Probing Structural Features and Binding Mode of 3-Arylpyrimidin-2,4-diones within Housefly γ-Aminobutyric Acid (GABA) Receptor

Abstract
:1. Introduction
2. Results and Discussion
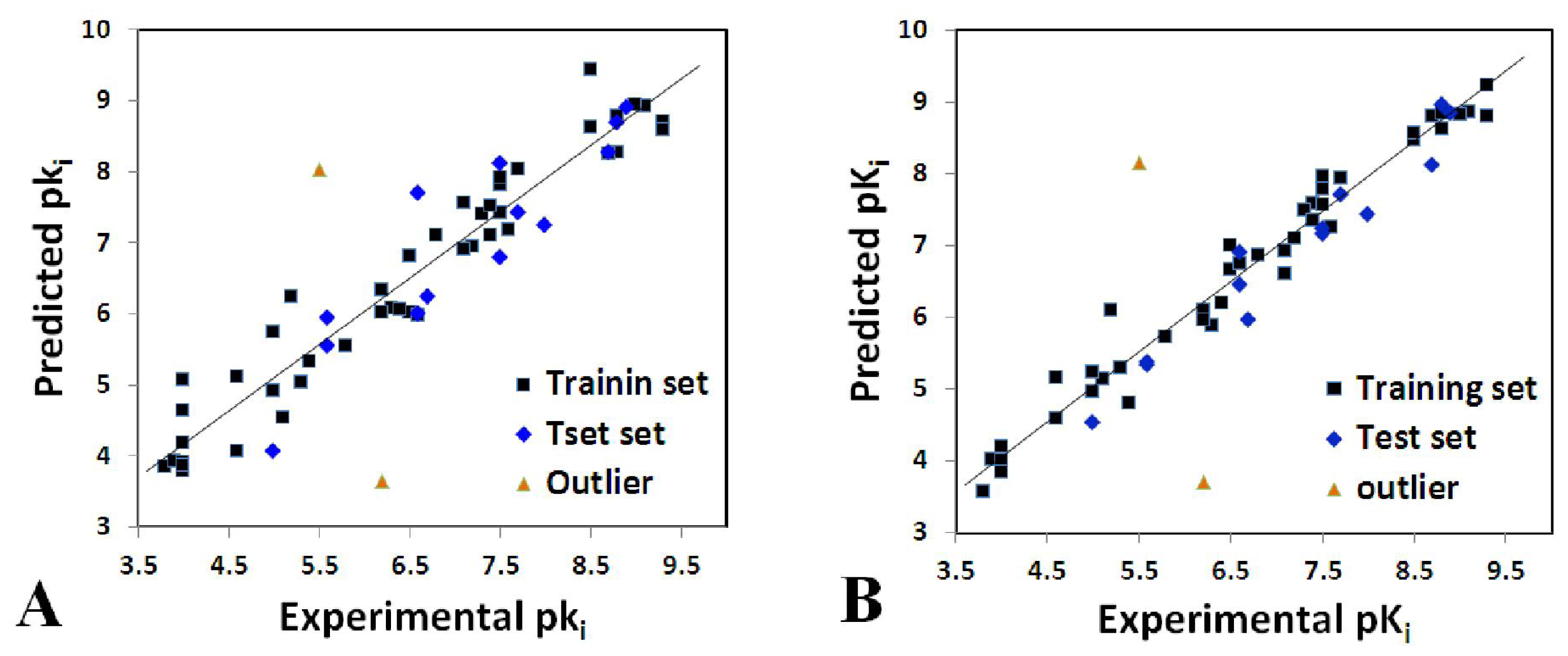
2.1. Statistical Analysis
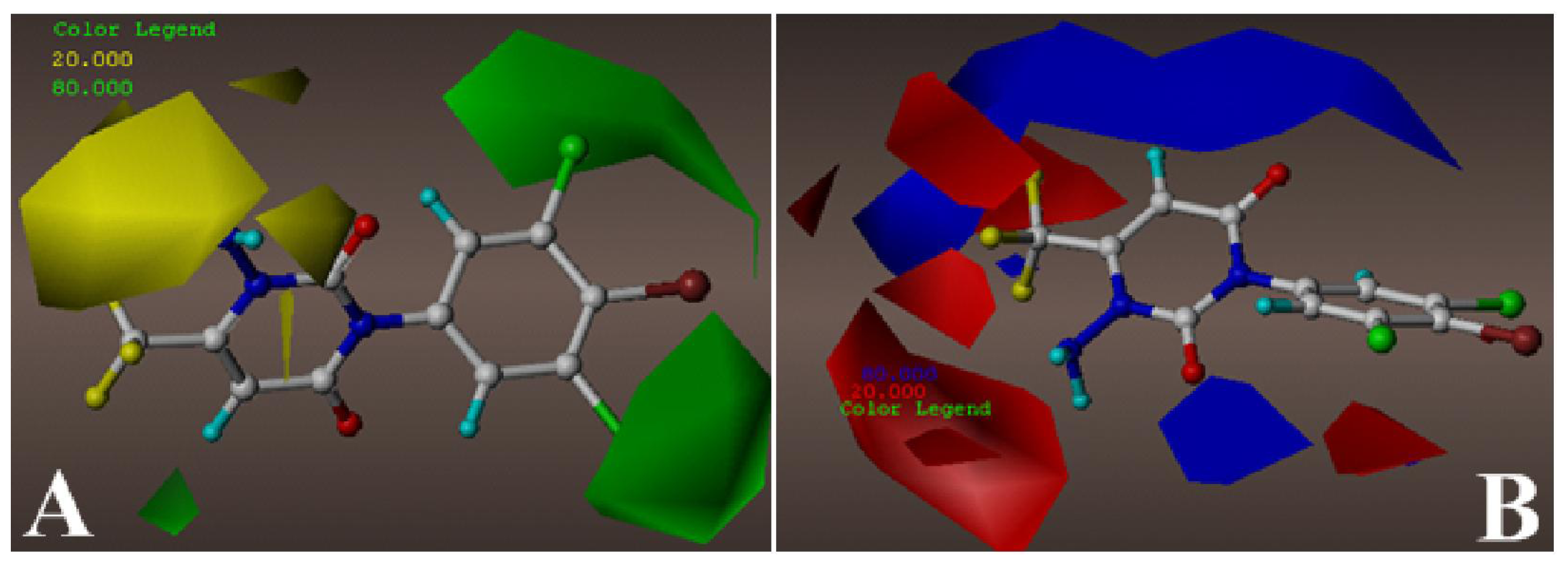
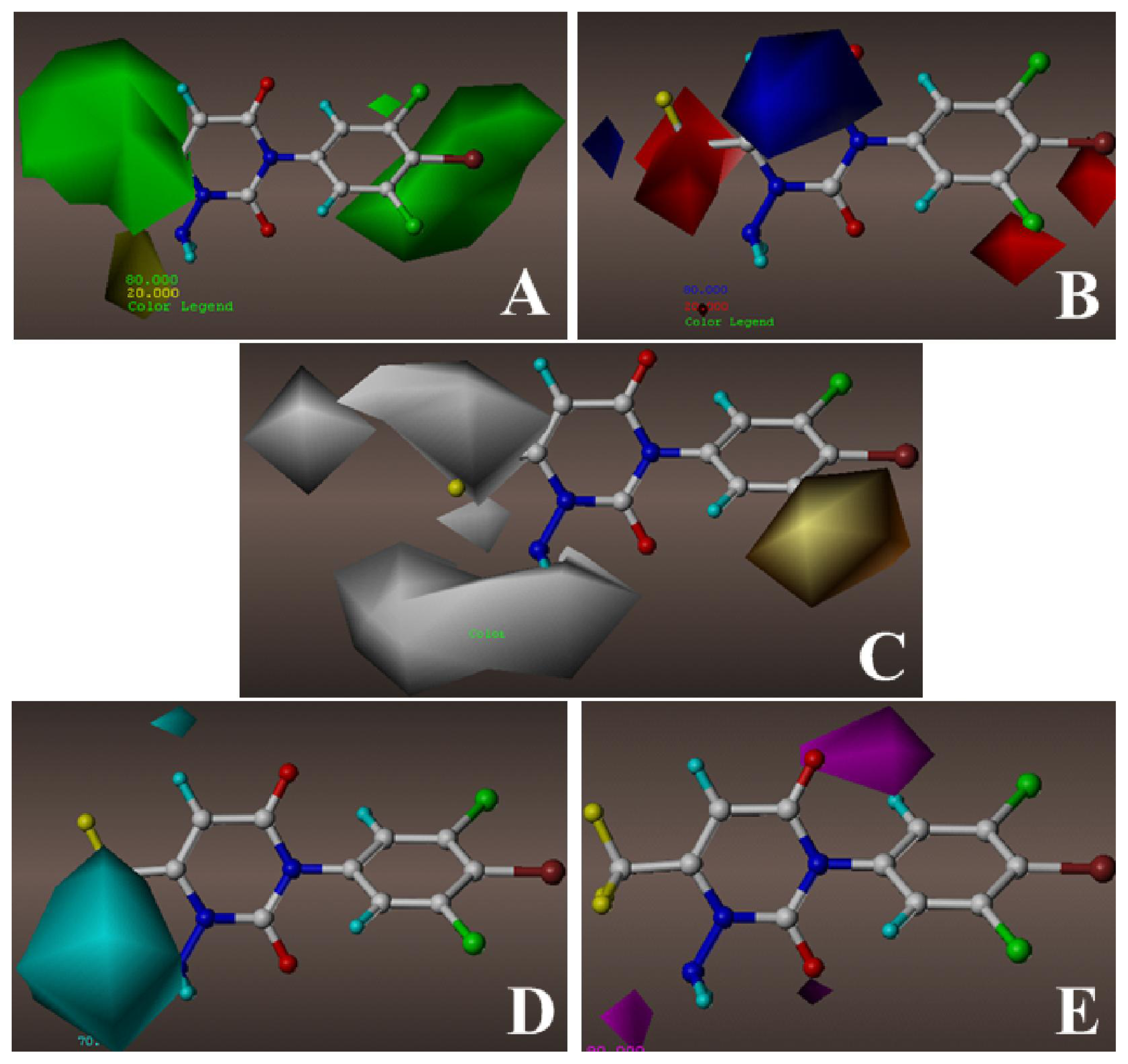
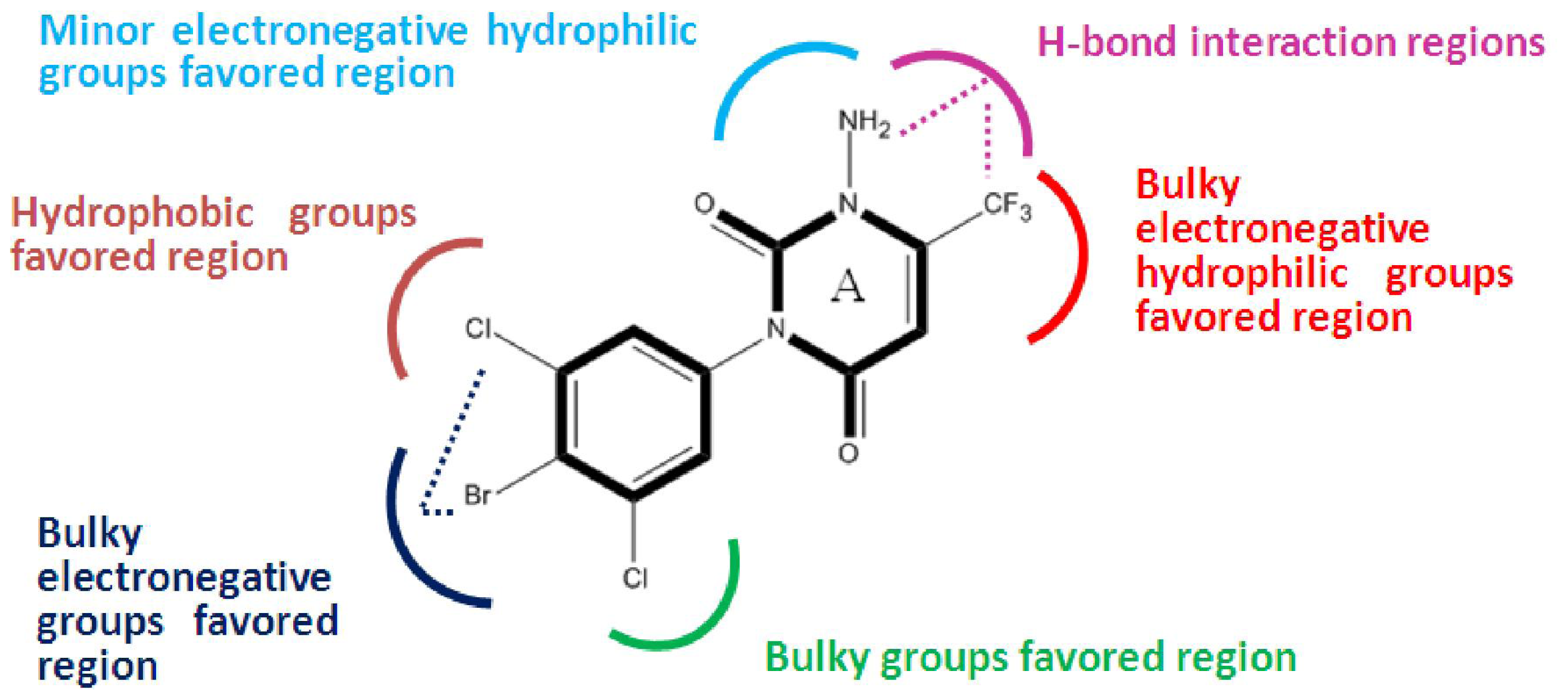
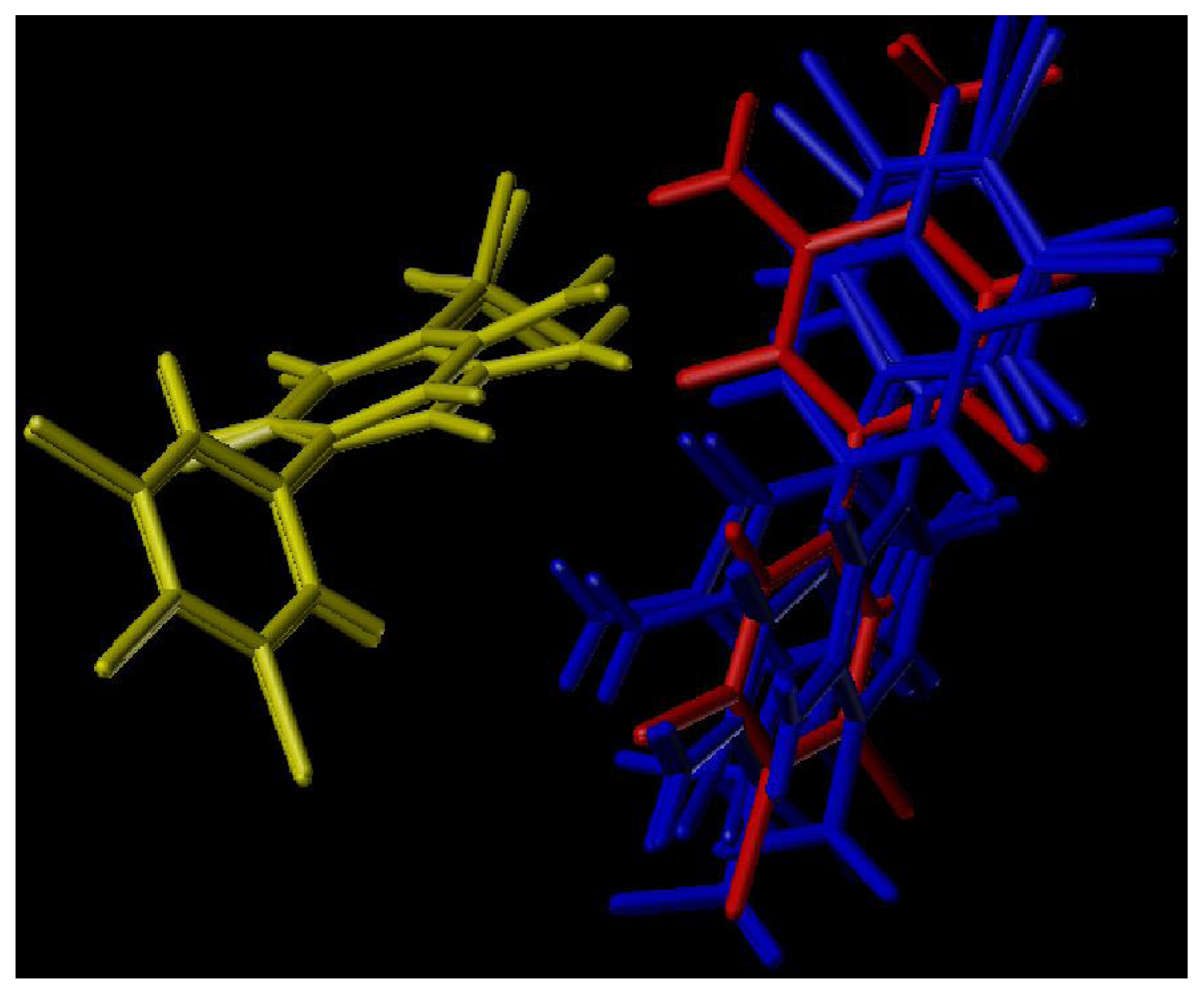
2.2. 3D-QSAR Contour Maps
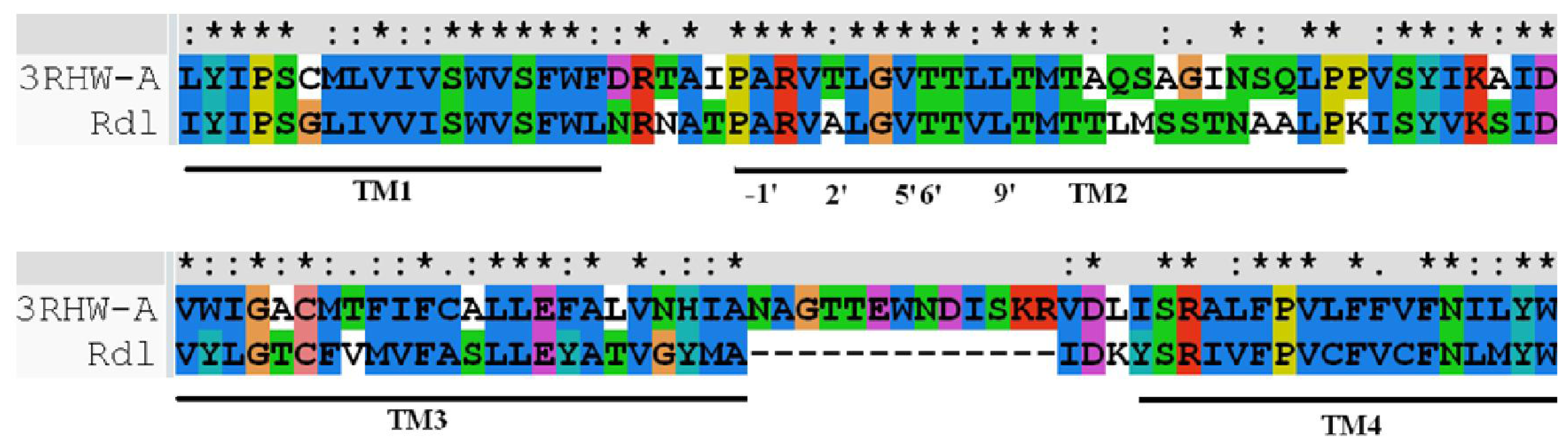
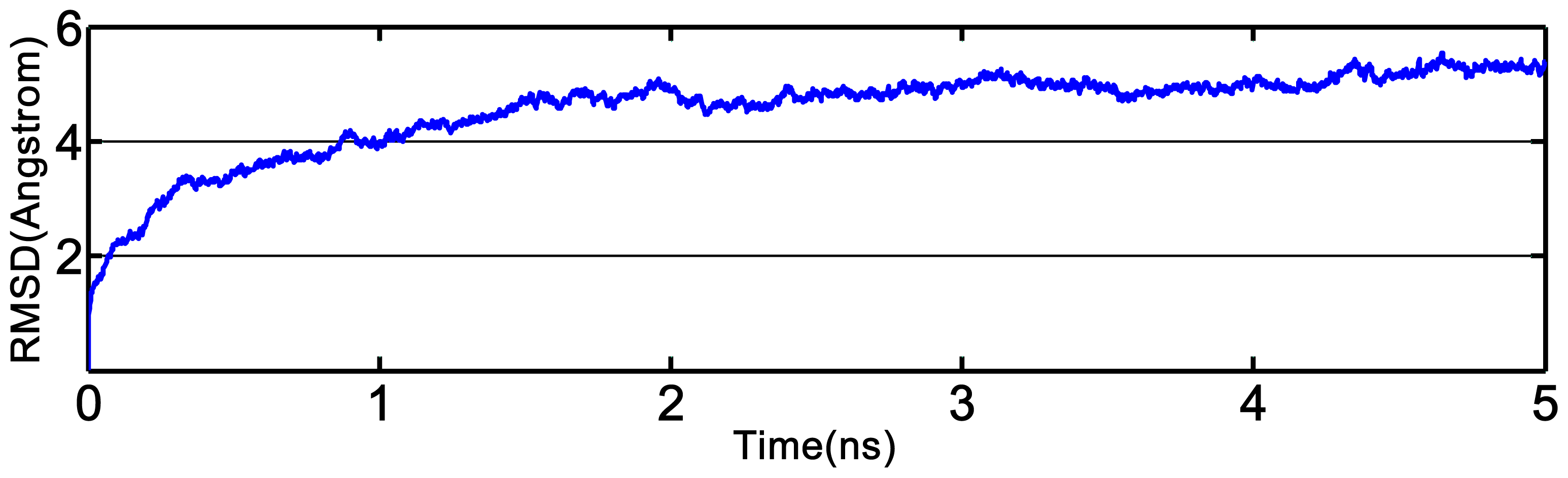
2.3. Homology Modeling and Molecular Dynamics Simulation
2.4. Docking Analysis and Comparisons with 3D-Contour Map
2.5. Binding Mode for APDs in the Housefly GABA Receptor
3. Materials and Methods
3.1. Dataset
3.2. Molecular Modeling and Alignment
3.3. Homology Modeling and Molecular Dynamics Simulation
3.4. Docking
3.5. 3D-QSAR Analysis
4. Conclusion
Acknowledgements
References
- Wolff, M; Wingate, V. Characterization and comparative pharmacological studies of a functional γ-aminobutyric acid (GABA) receptor cloned from the tobacco budworm, Heliothis virescens, (Noctuidae:Lepidoptera). Invertebr Neurosci 1998, 3, 305–315. [Google Scholar]
