Inhibitory Effect of Alisma canaliculatum Ethanolic Extract on NF-κB-Dependent CXCR3 and CXCL10 Expression in TNFα-Exposed MDA-MB-231 Breast Cancer Cells


Abstract

1. Introduction
2. Results and Discussion
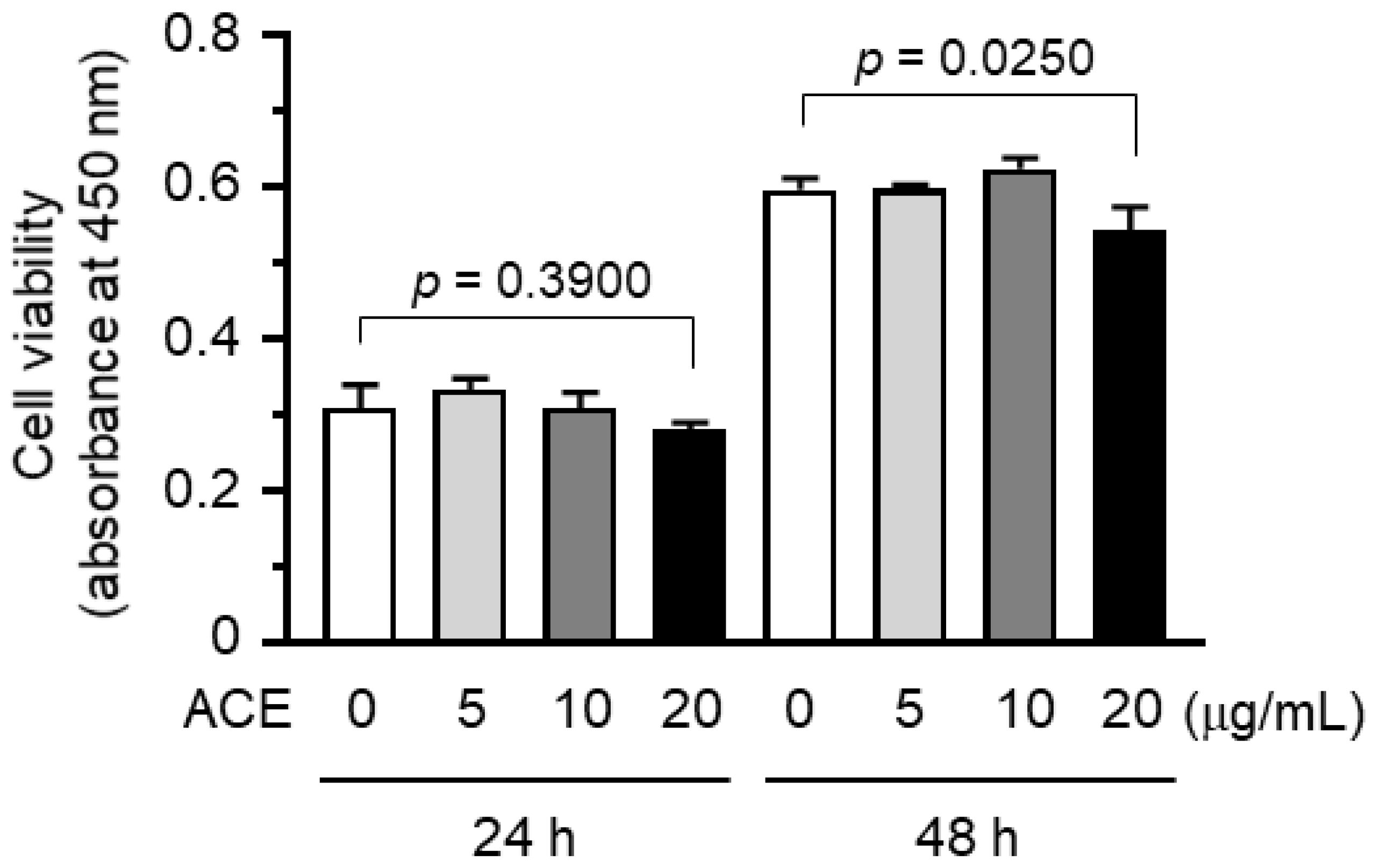
2.1. A. canaliculatum Ethanolic Extract (ACE) Showed no Cytotoxicity against MDA-MB-231 Breast Cancer Cells
2.2. A. canaliculatum Ethanolic Extract (ACE) Attenuates TNFα-Induced Motility of MDA-MB-231 Breast Cancer Cells
2.3. A. canaliculatum Ethanolic Extract (ACE) Inhibits TNFα-Induced CXCR3 mRNA Expression in MDA-MB-231 Cells
2.4. A. canaliculatum Ethanolic Extract (ACE) Inhibits TNFα-Induced CXCL10 mRNA Expression in MDA-MB-231 Cells
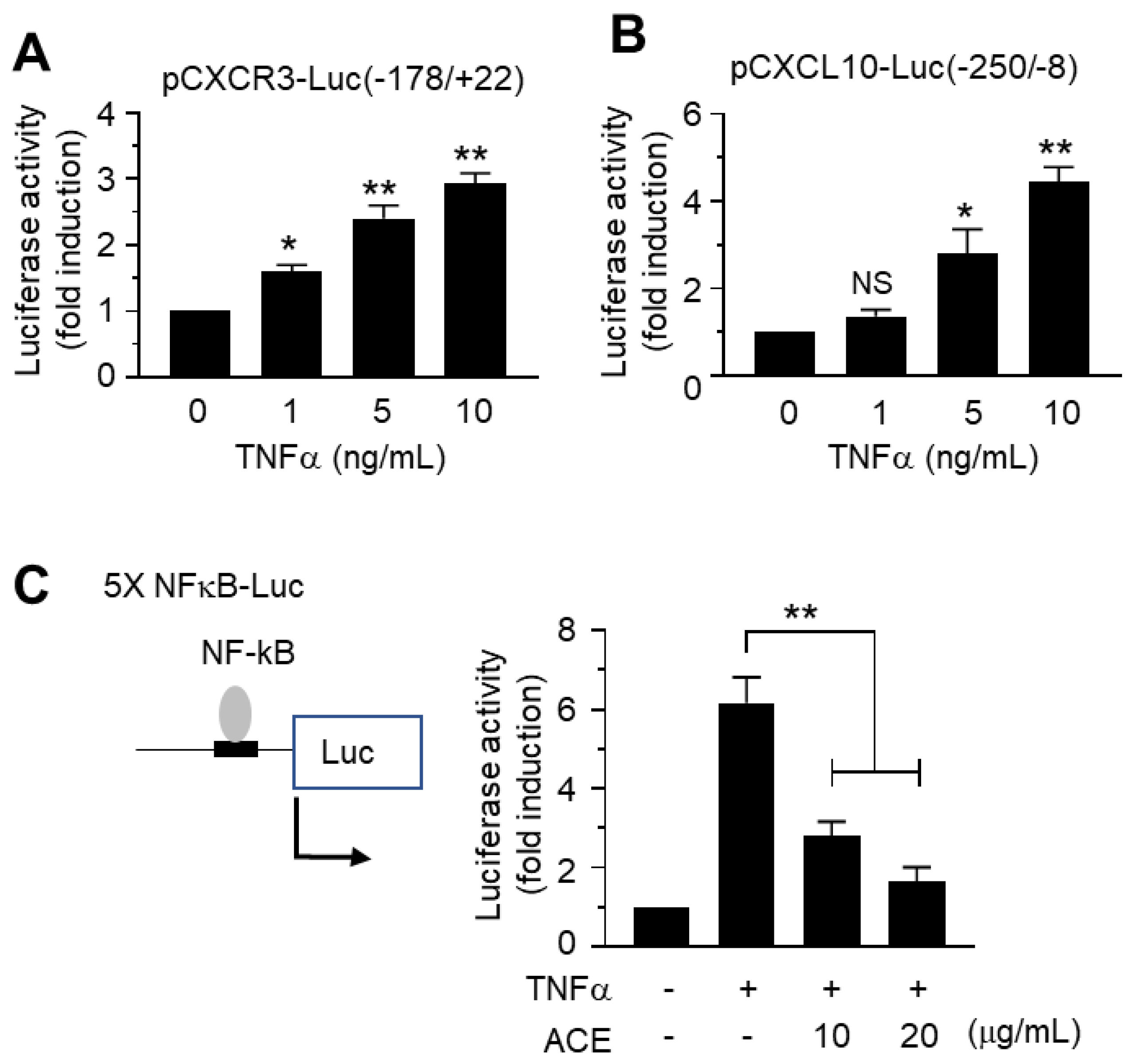
2.5. Inhibitory Effects of A. canaliculatum Ethanolic Extract (ACE) on NF-κB-Mediated CXCR3 and CXCL10 Gene Promoter Activation
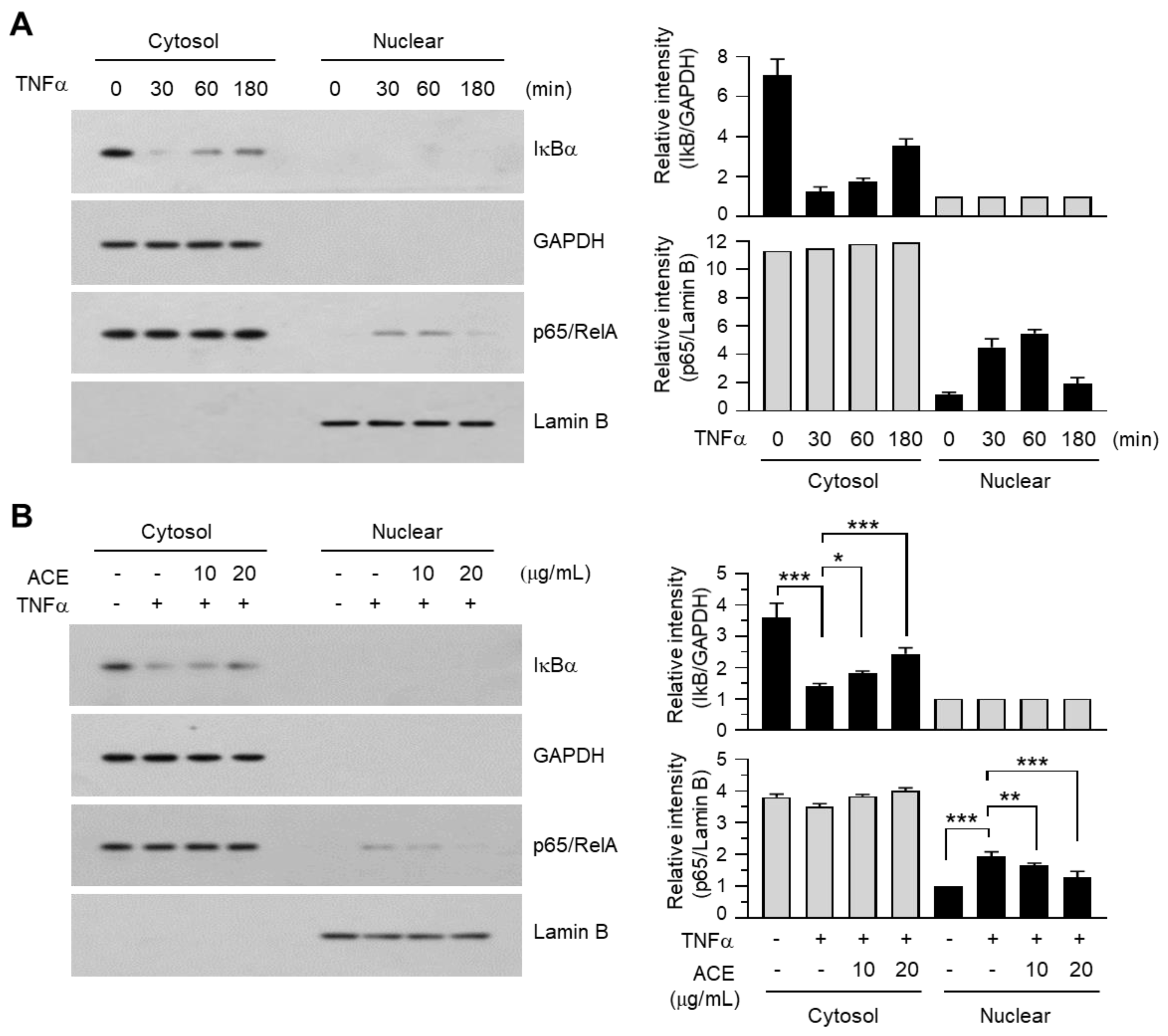
2.6. A. canaliculatum Ethanolic Extract (ACE) Suppresses NF-κB Activation via Inhibition of IKKα/β
2.7. A. canaliculatum Ethanolic Extract (ACE) Inhibits the Translocation of NF-κB into the Nucleus of MDA-MB-231 Cells
3. Materials and Methods
3.1. Preparation of A. canaliculatum Ethanolic Extract (ACE)
3.2. Cells and Reagents
3.3. Cytotoxicity Assay
3.4. Scratch Wound-Healing Assay
3.5. Actin Reorganization
3.6. Quantitative Real-Time PCR (qPCR) Analysis
3.7. Construction and Mutagenesis of the CXCR3 and CXCL10 Promoter Reporters
3.8. Gene Promoter Reporter Activity Assay
3.9. Immunoblot Analysis
3.10. NF-κB-Dependent Transcriptional Activity
3.11. Nuclear Extraction
3.12. Immunofluorescence Microscopic Analysis
3.13. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | A. canaliculatum ethanolic extract |
| TNFα | Tumor necrosis factor-alpha |
| IκB | Inhibitor of κB |
| IKK | IκB kinase |
| NF-κB | Nuclear factor kappa-B paired box gene 3 |
| CXCL10 | CXC motif chemokine ligand 10 |
