


Elucidation of Dithiol-yne Comb Polymer Architectures by Tandem Mass Spectrometry and Ion Mobility Techniques
 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
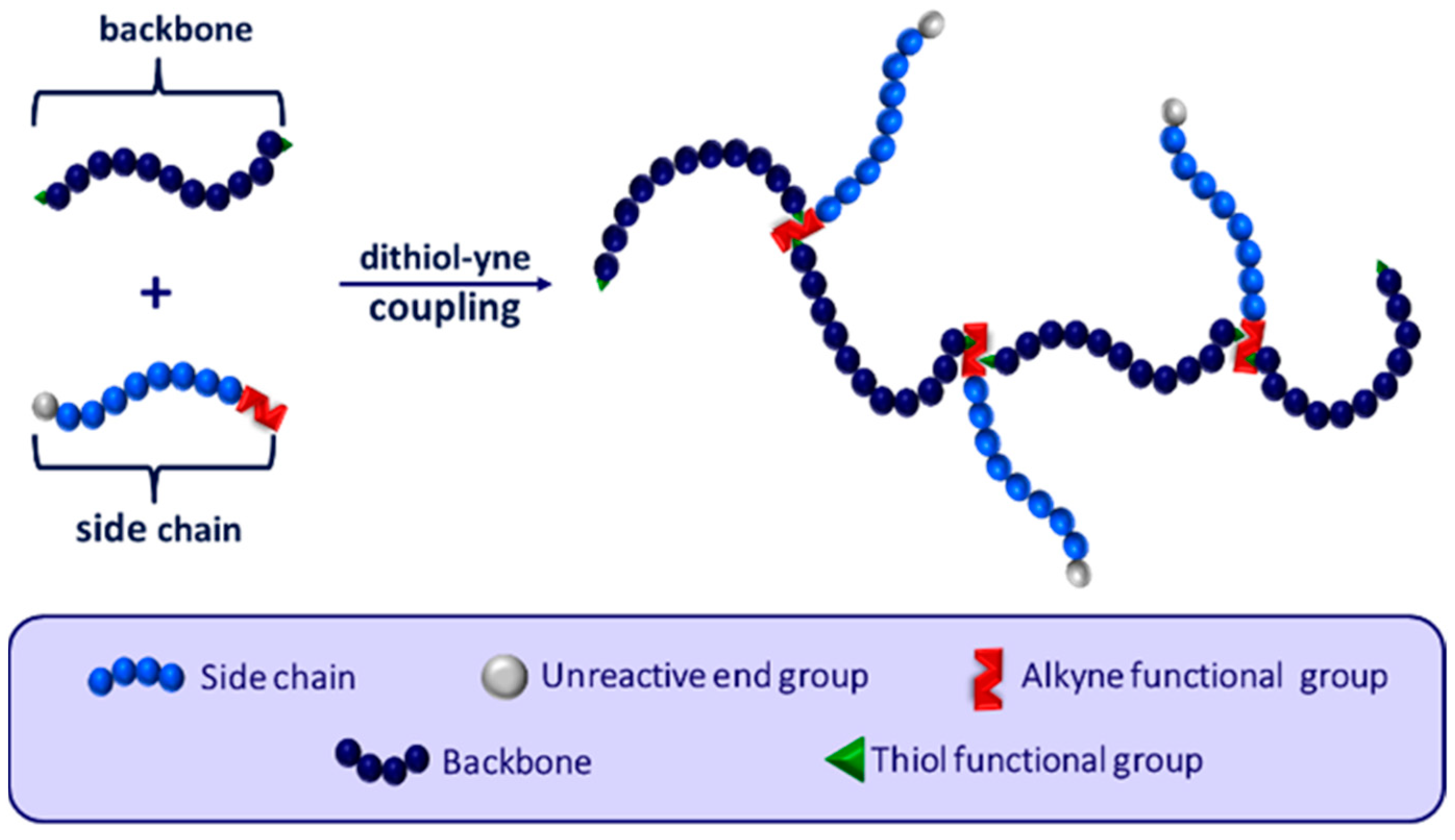
1. Introduction
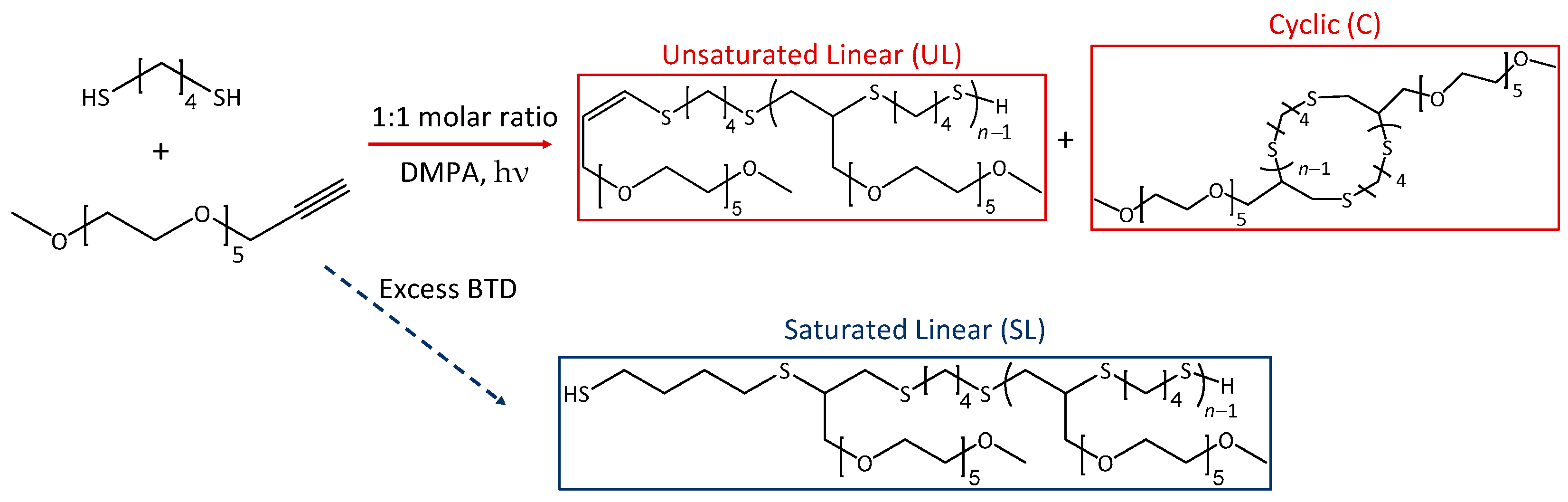
2. Materials and Methods
2.1. Materials
2.2. Synthesis of MeO-PEG5-Propargyl
2.3. Synthesis of CP MeO-PEG5-BDT
2.4. MS Instrument Conditions
2.5. Experimental Collision Cross-Sections
2.6. Computational Modeling and Theoretical Collision Cross-Section Calculations
3. Results and Discussion
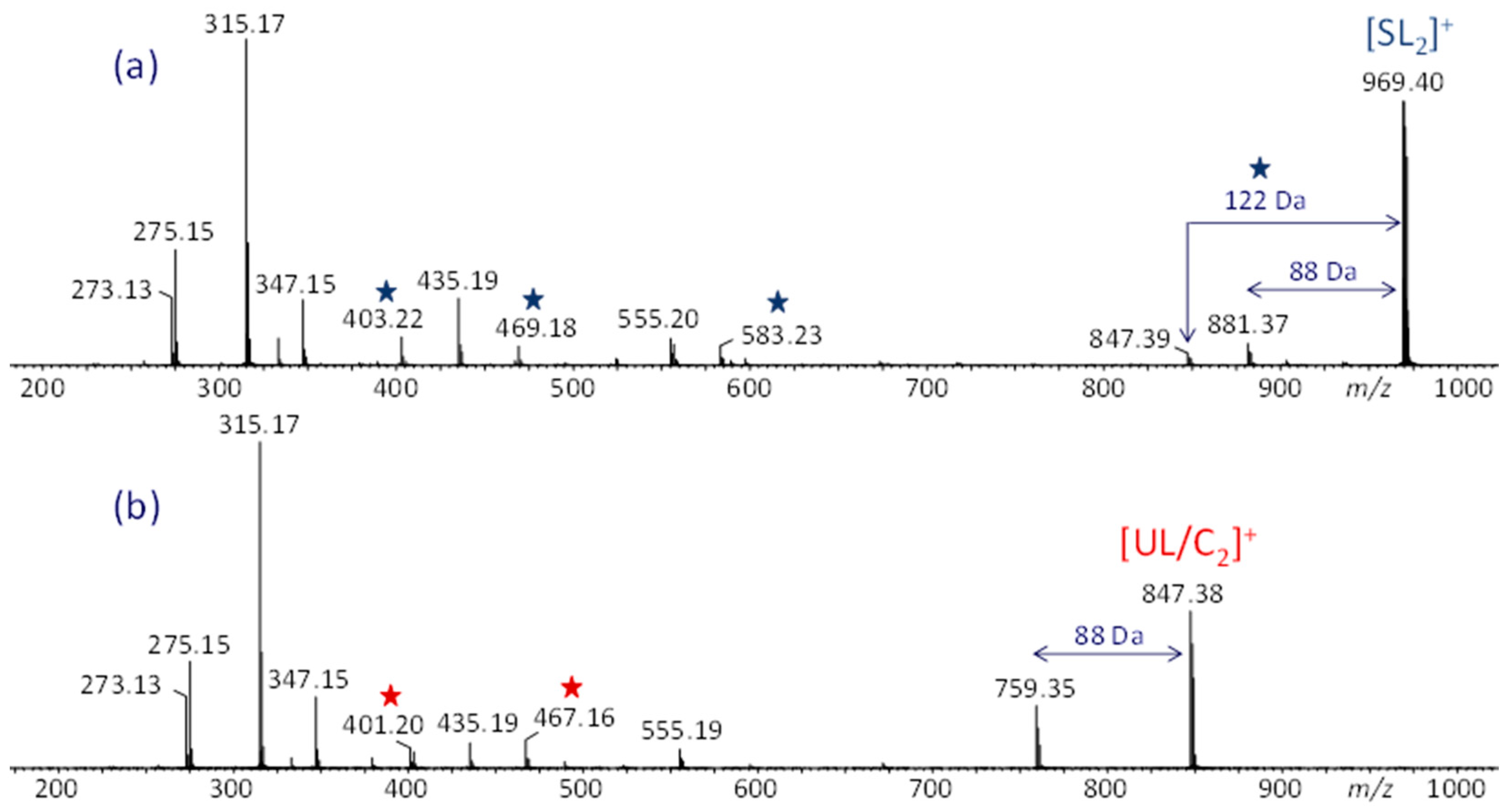
3.1. ESI-MS Analysis
3.2. ESI-MS/MS Analysis
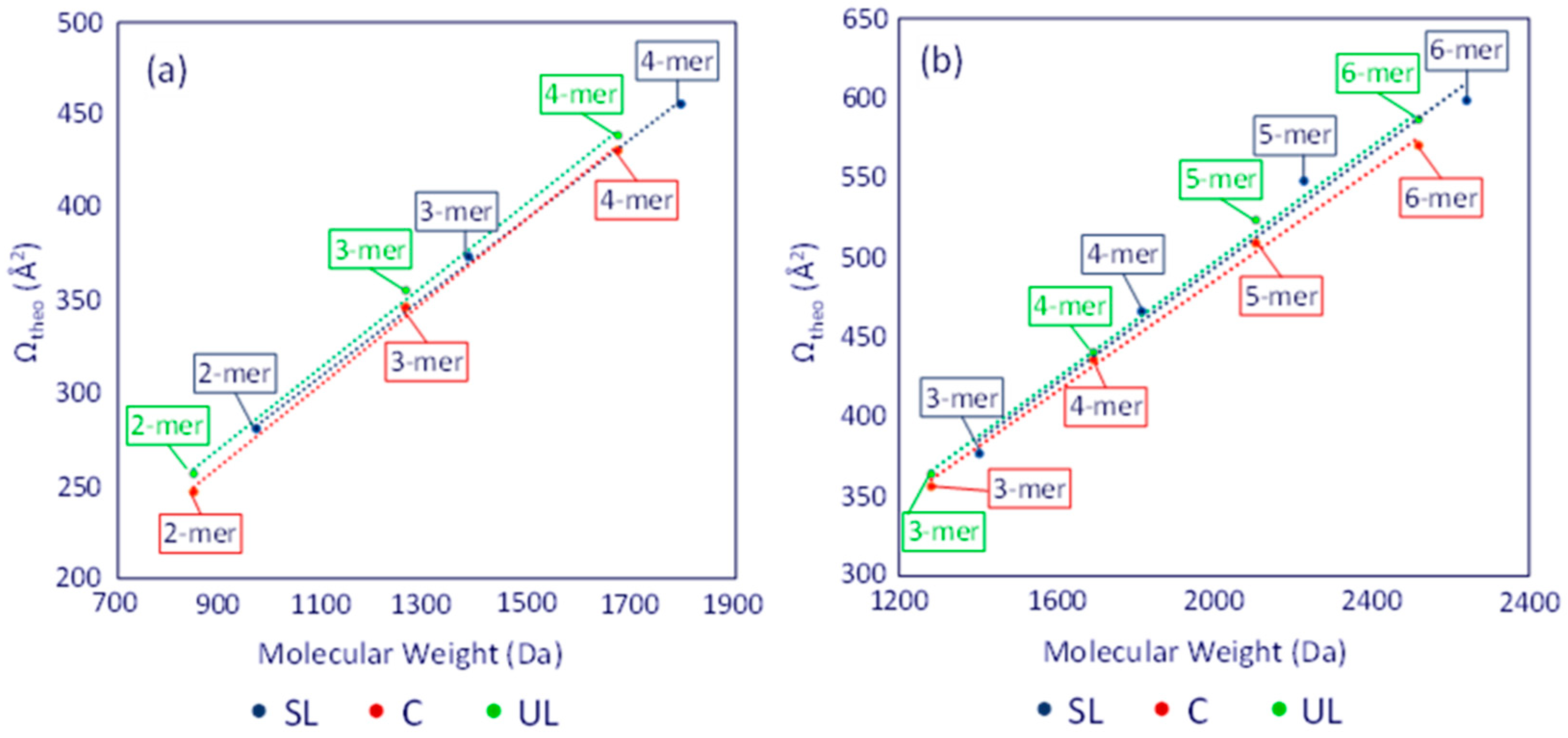
3.3. ESI-IM-MS Analysis
3.4. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Bochenek, S.; Rudov, A.A.; Sassmann, T.; Potemkin, I.I.; Richtering, W. Influence of architecture on the interfacial properties of polymers: Linear chains, stars, and microgels. Langmuir 2023, 39, 18354–18365. [Google Scholar] [CrossRef]
- Vicente-Alique, E.; Vega, J.F.; Robledo, N.; Nieto, J.; Martínez-Salazar, J. Study of the effect of the molecular architecture of the components on the melt rheological properties of polyethylene blends. J. Polym. Res. 2015, 22, 62. [Google Scholar] [CrossRef]
- Abbasi, M.; Faust, L.; Wilhelm, M. Comb and bottlebrush polymers with superior rheological and mechanical properties. Adv. Mater. 2019, 31, 1806484. [Google Scholar] [CrossRef]