- Shimotahira, H; Fusazaki, S; Ikeda, I; Ozoe, Y. A photoreactive probe that differentiates the binding sites of noncompetitive GABA receptor antagonists. Bioorg Med Chem Lett 2011, 21, 1598–1600. [Google Scholar]
- Tong, F; Coats, JR. Effects of monoterpenoid insecticides on [3H]-TBOB binding in house fly GABA receptor and 36Cl- uptake in American cockroach ventral nerve cord. Pestic Biochem Phys 2010, 98, 317–324. [Google Scholar]
- Vijayan, RSK; Ghoshal, N. Structural basis for ligand recognition at the benzodiazepine binding site of GABAA α3 receptor, and pharmacophore-based virtual screening approach. J Mol Graph Model 2008, 27, 286–298. [Google Scholar]
- Cheng, J; Ju, XL; Chen, XY; Liu, GY. Homology modeling of human α1β2γ2 and house fly β3 GABA receptor channels and Surflex-docking of fipronil. J Mol Model 2009, 15, 1145–1153. [Google Scholar]
- Buckingham, SD; Biggin, PC; Sattelle, BM; Brown, LA; Sattelle, DB. Insect GABA receptors: Splicing, editing, and targeting by antiparasitics and insecticides. Mol Pharmacol 2005, 68, 942–951. [Google Scholar]