| CXCR3 | CXC motif chemokine receptor 3 |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| qPCR | quantitative real time-PCR |
| GAPDH | Glyceraldehyde phosphate dehydrogenase |
References
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Artacho-Cordon, A.; Artacho-Cordon, F.; Rios-Arrabal, S.; Calvente, I.; Nunez, M.I. Tumor microenvironment and breast cancer progression: A complex scenario. Cancer Biol. Ther. 2012, 13, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Chemokine networks and breast cancer metastasis. Breast Dis. 2006, 26, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth 2. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Bai, Z.; Srinoulprasert, Y.; Yang, B.G.; Hayasaka, H.; Miyasaka, M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005, 96, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sadanandam, A.; Singh, R.K. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007, 26, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Burdick, M.D.; Mestas, J.; Gomperts, B.; Keane, M.P.; Belperio, J.A. Cancer CXC chemokine networks and tumour angiogenesis. Eur. J. Cancer 2006, 42, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Ben Baruch, A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006, 25, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Lazennec, G. Chemokines: Novel targets for breast cancer metastasis 2. Cancer Metastasis Rev. 2007, 26, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Janatpour, M.; Hudak, S.; Sathe, M.; Sedgwick, J.; McEvoy, L. Tumor necrosis factor-dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J. Exp. Med. 2001, 194, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.; Sozzani, S.; Adorini, L. Cutting edge: Selective usage of chemokine receptors by plasmacytoid dendritic cells. J. Immunol. 2001, 167, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, M.; Gerber, B.; Loetscher, P.; Jones, S.; Piali, L.