Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection



Abstract
:1. Introduction
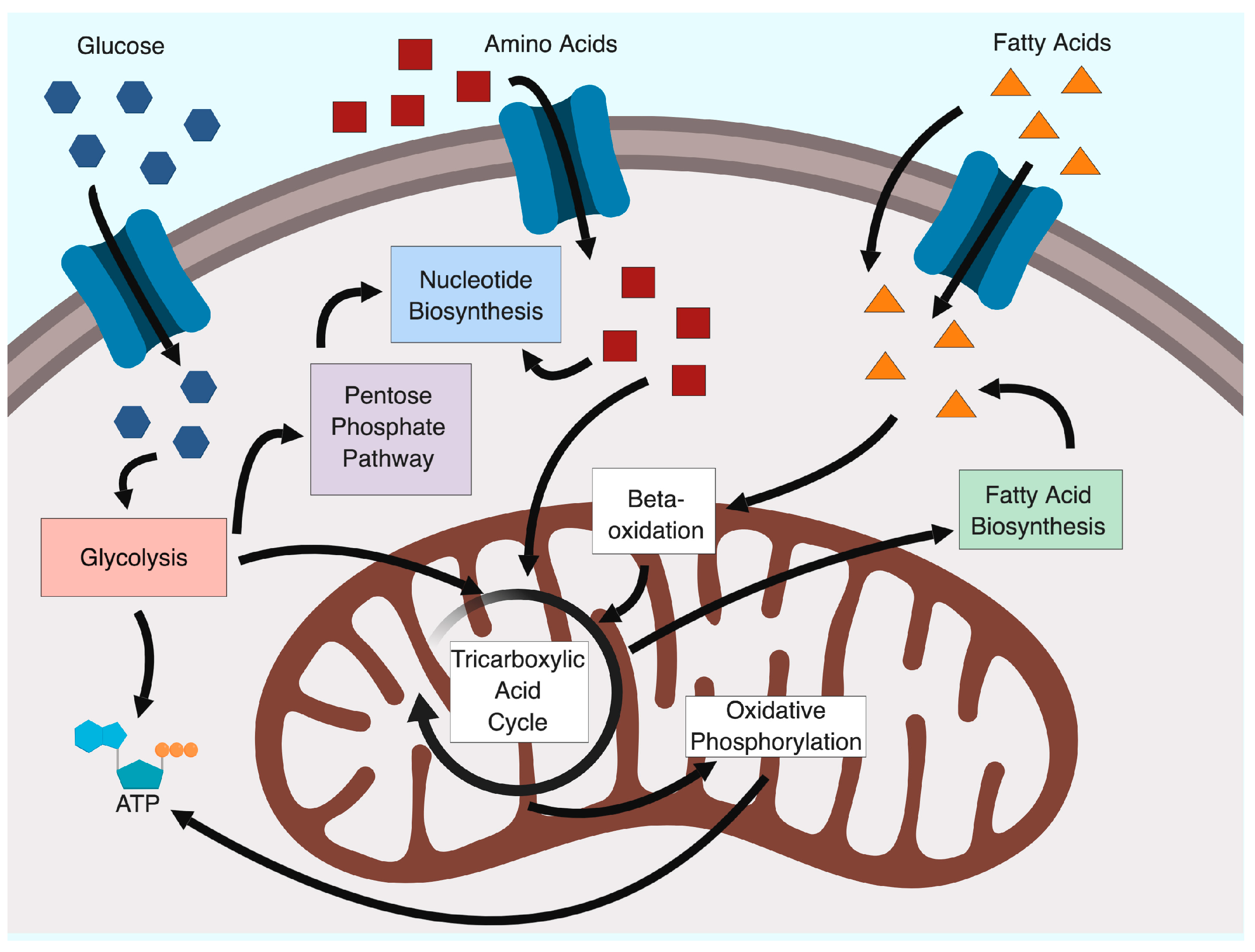
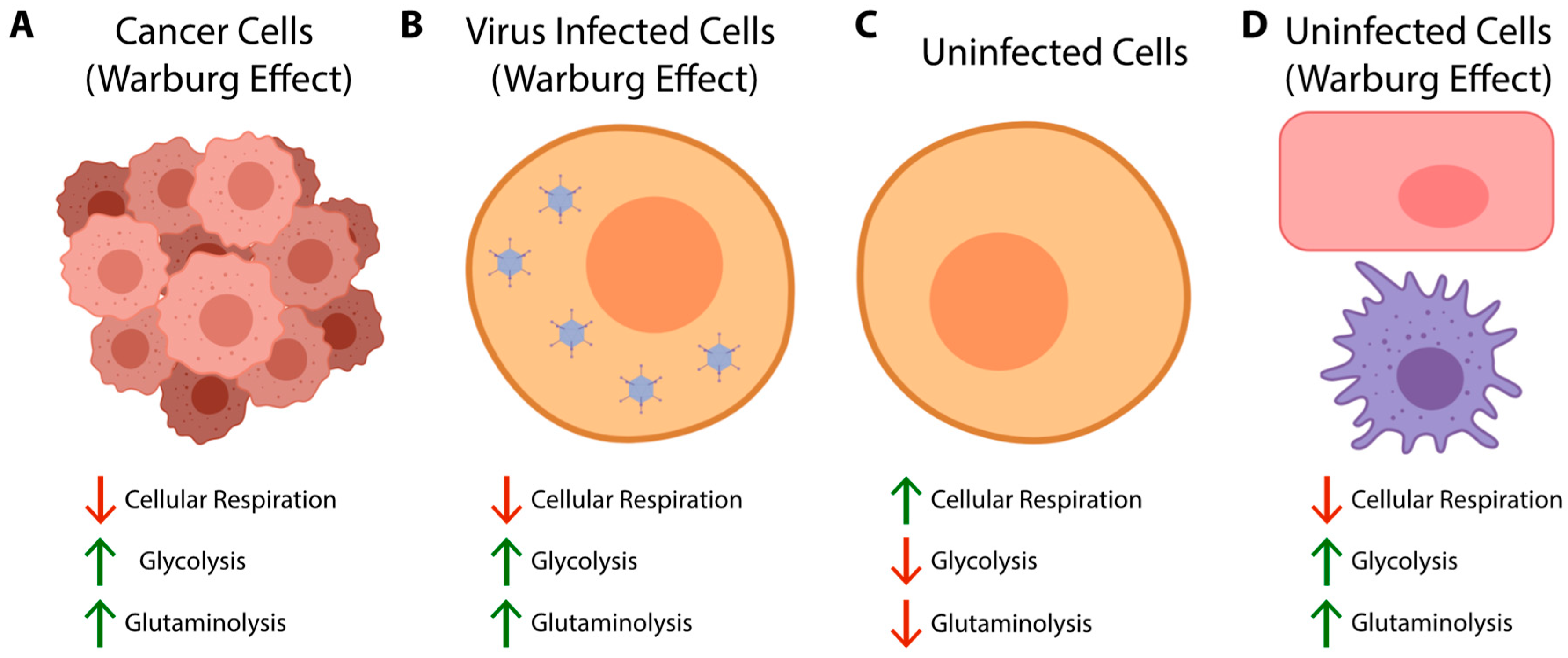
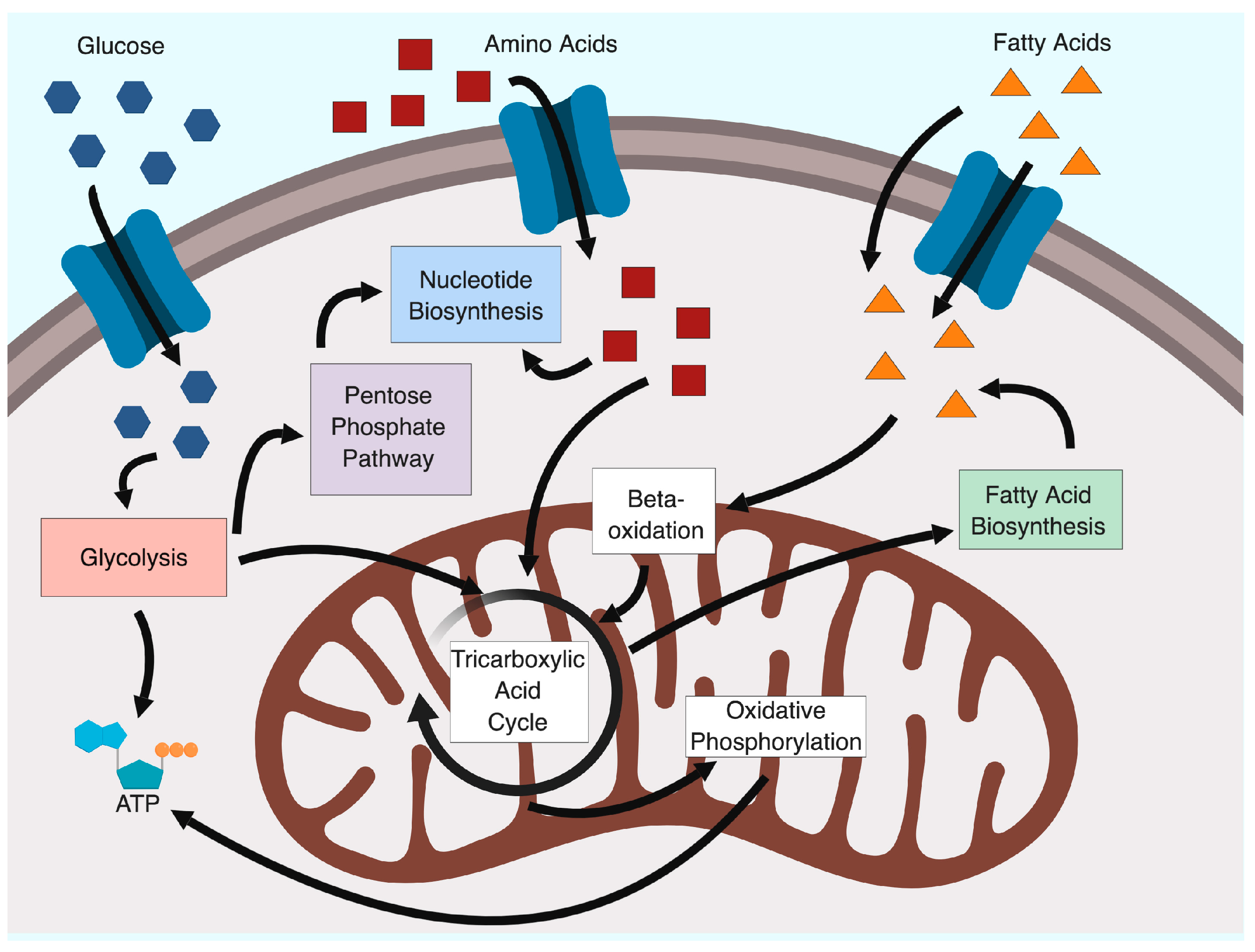
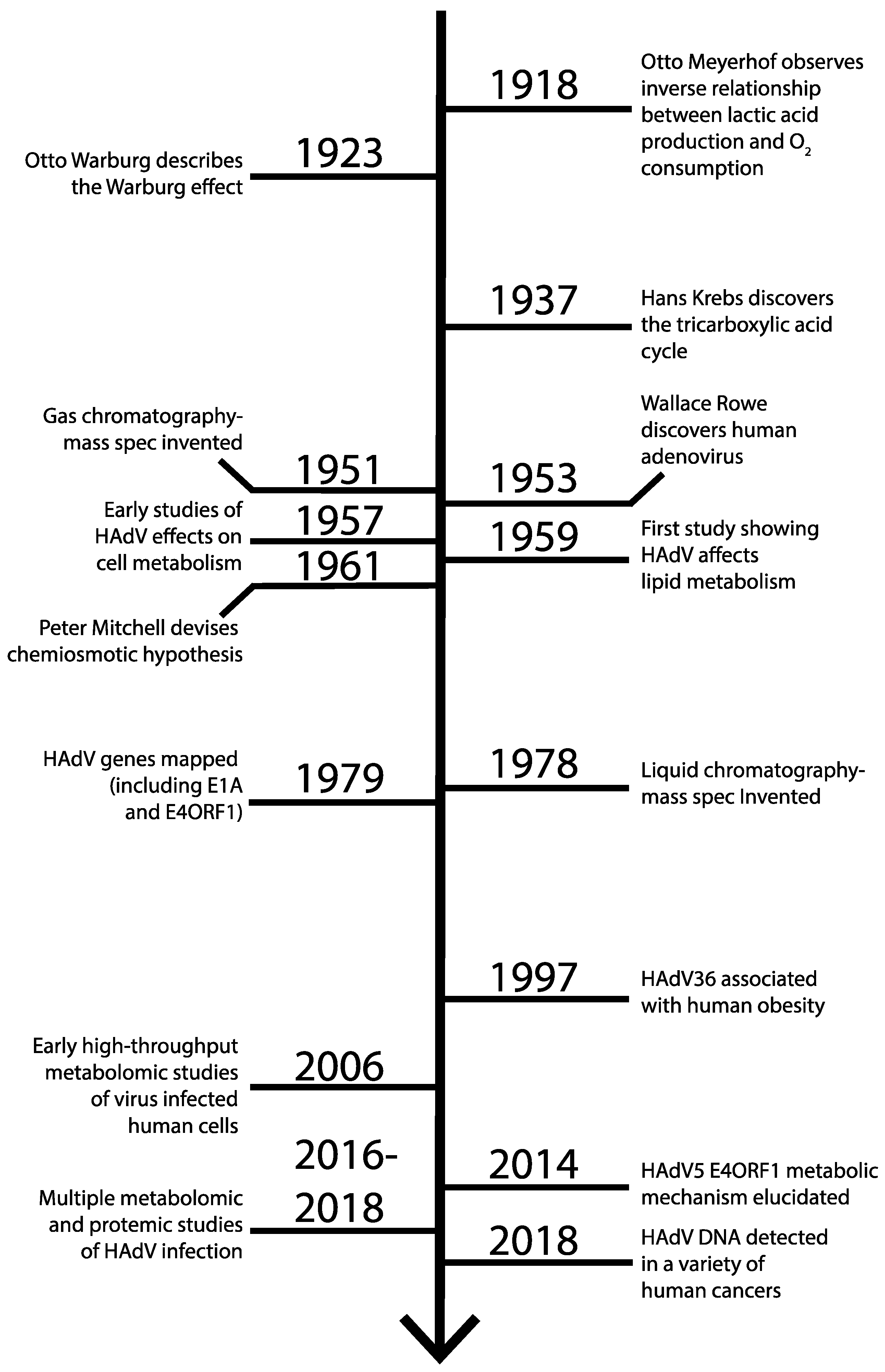
2. Glycolysis and the Warburg Effect
3. The Earliest Observations of Metabolic Changes due to HAdV Infection
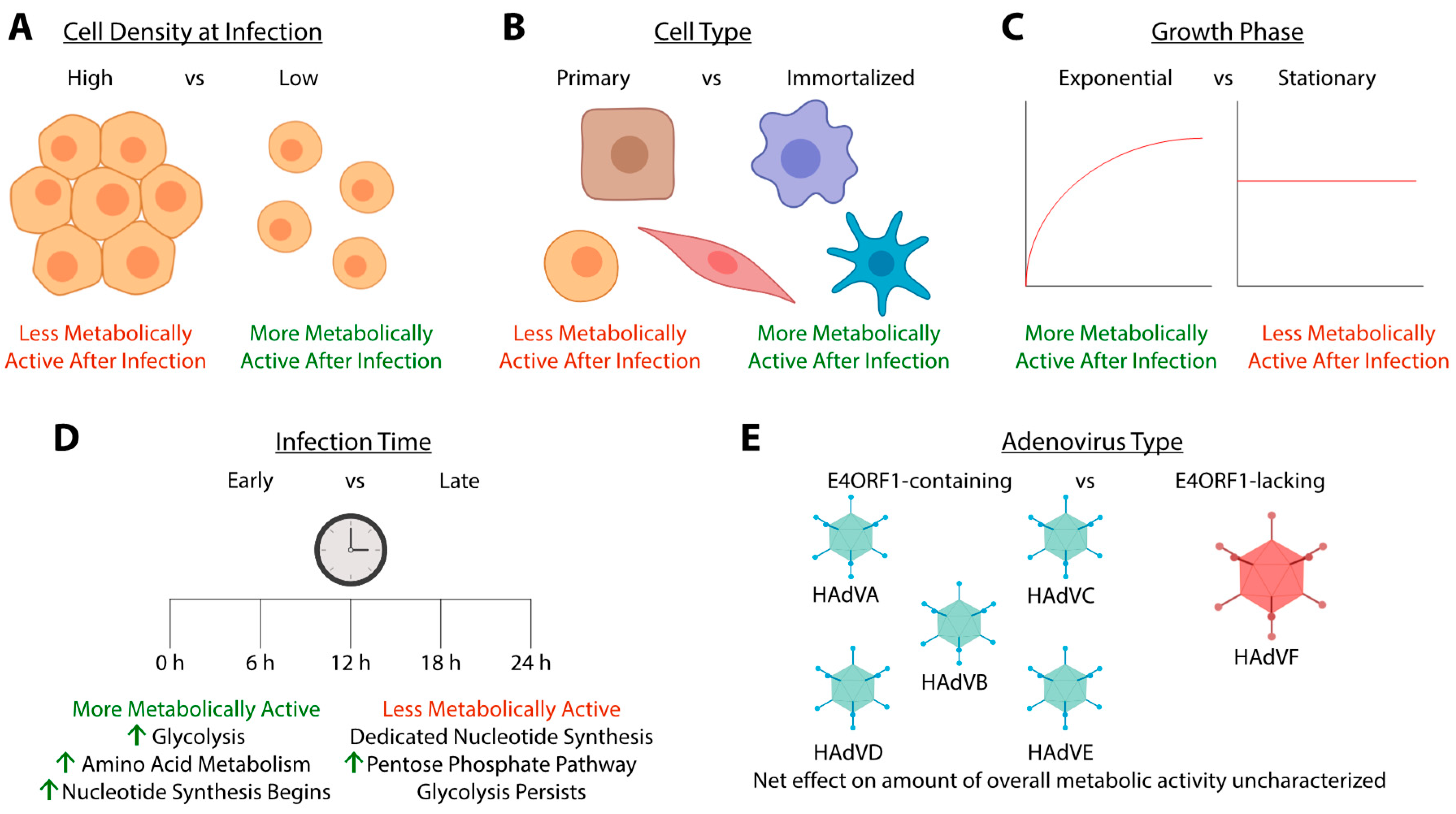
4. Metabolomic and Proteomic Analyses of Adenovirus Infection
5. E4ORF1 Positively Regulates Glycolysis and Glutamine Catabolism
6. Human Adenovirus 36 Influences Metabolism through E4ORF1
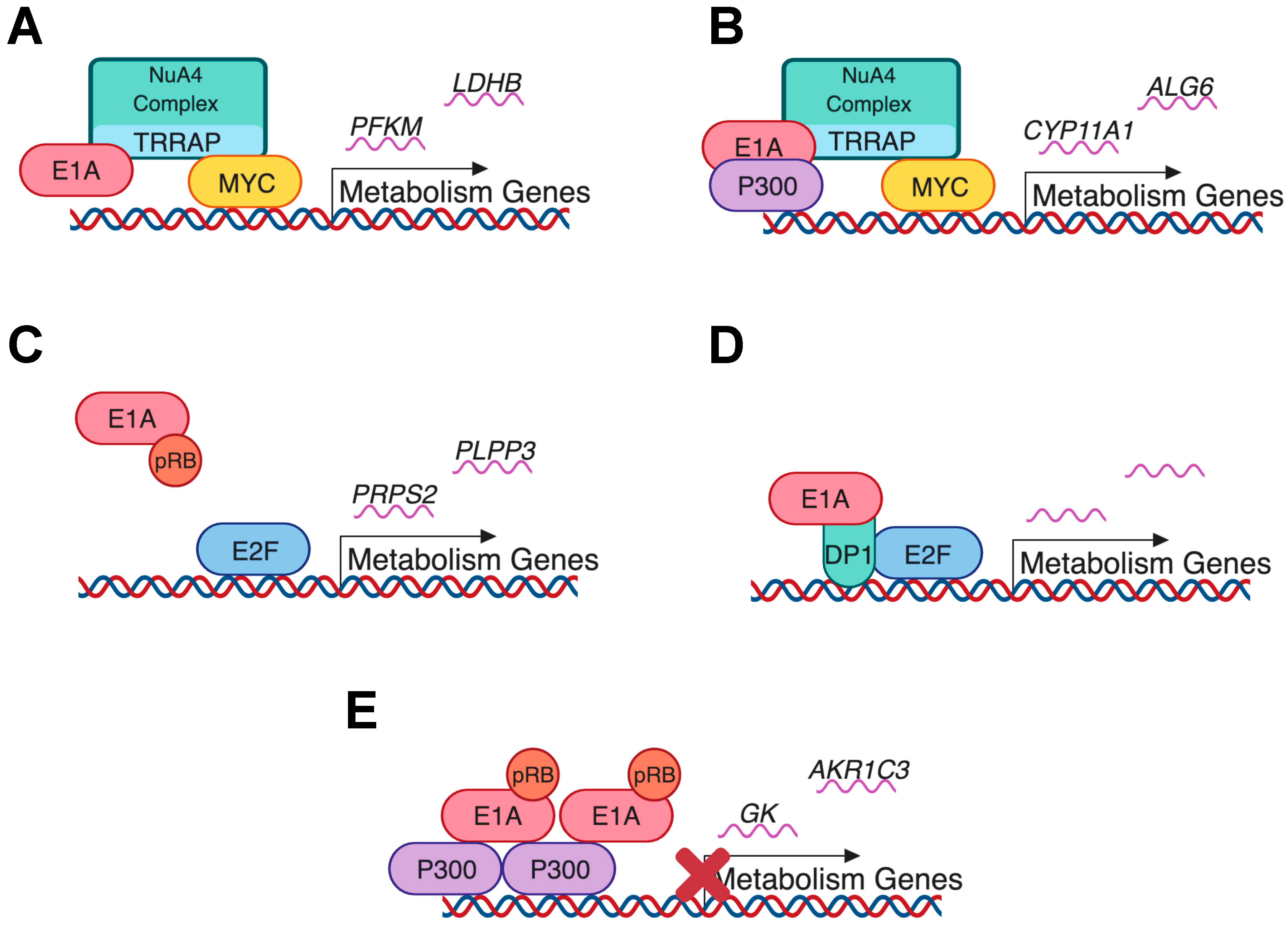
7. E1A as a Regulator of Cellular Metabolism During Infection
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lenaerts, L.; De Clercq, E.; Naesens, L. Clinical features and treatment of adenovirus infections. Pertanika J. Soc. Sci. Humanit. 2008, 18, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Nash, L.A.; Parks, R.J. Adenovirus Biology and Development as a Gene Delivery Vector. In Therapeutic Applications of Adenoviruses; Ng, P., Brunetti-Pierri, N., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 1–36. [Google Scholar]
- Pelka, P.; Ablack, J.N.G.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic Structural Disorder in Adenovirus E1A: A Viral Molecular Hub Linking Multiple Diverse Processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 2005, 10, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Whyte, P.; Ruley, H.E.; Harlow, E. Two regions of the adenovirus early region 1A proteins are required for transformation. J. Virol. 1988, 62, 257–265. [Google Scholar] [PubMed]
- Javier, R.T. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J. Virol. 1994, 68, 3917–3924. [Google Scholar] [PubMed]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef]
- Damania, B. Oncogenic γ-herpesviruses: Comparison of viral proteins involved in tumorigenesis. Nat. Rev. Microbiol. 2004, 2, 656–668. [Google Scholar] [CrossRef]
- Zhao, J.; Punj, V.; Matta, H.; Mazzacurati, L.; Schamus, S.; Yang, Y.; Yang, T.; Hong, Y.; Chaudhary, P.M. K13 blocks KSHV lytic replication and deregulates vIL6 nad hIL6 expression: A model of lytic replication induced clonal selection in viral oncogenesis. PLoS ONE 2007, 2, e1067. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A. The landscape of viral associations in human cancers. bioRxiv 2018, 465757. [Google Scholar] [CrossRef]