- Ozoe, Y; Akamatsu, M. Non-competitive GABA antagonists: Probing the mechanisms of their selectivity for insect versus mammalian receptors. Pest Manag Sci 2001, 57, 923–931. [Google Scholar]
- Sammelson, RE; Caboni, P; Durkin, KA; Casida, JE. GABA receptor antagonists and insecticides: Common structural features of 4-alkyl-1-phenylpyrazoles and 4-alkyl-1-phenyltrioxabicyclooctanes. Bioorg Med Chem 2004, 12, 3345–3355. [Google Scholar]
- Lyga, JW; Ali, SF; Kinne, LP; Marek, FL; Wusaty, MA; Staetz, CA; Willut, J. Discovery of 3-arylpyrimidin-2,4-diones as GABA-gated Chloride Channel Insecticides: Translation from Target Site to Field. In Synthesis and Chemistry of Agrochemicals, 7th ed; Lyga, JW, Theodoritis, G, Eds.; Oxford University Press: Cary, NC, USA, 2007; Volume 948, pp. 153–166. [Google Scholar]
- Chen, L; Durkin, KA; Casida, JE. Structural model for gamma-aminobutyric acid receptor noncompetitive antagonist binding: Widely diverse structures fit the same site. Proc Natl Acad Sci USA 2006, 103, 5185–5190. [Google Scholar]
- Bloomquist, JR. Chloride channels as tools for developing selective insecticides. Arch Insect Biochem 2003, 54, 145–156. [Google Scholar]