- Ahmad, F.; Yuvaraj, N.; Bajpai, P.K. Effect of reinforcement architecture on the macroscopic mechanical properties of fiberous polymer composites: A review. Polym. Compos. 2020, 41, 2518–2534. [Google Scholar] [CrossRef]
- Le, T.-H.; Kim, Y.; Yoon, H. Electrical and electrochemical properties of conducting polymers. Polymers 2017, 9, 150. [Google Scholar] [CrossRef]
- Lazarova, K.; Vasileva, M.; Ivanova, S.; Novakov, C.; Christova, D.; Babeva, T. Influence of macromolecular architecture on the optical and humidity-sensing properties of poly(N,N-dimethylacrylamide)-based block copolymers. Polymers 2018, 10, 769. [Google Scholar] [CrossRef]
- Young, R.J.; Lovell, P.A. Introduction to Polymers, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar] [CrossRef]
- Chremos, A.; Douglas, J.F. Influence of branching on the configurational and dynamical properties of entangled polymer melts. Polymers 2019, 11, 1045. [Google Scholar] [CrossRef]
- Curole, B.J.; Broussard, W.J.; Nadeem, A.; Grayson, S.M. Dithiol-yne polymerization: Comb polymers with poly(ethylene glycol) side chain. ACS Polym. Au 2023, 3, 70–81. [Google Scholar] [CrossRef]
- Beers, K.L.; Gaynor, S.G.; Matyjaszewski, K.; Sheiko, S.S.; Moeller, M. Synthesis of densely grafted copolymers by atom transfer radical polymerization. Macromolecules 1998, 31, 9413–9415. [Google Scholar] [CrossRef]