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Chemokine receptor specific for IP10 and mig: Structure, function and expression in activated T-lymphocytes. J. Exp. Med. 1996, 184, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Bianchi, G.; Bordignon, P.; D’Ambrosio, D.; Lang, R.; Borsatti, A.; Sozzani, S.; Allavena, P.; Gray, P.; Mantovani, A.; et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998, 187, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Mackay, C.; Lanzavecchia, A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 1998, 187, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Flaxenburg, J.A.; Laxmanan, S.; Geehan, C.; Grimm, M.; Waaga-Gasser, A.M.; Briscoe, D.M.; Pal, S. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: Relevance for the development of human breast cancer. Cancer Res. 2006, 66, 9509–9518. [Google Scholar] [CrossRef] [PubMed]
- Goldberg-Bittman, L.; Neumark, E.; Sagi-Assif, O.; Azenshtein, E.; Meshel, T.; Witz, I.P.; Ben Baruch, A. The expression of the chemokine receptor CXCR3 and its ligand, CXCL10, in human breast adenocarcinoma cell lines. Immunol. Lett. 2004, 92, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Suyama, T.; Usui, H.; Kasuya, Y.; Nishiyama, M.; Tanaka, N.; Ishiwata, I.; Nagai, Y.; Shozu, M.; Kimura, S. Up-regulation of CXC chemokines and their receptors: Implications for proinflammatory microenvironments of ovarian carcinomas and endometriosis. Hum. Pathol. 2007, 38, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Maru, S.V.; Holloway, K.A.; Flynn, G.; Lancashire, C.L.; Loughlin, A.J.; Male, D.K.; Romero, I.A. Chemokine production and chemokine receptor expression by human glioma cells: Role of CXCL10 in tumour cell proliferation. J. Neuroimmunol. 2008, 199, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Sonoshita, M.; Sakashita, H.; Takabayashi, A.; Yamaoka, Y.; Manabe, T.; Inaba, K.; Minato, N.; Oshima, M.; Taketo, M.M. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. 