- Racker, E. Bioenergetics and the problem of tumor growth: An understanding of the mechanism of the generation and control of biological energy may shed light on the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Eelen, G.; De Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Domblides, C.; Lartigue, L.; Faustin, B. Metabolic Stress in the Immune Function of T Cells, Macrophages and Dendritic Cells. Cells 2018, 7, 68. [Google Scholar] [CrossRef]
- Rowe, W.P.; Huebner, R.J.; Gilmore, L.K.; Parrott, R.H.; Ward, T.G. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc. Soc. Exp. Biol. Med. 1953, 84, 570–573. [Google Scholar] [CrossRef]
- Levy, H.B.; Baron, S.; Rowe, W.P. Metabolic effects of animal viruses in tissue culture. In Proceedings of the Federation of American Societies for Experimental Biology, Chicago, IL, USA, 15–19 April 1957; Volume 16, p. 422. [Google Scholar]
- Fisher, T.N.; Ginsberg, H.S. Accumulation of organic acids by HeLa cells infected with type 4 adenovirus. Proc. Soc. Exp. Biol. Med. 1957, 95, 47–51. [Google Scholar] [CrossRef]
- Rozee, K.R.; Ottey, L.J.; Van Rooyen, C.E. Some metabolic effects of adenovirus infection in HeLa cells. Can. J. Microbiol. 1957, 3, 1015–1020. [Google Scholar] [CrossRef]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell MeTable 2014, 19, 694–701. [Google Scholar] [CrossRef]
- Consigli, R.A.; Ginsberg, H.S. Activity of aspartate transcarbamylase in uninfected and type 5 adenovirus-infected HeLa cells. J. Bacteriol. 1964, 87, 1034–1043. [Google Scholar]
- Huang, M.; Graves, L.M. De novo synthesis of pyrimidine nucleotides; emerging interfaces with signal transduction pathways. Cell. Mol. Life Sci. C. 2003, 60, 321–336. [Google Scholar] [CrossRef]
- McIntosh, K.; Payne, S.; Russell, W.C. Studies on lipid metabolism in cells infected with adenovirus. J. Gen. Virol. 1971, 10, 251–265. [Google Scholar] [CrossRef]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, 1165–1175. [Google Scholar] [CrossRef]
- Silva, A.C.; Teixeira, A.P.; Alves, P.M. Impact of Adenovirus infection in host cell metabolism evaluated by1H-NMR spectroscopy. J. Biotechnol. 2016, 231, 16–23. [Google Scholar] [CrossRef]