- Ci, S; Ren, TR; Su, ZG. Modeling the interaction of fipronil-related non-competitive antagonists with the GABA β3-receptor. J Mol Model 2007, 13, 457–464. [Google Scholar]
- Kitulagodage, M; Buttemer, WA; Astheimer, LB. Adverse effects of fipronil on avian reproduction and development: maternal transfer of fipronil to eggs in zebra finch Taeniopygia guttata and in ovo exposure in chickens. Gallus domesticus Ecotoxicology 2011, 20, 653–660. [Google Scholar]
- Kitulagodage, M; Isanhart, J; Buttemer, WA; Hooper, MJ; Astheimer, LB. Fipronil toxicity in northern bobwhite quail Colinus virginianus: reduced feeding behaviour and sulfone metabolite formation. Chemosphere 2011, 83, 524–530. [Google Scholar]
- Overmyer, JP; Rouse, DR; Avants, JK; Garrison, AW; Delorenzo, ME; Chung, KW; Key, PB; Wilson, WA; Black, MC. Toxicity of fipronil and its enantiomers to marine and freshwater non-targets. J Environ Sci Health B 2007, 42, 471–480. [Google Scholar]
- Hisano, K; Ozoe, F; Huang, J; Kong, X; Ozoe, Y. The channel-lining 6′ amino acid in the second membrane-spanning region of ionotropic GABA receptors has more profound effects on 4′-ethynyl-4-n-propylbicycloorthobenzoate binding than the 2′ amino acid. Invert Neurosci 2007, 7, 39–46. [Google Scholar]
