (S1-S0) | |||
|---|---|---|---|---|---|---|
| Method | SCS-CC2 | FCFit | SCS-CC2 | FCFit | SCS-CC2 | FCFit |
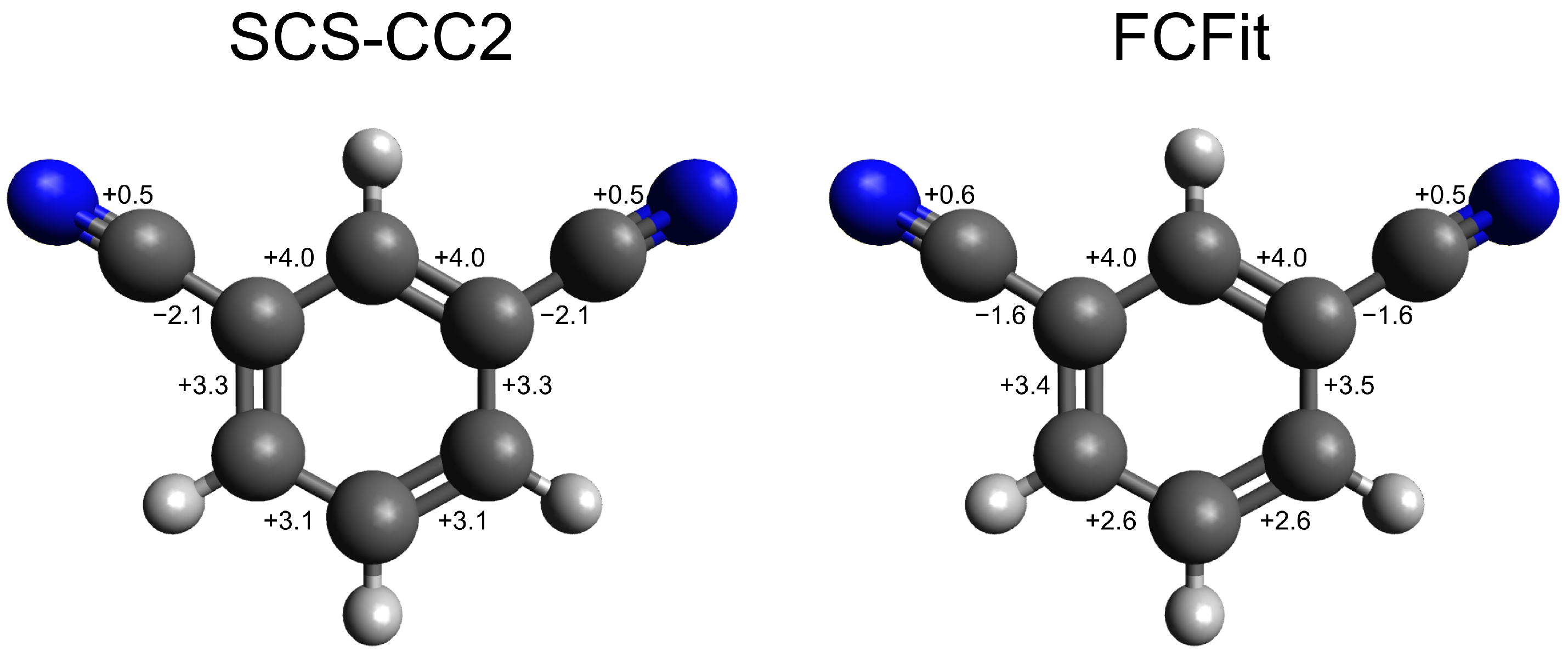
| C1-C2 | 139.4 | 139.4 | 142.5 | 142.0 | +3.1 | +2.6 |
| C2-C3 | 140.2 | 140.2 | 143.5 | 143.7 | +3.3 | +3.5 |
| C3-C4 | 139.9 | 139.9 | 143.9 | 143.9 | +4.0 | +4.0 |
| C4-C5 | 139.9 | 139.9 | 143.9 | 143.9 | +4.0 | +4.0 |
| C5-C6 | 140.2 | 140.2 | 143.5 | 143.6 | +3.3 | +3.4 |
| C6-C1 | 139.4 | 139.4 | 142.5 | 142.0 | +3.1 | +2.6 |
| C3-C8 | 143.8 | 143.8 | 141.7 | 142.2 | −2.1 | −1.6 |
| C8-N2 | 117.6 | 117.6 | 118.1 | 118.1 | +0.5 | +0.5 |
| C5-C7 | 143.8 | 143.8 | 141.7 | 142.2 | −2.1 | −1.6 |
| C7-N1 | 117.6 | 117.6 | 118.1 | 118.2 | +0.5 | +0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, J.; Amar, S.; Schmitt, M. Geometry Change of 1,3-Dicyanobenzene upon Electronic Excitation from a Franck–Condon Fit of Several Fluorescence Emission Spectra. Spectrosc. J. 2025, 3, 4. https://doi.org/10.3390/spectroscj3010004
Martini J, Amar S, Schmitt M. Geometry Change of 1,3-Dicyanobenzene upon Electronic Excitation from a Franck–Condon Fit of Several Fluorescence Emission Spectra. Spectroscopy Journal. 2025; 3(1):4. https://doi.org/10.3390/spectroscj3010004
Chicago/Turabian StyleMartini, Jascha, Simran Amar, and Michael Schmitt. 2025. "Geometry Change of 1,3-Dicyanobenzene upon Electronic Excitation from a Franck–Condon Fit of Several Fluorescence Emission Spectra" Spectroscopy Journal 3, no. 1: 4. https://doi.org/10.3390/spectroscj3010004
APA StyleMartini, J., Amar, S., & Schmitt, M. (2025). Geometry Change of 1,3-Dicyanobenzene upon Electronic Excitation from a Franck–Condon Fit of Several Fluorescence Emission Spectra. Spectroscopy Journal, 3(1), 4. https://doi.org/10.3390/spectroscj3010004