- Gey, G.O.; Coffman, W.D.; Kubicek, M.T. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 1952, 12, 264–265. [Google Scholar]
- Mayyasi, S.A.; Traul, K.A.; Garon, C.; Wright, B. Antigenic characteristics of Rauscher virus cultivated in human embryonic kidney cells. In Proceedings of the American Association for Cancer Research, Philadelphia, PA, USA, 9–11 April 1970; Volume 11, p. 54. [Google Scholar]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–66. [Google Scholar] [CrossRef]
- Silva, A.C.; Simão, D.; Küppers, C.; Lucas, T.; Sousa, M.F.Q.; Cruz, P.; Carrondo, M.J.T.; Kochanek, S.; Alves, P.M. Human amniocyte-derived cells are a promising cell host for adenoviral vector production under serum-free conditions. Biotechnol. J. 2015, 10, 760–771. [Google Scholar] [CrossRef]
- Nichols, W.W.; Murphy, D.G.; Cristofalo, V.J.; Toji, L.H.; Greene, A.E.; A, D.S. Characterization of a New Human Diploid Cell Strain, IMR-90. Science 1977, 196, 60–63. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Parks, W.P. In Vitro Cultivation of Human Tumors: Establishment of Cell Lines Derived From a Series of Solid Tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Fogh, J. Human Tumor Cells in vitro; Springer: New York, NY, USA, 1975. [Google Scholar]
- Line, E.C.; Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D.; Brenz, R.; Mcgrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; et al. Isolation and Characterization of a Spontaneously Immortalized Human Breast Epithelial Cell Line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Green, H.; Kehinde, O. Sublines of mouse 3T3 cells that accumulate lipid. Cell 1974, 1, 113–116. [Google Scholar] [CrossRef]
- Yateman, M.E. Regulation of human fibroblast insulin-like growth factor (IGF)-binding proteins by IGF-1 and cytokines, mechanisms of action and effects upon IGF bioactivity. PhD. Thesis, University of London, London, UK, 1995. [Google Scholar]
- Granberg, F.; Svensson, C.; Pettersson, U.; Zhao, H. Adenovirus-induced alterations in host cell gene expression prior to the onset of viral gene expression. Virology 2006, 353, 1–5. [Google Scholar] [CrossRef]
- Miller, D.L.; Myers, C.L.; Rickards, B.; Coller, H.A.; Flint, S.J. Adenovirus type 5 exerts genome-wide control over cellular programs governing proliferation, quiescence, and survival. Genome Biol. 2007, 8, R58. [Google Scholar] [CrossRef]