2004, 64, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer. 2015, 113, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Nam, J.S.; Lim, Y.; Lee, Y.H. TNFα-exposed bone marrow-derived mesenchymal stem cells promote locomotion of MDA-MB-231 breast cancer cells through transcriptional activation of CXCR3 ligand chemokines. J. Biol. Chem. 2010, 285, 30731–30740. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Han, L.; Bi, X.; Wang, X.; Mu, Y.; Guan, P.; Li, L.; Huang, X. Structures and biological activities of the triterpenoids and sesquiterpenoids from Alisma orientale. Phytochemistry 2016, 131, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Huang, D.M.; Chueh, S.C.; Teng, C.M.; Guh, J.H. Alisol B acetate, a triterpene from Alismatis rhizoma, induces Bax nuclear translocation and apoptosis in human hormone-resistant prostate cancer PC-3 cells. Cancer Lett. 2006, 231, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.F.; Wang, C.; Zhu, G.Y.; Leung, C.H.; Yang, M.S.; Cheung, H.Y. Reversal of multidrug resistance in cancer cells by Rhizoma Alismatis extract. Phytomedicine 2007, 14, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Han, Y.R.; Nam, J.S.; Han, C.W.; Kim, B.J.; Jeong, H.S.; Ha, K.T.; Jung, M.H. Protective Effects of Alisma orientale Extract against Hepatic Steatosis via Inhibition of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2015, 16, 26151–26165. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.P.; Tian, G.; Huang, X.F.; Lou, F.C. Guaiane-type sesquiterpenoids from Alisma orientalis. Phytochemistry 2003, 63, 877–881. [Google Scholar] [CrossRef]
- Zhang, A.; Sheng, Y.; Zou, M. Antiproliferative activity of Alisol B in MDA-MB-231 cells is mediated by apoptosis, dysregulation of mitochondrial functions, cell cycle arrest and generation of reactive oxygen species. Biomed. Pharmacother. 