- Le Goff, G; Hamon, A; Berge, JB; Amichot, M. Resistance to fipronil in Drosophila simulans: Influence of two point mutations in the RDL GABA receptor subunit. J Neurochem 2005, 92, 1295–1305. [Google Scholar]
- Hosie, AM; Buckingham, SD; Hamon, A; Sattelle, DB. Replacement of asparagine with arginine at the extracellular end of the second transmembrane (M2) region of insect GABA receptors increases sensitivity to penicillin G. Invert Neurosci 2006, 6, 75–79. [Google Scholar]
- Akamatsu, M; Ozoe, Y; Ueno, T; Fujita, T; Mochida, K; Nakamura, T; Matsumura, F. Sites of action of noncompetitive GABA antagonists in houseflies and rats: Three-dimensional QSAR analysis. Pestic Sci 1997, 49, 319–332. [Google Scholar]
- Anthony, N; Unruh, T; Ganser, D; ffrench-Constant, R. Duplication of the Rdl GABA receptor subunit gene in an insecticide-resistant aphid. Myzus persicae Mol Gen Genet 1998, 260, 165–175. [Google Scholar]
- Alam, MS; Huang, J; Ozoe, F; Matsumura, F; Ozoe, Y. Synthesis, 3D-QSAR, and docking studies of 1-phenyl-1H-1,2,3-triazoles as selective antagonists for β3 over α1β2γ2 GABA receptors. Bioorg Med Chem 2007, 15, 5090–5104. [Google Scholar]
- Wang, G; Li, Y; Liu, X; Wang, Y. Understanding the aquatic toxicity of pesticide: Structure-activity relationship and molecular descriptors to distinguish the ratings of toxicity. QSAR Comb Sci 2009, 28, 1418–1431. [Google Scholar]
- McGonigle, I; Lummis, SCR. Molecular characterization of agonists that bind to an insect GABA receptor. Biochemistry 2010, 49, 2897–2902. [Google Scholar]
- Penuel, E; Han, D; Favero, K; Lam, E; Liu, Y; Parkin, NT. Development of a rapid phenotypic susceptibility assay for HCV polymerase inhibitors. Antiviral Ther 2006, 11, S12. [Google Scholar]
- Sedelnikova, A; Erkkila, BE; Harris, H; Zakharkin, SO; Weiss, DS. Stoichiometry of a pore mutation that abolishes picrotoxin-mediated antagonism of the GABA(A) receptor. J Physiol 2006, 577, 569–577. [Google Scholar]
- Clark, M; Cramer, RD; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J Comput Chem 1989, 10, 982–1012. [Google Scholar]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov (accessed on 1 April 2011).