- Zdyrko, B.; Luzinov, I. Polymer brushes by the “grafting to” method. Macromol. Rapid Commun. 2011, 32, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Verduzco, R.; Li, X.; Pesek, S.L.; Stein, G.E. Structure, function, self-assembly, and applications of bottlebrush copolymers. Chem. Soc. Rev. 2015, 44, 2405–2420. [Google Scholar] [CrossRef]
- Williams-Pavlantos, K. Multidimensional Mass Spectrometry Analysis and Imaging of Macromolecules and Material Surfaces. Ph.D. Dissertation, The University of Akron, Akron, OH, USA, May 2023. Available online: http://rave.ohiolink.edu/etdc/view?acc_num=akron1682339700569807 (accessed on 6 June 2024).
- Barth, H.G.; Boyes, B.E. Size exclusion chromatography. Anal. Chem. 1990, 62, 268–303. [Google Scholar] [CrossRef]
- Kissin, Y.V. Molecular weight distributions of linear polymers: Detailed analysis from GPC data. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 227–237. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer molecular weight analysis by 1H NMR spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Hanton, S.D. Mass spectrometry of polymers and polymer surfaces. Chem. Rev. 2001, 101, 527–570. [Google Scholar] [CrossRef]
- Gruendling, T.; Weidner, S.; Falkenhagen, J.; Barner-Kowollik, C. Mass spectrometry in polymer chemistry: A state-of-the-art up-date. Polym. Chem. 2010, 1, 599–617. [Google Scholar] [CrossRef]
- De Bruycker, K.; Welle, A.; Hirth, S.; Blanksby, S.J.; Barner-Kowollik, C. Mass spectrometry as a tool to advance polymer science. Nat. Rev. Chem. 2020, 4, 257–268. [Google Scholar] [CrossRef]