- Crisostomo, L.; Soriano, A.M.; Mendez, M.; Graves, D.; Pelka, P. Temporal dynamics of adenovirus 5 gene expression in normal human cells. PLoS ONE 2019, 14, e0211192. [Google Scholar] [CrossRef]
- Dyer, A.; Schoeps, B.; Frost, S.; Jakeman, P.G.; Scott, E.M.; Freedman, J.; Seymour, L.W. Antagonism of glycolysis and reductive carboxylation of glutamine potentiates activity of oncolytic adenoviruses in cancer cells. Cancer Res. 2019, 79, 331–345. [Google Scholar] [CrossRef]
- Carinhas, N.; Koshkin, A.; Pais, D.A.M.; Alves, P.M.; Teixeira, A.P. 13C-metabolic flux analysis of human adenovirus infection: Implications for viral vector production. Biotechnol. Bioeng. 2017, 114, 195–207. [Google Scholar] [CrossRef]
- Valdés, A.; Zhao, H.; Pettersson, U.; Lind, S.B. Time-resolved proteomics of adenovirus infected cells. PLoS ONE 2018, 13, 1–23. [Google Scholar] [CrossRef]
- Guissoni, A.C.P.; Soares, C.M.A.; Badr, K.R.; Ficcadori, F.S.; Parente, A.F.A.; Parente, J.A.; Baeza, L.C.; Souza, M.; Cardoso, D.D.P. Proteomic analysis of A-549 cells infected with human adenovirus 40 by LC-MS. Virus Genes 2018, 54, 351–360. [Google Scholar] [CrossRef]
- Zhao, L.; Loewenstein, P.M.; Green, M. Enhanced MYC association with the NuA4 histone acetyltransferase complex mediated by the adenovirus E1A N-terminal domain activates a subset of MYC target genes highly expressed in cancer cells. Genes Cancer 2017, 8, 752. [Google Scholar]
- Pelka, P.; Miller, M.S.; Cecchini, M.; Yousef, A.F.; Bowdish, D.M.; Dick, F.; Whyte, P.; Mymryk, J.S. Adenovirus E1A Directly Targets the E2F/DP-1 Complex. J. Virol. 2011, 85, 8841–8851. [Google Scholar] [CrossRef]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor RB to repress select host genes and promote productive virus infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Heaton, N.S.; Randall, G. Lipids at the interface of virus-host interactions. Curr. Opin. Microbiol. 2012, 15, 512–518. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Hidalgo, P.; Anzures, L.; Hernández-Mendoza, A.; Guerrero, A.; Wood, C.D.; Valdés, M.; Dobner, T.; Gonzalez, R.A. Morphological, Biochemical, and Functional Study of Viral Replication Compartments Isolated from Adenovirus-Infected Cells. J. Virol. 2016, 90, 3411–3427. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2017, 162, 603–610. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650. [Google Scholar] [CrossRef]