- Sali, A; Blundell, TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993, 234, 779–815. [Google Scholar]
- Thompson, JD; Higgins, DG; Gibson, TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Res 1994, 22, 4673–4680. [Google Scholar]
- Eramian, D; Shen, MY; Devos, D; Melo, F; Sali, A; Marti-Renom, MA. A composite score for predicting errors in protein structure models. Protein Sci 2006, 15, 1653–1666. [Google Scholar]
- Lindahl, E; Hess, B; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Model 2001, 7, 306–317. [Google Scholar]
- van der Spoel, D; van Buuren, AR; Tieleman, DP; Berendsen, HJ. Molecular dynamics simulations of peptides from BPTI: A closer look at amide-aromatic interactions. J Biomol NMR 1996, 8, 229–238. [Google Scholar]
- Berendsen, HJC; Grigera, JR; Strsstsma, TP. The missing term in effective pair potentials. J Phys Chem 1987, 91, 6269–6271. [Google Scholar]
- Hess, B; Bekker, H; Berendsen, HJC; Fraaije, JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem 1997, 18, 1463–1472. [Google Scholar]
- Darden, T; York, D; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys 1993, 98, 10089–10092. [Google Scholar]
- Berendsen, HJC; Postma, JPM; van Gunsteren, WF; DiNola, A; Haak, JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984, 81, 3684–3690. [Google Scholar]
- Liu, JL; Wang, FF; Ma, Z; Wang, X; Wang, YH. Structural determination of three different series of compounds as Hsp90 inhibitors using 3D-QSAR modeling, molecular docking and molecular dynamics methods. Int J Mol Sci 2011, 12, 946–970. [Google Scholar]
- Welch, W; Ruppert, J; Jain, AN. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol 1996, 3, 449–462. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Ligand-Based | Receptor-Based | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| q2 a | 0.60 | 0.62 | 0.34 | 0.55 |
| r2ncv b | 0.94 | 0.97 | 0.81 | 0.85 |
| SEE c | 0.48 | 0.32 | 0.78 | 0.70 |
| F d | 90.71 | 126.18 | 56.61 | 45.23 |
| r2pred e | 0.82 | 0.95 | 0.85 | 0.90 |
| SEP f | 1.17 | 1.22 | 1.44 | 1.21 |
| Nc g | 6 | 10 | 3 | 5 |
| Field contribution
| ||||
| S | 0.573 | 0.139 | 0.533 | 0.222 |
| E | 0.427 | 0.338 | 0.467 | - |
| H | - | 0.383 | - | 0.515 |
| D | - | 0.059 | - | 0.091 |
| A | - | 0.081 | - | 0.173 |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No | X | R1 | R2 | pKi a | No | X | R1 | R2 | pKi a |
| 1 | 4-Cl | CH3 | CF3 | 5.4 | 31 | 3-Cl,4-Cl,5-Cl | CH3 | CH2SCH3 | 4.6 |
| 2 | 3-Cl | CH3 | CF3 | 5.8 | 32 | 3-Cl,4-Cl,5-Cl | CH3 | COOH | 4.0 |
| 3 | 3-Cl,5-Cl | CH3 | CF3 | 6.8 | 33 | 3-Cl,4-Cl,5-Cl | CH3 | CN | 6.2 |
| 4# | 3-Cl,4-Cl | CH3 | CF3 | 6.6 | 34 | 3-Cl,4-Cl,5-Cl | CH3 | CH2CH2CF3 | 6.4 |
| 5# | 2-Cl,3-Cl | CH3 | CF3 | 6.6 | 35 | 3-Cl,4-Cl,5-Cl | CH3 | CH2CH2CH3 | 6.2 |
| 6# | 2-Cl,6-Cl | CH3 | CF3 | 5.6 | 36 | 3-Cl,4-Cl,5-Cl | CH3 | CF2CF2CF2CF3 | 5.0 |
| 7# | 3-Cl,4-Cl,5-Cl | CH3 | CF3 | 8.0 | 37 | 3-Cl,4-Cl,5-Cl | CH3 | 2-F-phenyl | 4.0 |
| 8 | 2-Cl,3-Cl,5-Cl | CH3 | CF3 | 7.4 | 38# | 3-Cl,4-Cl,5-Cl | CH3 | 4-F-phenyl | 5.0 |
| 9 | 2-Cl,4-Cl,6-Cl | CH3 | CF3 | 5.2 | 39# | 3-Cl,4-Cl,5-Cl | CH3 | Cl | 5.6 |