- Wesdemiotis, C.; Williams-Pavlantos, K.N.; Keating, A.R.; McGee, A.S.; Bochenek, C. Mass spectrometry of polymers: A tutorial review. Mass Spectrom. Rev. 2024, 43, 427–476. [Google Scholar] [CrossRef]
- Wesdemiotis, C.; Solak, N.; Polce, M.J.; Dabney, D.E.; Chaicharoen, K.; Katzenmeyer, B.C. Fragmentation pathways of polymer ions. Mass Spectrom. Rev. 2011, 30, 523–559. [Google Scholar] [CrossRef] [PubMed]
- Girod, M.; Phan, T.N.T.; Charles, L. Microstructural study of a nitroxide-mediated poly(Ethylene oxide)/polystyrene block copolymer (PEO-b-PS) by electrospray tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Crecelius, A.C.; Becer, C.R.; Knop, K.; Schubert, U.S. Block length determination of the block copolymer mPEG-b-PS using MALDI-TOF MS/MS. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4375–4384. [Google Scholar] [CrossRef]
- Yol, A.M.; Wesdemiotis, C. Multidimensional mass spectrometry methods for the structural characterization of cyclic polymers. React. Funct. Polym. 2014, 80, 95–108. [Google Scholar] [CrossRef]
- O’Neill, J.M.; Grayson, S.M.; Wesdemiotis, C. Separation and structural differentiation of tadpole and macrocycle isomers by ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS). Analyst 2022, 147, 2089–2096. [Google Scholar] [CrossRef]
- Gies, A.P.; Heath, W.H.; Keaton, R.J.; Jimenez, J.J.; Zupancic, J.J. MALDI-TOF/TOF CID Study of polycarbodiimide branching reactions. Macromolecules 2013, 46, 7616–7637. [Google Scholar] [CrossRef]