2017, 87, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Xu, Y.L.; Tang, Z.H.; Xu, X.H.; Chen, X.; Li, T.; Ding, C.Y.; Huang, M.Q.; Chen, X.P.; Wang, Y.T.; et al. Effects of alisol B 23-acetate on ovarian cancer cells: G1 phase cell cycle arrest, apoptosis, migration and invasion inhibition. Phytomedicine 2016, 23, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Wang, P.; Ma, Q.; Han, L.; Wang, X.; Mu, Y.; Guan, P.; Qu, X.; Wang, Z.; Huang, X. Anti-inflammatory activities and liver protection of alisol F and 25-anhydroalisol F through the inhibition of MAPK, STAT3 and NF-κB activation in vitro and in vivo. Molecules 2017, 22, 951. [Google Scholar]
- Lacroix, M. Significance, detection and markers of disseminated breast cancer cells. Endocr. Relat. Cancer 2006, 13, 1033–1067. [Google Scholar] [CrossRef] [PubMed]
- Ham, M.; Moon, A. Inflammatory and microenvironmental factors involved in breast cancer progression. Arch. Pharm. Res. 2013, 36, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Singh, R.; Sabharwal, L.; Bando, H.; Meng, J.; Arima, Y.; Yamada, M.; Harada, M.; Jiang, J.J.; Kamimura, D.; et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014, 74, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Kim, C.G.; Jung, Y.J.; Lim, Y.; Lee, Y.H. The UPR inducer DPP23 inhibits the metastatic potential of MDA-MB-231 human breast cancer cells by targeting the Akt-IKK-NF-kappaB-MMP-9 axis. Sci. Rep. 2016, 6, 34134. [Google Scholar] [CrossRef] [PubMed]
- Son, S.W.; Min, B.W.; Lim, Y.; Lee, Y.H.; Shin, S.Y. Regulatory mechanism of TNF-α autoregulation in HaCaT cells: The role of the transcription factor EGR-1. Biochem. Biophys. Res. Commun. 2008, 374, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Song, H.; Kim, C.G.; Choi, Y.K.; Lee, K.S.; Lee, S.J.; Lee, H.J.; Lim, Y.; Lee, Y.H. Egr-1 is necessary for fibroblast growth factor-2-induced transcriptional activation of the glial cell line-derived neurotrophic factor in murine astrocytes. J. Biol. Chem. 2009, 284, 30583–30593. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer |
|---|---|
| CXCL10 | Forward, 5′-AGCAAGGAAAGGTCTAAAAGATCTCC-3′ |