- Ichikawa, M.; Scott, D.A.; Losfeld, M.E.; Freeze, H.H. The metabolic origins of mannose in glycoproteins. J. Biol. Chem. 2014, 289, 6751–6761. [Google Scholar] [CrossRef]
- Sharma, V.; Ichikawa, M.; Freeze, H.H. Mannose metabolism: More than meets the eye. Biochem. Biophys. Res. Commun. 2014, 453, 220–228. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional Regulation of Metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Flint, J.; Shenk, T. Adenovirus E1A protein paradigm viral transactivator. Annu. Rev. Genet. 1989, 23, 141–161. [Google Scholar] [CrossRef]
- Liu, F.; Green, M.R. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell 1990, 61, 1217–1224. [Google Scholar] [CrossRef]
- Lee, J.S.; Zhang, X.; Shin, Y. Differential interactions of the CREB/ATF family of transcription factors with p300 and adenovirus E1A. J. Biol. Chem. 1996, 271, 17666–17674. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Chung, S.-H.; Frese, K.K.; Weiss, R.S.; Prasad, B.V.V.; Javier, R.T. A new crucial protein interaction element that targets the adenovirus E4-ORF1 oncoprotein to membrane vesicles. J. Virol. 2007, 81, 4787–4797. [Google Scholar] [CrossRef]
- Frese, K.K.; Lee, S.S.; Thomas, D.L.; Latorre, I.J.; Weiss, R.S.; Glaunsinger, B.A.; Javier, R.T. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 2003, 22, 710–721. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Hanover, J.A.; Chen, W.; Bond, M.R. O-GlcNAc in cancer: An Oncometabolism-fueled vicious cycle. J. Bioenerg. Biomembr. 2018, 50, 155–173. [Google Scholar] [CrossRef]
- Atkinson, R.L.; Dhurandhar, N.V.; Allison, D.B.; Bowen, R.L.; Israel, B.A.; Albu, J.B.; Augustus, A.S. Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. Int. J. Obes. 2005, 29, 281–286. [Google Scholar] [CrossRef]
- Bil-Lula, I.; Stąpor, S.; Sochocka, M.; Wołyniec, M.; Zatońska, K.; Ilow, R.; Szuba, A.; Sawicki, G.; Woźniak, M. Infectobesity in the Polish population—Evaluation of an association between adenoviruses type 5, 31 36 and human obesity. Int. J. Virol. Mol. 2014, 3, 1–8. [Google Scholar]
- Bil-Lula, I.; Krzywonos-Zawadzka, A.; Sawicki, G.; Woźniak, M. An infection of human adenovirus 31 affects the differentiation of preadipocytes into fat cells, its metabolic profile and fat accumulation. J. Med. Virol. 2016, 88, 400–407. [Google Scholar] [CrossRef]