- Clemmer, D.E.; Jarrold, M.F. Ion mobility measurements and their applications to clusters and biomolecules. J. Mass Spectrom. 1997, 31, 577–592. [Google Scholar] [CrossRef]
- Bush, M.F.; Hall, Z.; Giles, K.; Hoyes, J.; Robinson, C.V.; Ruotolo, B.T. Collision cross sections of proteins and their complexes: A calibration framework and database for gas-phase structural biology. Anal. Chem. 2010, 82, 9557–9565. [Google Scholar] [CrossRef]
- Salbo, R.; Bush, M.F.; Naver, H.; Campuzano, I.; Robinson, C.V.; Pettersson, I.; Jørgensen, T.J.D.; Haselmann, K.F. Traveling-wave ion mobility mass spectrometry of protein complexes: Accurate calibrated collision cross-sections of human insulin oligomers. Rapid Commun. Mass Spectrom. 2012, 26, 1181–1193. [Google Scholar] [CrossRef]
- Christofi, E.; Barran, P. Ion mobility mass spectrometry (IM-MS) for structural biology: Insights gained by measuring mass, charge, and collision cross section. Chem. Rev. 2023, 123, 2902–2949. [Google Scholar] [CrossRef]
- Morsa, D.; Defize, T.; Dehareng, D.; Jérôme, C.; De Pauw, E. Polymer topology revealed by ion mobility coupled with mass spectrometry. Anal. Chem. 2014, 86, 9693–9700. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Marklund, E. Fundamentals of ion mobility spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.P.; Giles, K.; Bateman, R.H.; Radford, S.E.; Ashcroft, A.E. Monitoring copopulated conformational states during protein folding events using electrospray ionization-ion mobility spectrometry-mass spectrometry. J. Am. Soc. Mass Spectrom. 2007, 18, 2180–2190. [Google Scholar] [CrossRef]