| 10 | 2-Cl,3-Cl,6-Cl | CH3 | CF3 | 7.6 | 40# | 4-Cl | NH2 | CF3 | 6.7 |
| 11 | 2-Cl,4-Cl,5-Cl | CH3 | CF3 | 7.1 | 41# | 2-Cl,4-Cl | NH2 | CF3 | 7.5 |
| 12 | -H | CH3 | CF3 | 4.0 | 42 | 3-Cl,5-Cl | NH2 | CF3 | 7.7 |
| 13 | 3-Cl,4-Cl,5-Cl | CH(CH3)2 | CF3 | 6.5 | 43# | 3-Cl,4-Cl | NH2 | CF3 | 7.7 |
| 14# | 3-Cl,4-Cl,5-Cl | CH2C(CH3)3 | CF3 | 6.2 | 44 | 2-Cl,4-Cl,5-Cl | NH2 | CF3 | 7.5 |
| 15 | 3-Cl,4-Cl,5-Cl | CH2CF3 | CF3 | 7.2 | 45# | 2-Cl,4-Cl,6-Cl | NH2 | CF3 | 7.5 |
| 16 | 3-Cl,4-Cl,5-Cl | CH2COCH3 | CF3 | 3.8 | 46 | 2-Cl,6-Cl | NH2 | CF3 | 7.1 |
| 17 | 3-Cl,4-Cl,5-Cl | CH2CN | CF3 | 7.4 | 47 | 3-Cl | NH2 | CF3 | 6.5 |
| 18 | 3-Cl,4-Cl,5-Cl | CH2Ph | CF3 | 3.9 | 48 | 2-Cl,3-Cl,4-Cl | NH2 | CF3 | 8.5 |
| 19 | 3-Cl,4-Cl,5-Cl | CH2CH2OCH3 | CF3 | 6.6 | 49 | -H | NH2 | CF3 | 5 |
| 20 | 3-Cl,4-Cl,5-Cl | CH2CH2CH2CH3 | CF3 | 4.0 | 50 | 3-Cl,4-I,5-Cl | NH2 | CF3 | 9.3 |
| 21# | 3-Cl,4-Cl,5-Cl | H | CF3 | 5.5 | 51 | 3-Cl,4-SOCH3,5-Cl | NH2 | CF3 | 7.5 |
| 22 | 3-Cl,4-Cl,5-Cl | NH2 | CF3 | 8.8 | 52 | 3-Cl,4-SCH3,5-Cl | NH2 | CF3 | 8.7 |
| 23 | 3-Cl,4-Cl,5-Cl | NHCOCH3 | CF3 | 5.3 | 53# | 3-Cl,4-Ph,5-Cl | NH2 | CF3 | 8.9 |
| 24 | 3-Cl,4-Cl,5-Cl | CH3 | CH(CH3)2 | 6.3 | 54 | 3-Cl,4-OCH3,5-Cl | NH2 | CF3 | 8.5 |
| 25 | 3-Cl,4-Cl,5-Cl | CH3 | CF2CF2 CF3 | 7.3 | 55 | 3-Cl,4-(2-thienyl),5-Cl | NH2 | CF3 | 9.1 |
| 26 | 3-Cl,4-Cl,5-Cl | CH3 | CH2O CH3 | 5.1 | 56# | 3-Cl,4-N(CH3)2,5-Cl | NH2 | CF3 | 8.8 |
| 27 | 3-Cl,4-Cl,5-Cl | CH3 | Ph | 4.0 | 57 | 3-Cl,4-OCH(CH3)2,5-Cl | NH2 | CF3 | 9.0 |
| 28 | 3-Cl,4-Cl,5-Cl | CH3 | CH2SO2 CH3 | 42 | 58 | 3-Cl,4-Br,5-Cl | NH2 | CF3 | 9.3 |
| 29 | 3-Cl,4-Cl,5-Cl | CH3 | CH3 | 4.6 | 59 | 3-Cl,4-CF2CF3,5-Cl | NH2 | CF3 | 8.8 |
| 30 | 3-Cl,4-Cl,5-Cl | CH3 | C(CH3)3 | 7.5 | 60# | 3-F,4-CF3,5-F | NH2 | CF3 | 8.7 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Q.; Zhang, L.; Ma, Z.; Kong, X.; Wang, F.; Zhang, H.; Wang, Y. Probing Structural Features and Binding Mode of 3-Arylpyrimidin-2,4-diones within Housefly γ-Aminobutyric Acid (GABA) Receptor. Int. J. Mol. Sci. 2011, 12, 6293-6311. https://doi.org/10.3390/ijms12096293
Li Q, Zhang L, Ma Z, Kong X, Wang F, Zhang H, Wang Y. Probing Structural Features and Binding Mode of 3-Arylpyrimidin-2,4-diones within Housefly γ-Aminobutyric Acid (GABA) Receptor. International Journal of Molecular Sciences. 2011; 12(9):6293-6311. https://doi.org/10.3390/ijms12096293
Chicago/Turabian StyleLi, Qinfan, Lihui Zhang, Zhi Ma, Xiangya Kong, Fangfang Wang, Hong Zhang, and Yonghua Wang. 2011. "Probing Structural Features and Binding Mode of 3-Arylpyrimidin-2,4-diones within Housefly γ-Aminobutyric Acid (GABA) Receptor" International Journal of Molecular Sciences 12, no. 9: 6293-6311. https://doi.org/10.3390/ijms12096293
APA StyleLi, Q., Zhang, L., Ma, Z., Kong, X., Wang, F., Zhang, H., & Wang, Y. (2011). Probing Structural Features and Binding Mode of 3-Arylpyrimidin-2,4-diones within Housefly γ-Aminobutyric Acid (GABA) Receptor. International Journal of Molecular Sciences, 12(9), 6293-6311. https://doi.org/10.3390/ijms12096293