- Karlsson, E.A.; Beck, M.A. The burden of obesity on infectious disease. Exp. Biol. Med. 2010, 235, 1412–1424. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Whigham, L.D.; Abbott, D.H.; Schultz-darken, N.J.; Israel, B.A.; Bradley, S.M.; Kemnitz, J.W.; Allison, D.B.; Atkinson, R.L. Human Adenovirus Ad-36 Promotes Weight Gain in Male Rhesus and. J. Nutr. 2002, 132, 3155–3160. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Israel, B.A.; Kolesar, J.M.; Mayhew, G.F.; Cook, M.E.; Atkinson, R.L. Increased adiposity in animals due to a human virus. Int. J. Obes. 2000, 24, 989–996. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Na, H.N.; Dubuisson, O.; Hegde, V.; Nam, J.H.; Dhurandhar, N. V Human adenovirus Ad36 and its E4orf1 gene enhance cellular glucose uptake even in the presence of inflammatory cytokines. Biochimie 2016, 124, 3–10. [Google Scholar] [CrossRef]
- McMurphy, T.B.; Huang, W.; Xiao, R.; Liu, X.; Dhurandhar, N.V.; Cao, L. Hepatic expression of adenovirus 36 E4ORF1 improves glycemic control and promotes glucose metabolism through AKT activation. Diabetes 2017, 66, 358–371. [Google Scholar] [CrossRef]
- King, C.R.; Zhang, A.; Tessier, T.M.; Gameiro, S.F.; Mymryk, J.S. Hacking the Cell: Network Intrusion and Exploitation by Adenovirus E1A. MBio 2018, 9, e00390-18. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Lillie, J.W.; Daouk, G.H.; Green, M.R.; Kingston, R.; Schimmel, P. Induction of a cellular enzyme for energy metabolism by transforming domains of adenovirus E1a. Mol. Cell. Biol. 1990, 10, 1476–1483. [Google Scholar] [CrossRef]
- Schlattner, U.; Klaus, A.; Ramirez Rios, S.; Guzun, R.; Kay, L.; Tokarska-Schlattner, M. Cellular compartmentation of energy metabolism: Creatine kinase microcompartments and recruitment of B-type creatine kinase to specific subcellular sites. Amino Acids 2016, 48, 1751–1774. [Google Scholar] [CrossRef]
- Zerler, B.; Roberts, R.; Mathews, M.; Moran, E. Different functional domains of the adenovirus E1A gene are involved in regulation of host cell cycle products. Mol. Cell. Biol. 1987, 7, 821–829. [Google Scholar] [CrossRef]