- Charles, L.; Chendo, C.; Poyer, S. Ion mobility spectrometry–mass spectrometry coupling for synthetic polymers. Rapid Commun. Mass Spectrom. 2020, 34, e8624. [Google Scholar] [CrossRef]
- Duez, A.; Hoyas, S.; Josse, T.; Cornil, J.; Gerbaux, P.; De Winter, J. Gas-phase structure of polymer ions: Tying together theoretical approaches and ion mobility spectrometry. Mass Spectrom. Rev. 2023, 42, 1129–1151. [Google Scholar] [CrossRef]
- Liénard, R.; Dues, Q.; Grayson, S.M.; Gerbaux, P.; Coulembier, O.; De Winter, J. Limitations of ion mobility spectrometry-mass spectrometry for the relative quantification of architectural isomeric polymers: A case study. Rapid Commun. Mass Spectrom. 2020, 34, e8660. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, B.T.; Benesch, J.L.P.; Sandercock, A.M.; Hyung, S.-J.; Robinson, C.V. Ion mobility–mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.F.; Campuzano, I.D.G.; Robinson, C.V. Ion mobility mass spectrometry of peptide ions: Effects of drift gas and calibration strategies. Anal. Chem. 2012, 84, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility mass spectrometry measurement. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Material Studio™, by Dassault Systèmes BIOVIA, UK (Accelrys®), (License Purchased by The University of Akron). Available online: https://www.3ds.com/products/biovia/materials-studio (accessed on 6 June 2024).