| Reverse, 5′-GGCTTGACATATACTCCATGTAGGG-3′ | |
| TaqMan probe, 5′-FAM-AGGCAGCCTCTGTGTGGTCCATCCTT-BHQ-3′ | |
| CXCR3 | Forward, 5′-GCTCTGAGGACTGCACCATTG-3′ |
| Reverse, 5′-TGAAGTTTTAGTTTCCAAATGAGAAGGG-3′ | |
| TaqMan probe, 5′-FAM-CTGCCAAGCCCCATCCTGCCGCC-BHQ-3′ | |
| GAPDH | Forward, 5′-TCGACAGTCAGCCGCATCTTC-3′ |
| Reverse, 5′-CGCCCAATACGACCACCTCCG-3′ | |
| TaqMan probe, 5′-Yakima Yellow TM-CGTCGCCAGCCCAGCCACGC-BHQ-1-3′ |
| Mutant Name | Primer |
|---|---|
| mtNF-κB(I) | Forward, 5′-CCTGGAAGAGGCTGCTGC-3′ |
| Reverse, 5′-TAGTTACCTCTACCAGACCTCCCTAAA-3′ | |
| mtNF-κB(II) | Forward, 5′-CCACTTCCTCTGTGACTGCAG-3′ |
| Reverse, 5′-TAGTTTCAGGCAGTTCTCAGCAG-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Ahn, S.S.; Lim, Y.; Lee, Y.H.; Shin, S.Y. Inhibitory Effect of Alisma canaliculatum Ethanolic Extract on NF-κB-Dependent CXCR3 and CXCL10 Expression in TNFα-Exposed MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2607. https://doi.org/10.3390/ijms19092607
Choi J, Ahn SS, Lim Y, Lee YH, Shin SY. Inhibitory Effect of Alisma canaliculatum Ethanolic Extract on NF-κB-Dependent CXCR3 and CXCL10 Expression in TNFα-Exposed MDA-MB-231 Breast Cancer Cells. International Journal of Molecular Sciences. 2018; 19(9):2607. https://doi.org/10.3390/ijms19092607
Chicago/Turabian StyleChoi, Jihye, Sung Shin Ahn, Yoongho Lim, Young Han Lee, and Soon Young Shin. 2018. "Inhibitory Effect of Alisma canaliculatum Ethanolic Extract on NF-κB-Dependent CXCR3 and CXCL10 Expression in TNFα-Exposed MDA-MB-231 Breast Cancer Cells" International Journal of Molecular Sciences 19, no. 9: 2607. https://doi.org/10.3390/ijms19092607
APA StyleChoi, J., Ahn, S. S., Lim, Y., Lee, Y. H., & Shin, S. Y. (2018). Inhibitory Effect of Alisma canaliculatum Ethanolic Extract on NF-κB-Dependent CXCR3 and CXCL10 Expression in TNFα-Exposed MDA-MB-231 Breast Cancer Cells. International Journal of Molecular Sciences, 19(9), 2607. https://doi.org/10.3390/ijms19092607