- Madhu, B.; Narita, M.; Jauhiainen, A.; Menon, S.; Stubbs, M.; Tavaré, S.; Narita, M.; Griffiths, J.R. Metabolomic changes during cellular transformation monitored by metabolite–metabolite correlation analysis and correlated with gene expression. Metabolomics 2015, 11, 1848–1863. [Google Scholar] [CrossRef]
- Urbanczyk-Wochniak, E.; Willmitzer, L.; Fernie, A.R. Integrating profiling data: Using linear correlation to reveal coregulation of transcript and metabolites. Methods Mol. Biol. 2007, 358, 77–85. [Google Scholar]
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38. [Google Scholar] [CrossRef]
- Nevins, J.R. Mechanism of activation of early viral transcription by the adenovirus E1A gene product. Cell 1981, 26, 213–220. [Google Scholar] [CrossRef]
- Whyte, P.; Buchkovich, K.J.; Horowitz, J.M.; Friend, S.H.; Raybuck, M.; Weinberg, R.A.; Harlow, E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988, 334, 124. [Google Scholar] [CrossRef]
- Ferrari, R.; Pellegrini, M.; Horwitz, G.A.; Xie, W.; Berk, A.J.; Kurdistani, S.K. Epigenetic reprogramming by adenovirus e1a. Science 2008, 321, 1086–1088. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Types | Tissue Tropism (Types) | Associated Infections | Contains E4ORF1 (Y/N) |
|---|---|---|---|---|
| A | 12, 18, 31, 61 | Gastrointestinal | Gastroenteritis | Yes |
| B | 3, 7, 11, 14, 16, 21, 34, 35, 50, 55, 66, 68, 72, 79 | Respiratory (3, 7, 16, 21, 50) Urinary/Renal (11, 14, 34, 35) Ocular (3, 7, 11, 14) | Acute respiratory disease, conjunctivitis, nephritis | Yes |
| C | 1, 2, 5, 6, 57 | Respiratory, Ocular (5) | Acute respiratory disease, conjunctivitis | Yes |
| D | 8–10, 13, 15, 17, 19, 20, 22–30, 32, 33, 36–39, 42–49, 51, 53, 54, 56, 58–60, 62–65, 67, 69, 70, 71, 73–75, 81, 83–85, 90 | Ocular, Gastrointestinal (36, 67) | Follicular conjunctivitis, pharyngeal conjunctival fever, epidemic keratoconjunctivitis, gastroenteritis | Yes |
| E | 4 | Respiratory, Ocular | Acute respiratory disease, conjunctivitis | Yes |
| F | 40, 41 | Gastrointestinal | Gastroenteritis | No |
| G | 52 | Gastrointestinal | Gastroenteritis | Yes |
| Unclassified/No record | 76–78, 80, 82, 86–89 | - | - | - |
| Cell Line | Donor Characteristics | Date Established | Cell Morphology | Tissue of Origin | Transformation Status |
|---|---|---|---|---|---|
| HeLa | Female—31 years old | 1951 [32] | Epithelial | Cervical adenocarcinoma | HPV transformed |