- Sun, H.; Jin, Z.; Yang, C.; Akkermans, R.L.C.; Robertson, S.H.; Spenley, N.A.; Miller, S.; Todd, S.M. COMPASS II: Extended coverage for polymer and drug-like molecule databases. J. Mol. Model. 2016, 22, 47. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.H.; Bhotika, H.; Zheng, X.; Smith, R.D.; Burnum-Johnson, K.E.; Bilbao, A. Computational tools and algorithms for ion mobility spectrometry-mass spectrometry. Proteomics 2024, e2200436. [Google Scholar] [CrossRef]
- Mesleh, M.F.; Hunter, J.M.; Shvartsburg, A.A.; Schatz, G.C.; Jarrold, M.F. Structural information from ion mobility measurements: Effects of the long-range potential. J. Phys. Chem. 1996, 100, 16082–16086, reprinted in J. Phys. Chem. A 1997, 101, 968. [Google Scholar] [CrossRef]
- Beveridge, R.; Migas, L.G.; Das, R.K.; Pappu, R.V.; Kriwacki, R.W.; Barran, P.E. Ion mobility mass spectrometry uncovers the impact of the patterning of oppositely charged residues on the conformational distributions of intrinsically disordered proteins. J. Am. Chem. Soc. 2019, 141, 4908–4918. [Google Scholar] [CrossRef]
- Rainey, M.A.; Watson, C.A.; Asef, C.K.; Foster, M.R.; Baker, E.S.; Fernández, F.M. CCS predictor 2.0: An open-source Jupyter notebook tool for filtering out false positives in metabolomics. Anal. Chem. 2022, 94, 17456–17466. [Google Scholar] [CrossRef]
- Geue, N.; Bennett, T.S.; Arama, A.-A.-M.; Ramakers, A.I.; Whitehead, G.F.S.; Timco, G.A.; Armentrout, P.B.; McInnes, E.J.; Burton, N.A.; Winpenny, R.E.P.; et al. Disassembly mechanisms and energetics of polymetallic rings and rotaxanes. J. Am. Chem. Soc. 2022, 144, 22528–22539. [Google Scholar] [CrossRef]
- Geue, N.; Winpenny, R.E.P.; Barran, P.E. Ion mobility mass spectrometry for large Synthetic molecules: Expanding the analytical toolbox. J. Am. Chem. Soc. 2024, 46, 8800–8819. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams-Pavlantos, K.; Mokarizadeh, A.H.; Curole, B.J.; Grayson, S.M.; Tsige, M.; Wesdemiotis, C. Elucidation of Dithiol-yne Comb Polymer Architectures by Tandem Mass Spectrometry and Ion Mobility Techniques. Polymers 2024, 16, 1665. https://doi.org/10.3390/polym16121665
Williams-Pavlantos K, Mokarizadeh AH, Curole BJ, Grayson SM, Tsige M, Wesdemiotis C. Elucidation of Dithiol-yne Comb Polymer Architectures by Tandem Mass Spectrometry and Ion Mobility Techniques. Polymers. 2024; 16(12):1665. https://doi.org/10.3390/polym16121665
Chicago/Turabian StyleWilliams-Pavlantos, Kayla, Abdol Hadi Mokarizadeh, Brennan J. Curole, Scott M. Grayson, Mesfin Tsige, and Chrys Wesdemiotis. 2024. "Elucidation of Dithiol-yne Comb Polymer Architectures by Tandem Mass Spectrometry and Ion Mobility Techniques" Polymers 16, no. 12: 1665. https://doi.org/10.3390/polym16121665
APA StyleWilliams-Pavlantos, K., Mokarizadeh, A. H., Curole, B. J., Grayson, S. M., Tsige, M., & Wesdemiotis, C. (2024). Elucidation of Dithiol-yne Comb Polymer Architectures by Tandem Mass Spectrometry and Ion Mobility Techniques. Polymers, 16(12), 1665. https://doi.org/10.3390/polym16121665