| HEK 1 | Fetus | 1970 [33] | Epithelial | Embryonic kidney | Primary |
| HEK293 | Female—Fetus | 1977 [34] | Epithelial | Embryonic kidney | HAdV5 E1A transformed |
| 1G3 2 | Fetus | 2015 [35] | Amniocyte | Amniotic fluid | HAdV5 E1A transformed |
| IMR-90 | Female—Fetus (16 weeks) | 1977 [36] | Fibroblast | Lung | Primary |
| A549 | Male—58 years old | 1973 [37] | Epithelial | Lung adenocarcinoma | Transformed |
| SKOV3 | Female—64 years old | 1973 [38] | Epithelial | Ovarian adenocarcinoma ascites | Transformed |
| MCF10A | Female—36 years old | 1990 [39] | Epithelial | Fibrocystic breast mammary gland | Spontaneously immortalized |
| NHBE | Human 3 | N.A. 3 | Epithelial | Bronchial | Primary |
| 3T3-L1 | Mouse—Fetus | 1973 [40] | Fibroblast | Embryonic–pre-adipose | Spontaneously immortalized |
| BRK | Rat—Neonate | N.A. 3 | Epithelial | Kidney | Primary |
| HS68 | Newborn | 1969 [41] | Fibroblast | Foreskin | Primary |
| HAdVD-36 E4ORF1 | HAdVC-5 E4ORF1 | |
|---|---|---|
Diabetic mice | ↑ glycemic control ↑ glucose disposal ↓ non-fasting blood glucose ↓ adiponectin ↓ weight ↓ lipid metabolism gene expression | ∅ glycemic control ∅ blood glucose effects ↓ weight ↓ lipid metabolism gene expression |
Diet-induced obese mice | ↑ glycemic control ↑ glycolytic gene expression ↑ lipid metabolism gene expression ↓ non-fasting blood glucose ↓ weight | ↑ lipid metabolism gene expression ∅ glycemic control ∅ blood glucose effects ∅ weight change |
Wild type mice | ↓ non-fasting blood glucose ↑ glycolytic gene expression ↑ p-AKT and p-FoxO1 ↑ inflammation (high dose) | ∅ blood glucose effects ∅ glycolytic gene expression * ↑ lipid metabolism gene expression ↑ p-AKT ∅ MYC activity changes * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection. Viruses 2019, 11, 141. https://doi.org/10.3390/v11020141
Prusinkiewicz MA, Mymryk JS. Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection. Viruses. 2019; 11(2):141. https://doi.org/10.3390/v11020141
Chicago/Turabian StylePrusinkiewicz, Martin A., and Joe S. Mymryk. 2019. "Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection" Viruses 11, no. 2: 141. https://doi.org/10.3390/v11020141
APA StylePrusinkiewicz, M. A., & Mymryk, J. S. (2019). Metabolic Reprogramming of the Host Cell by Human Adenovirus Infection. Viruses, 11(2), 141. https://doi.org/10.3390/v11020141