The Astrocytic cAMP Pathway in Health and Disease

Abstract
:
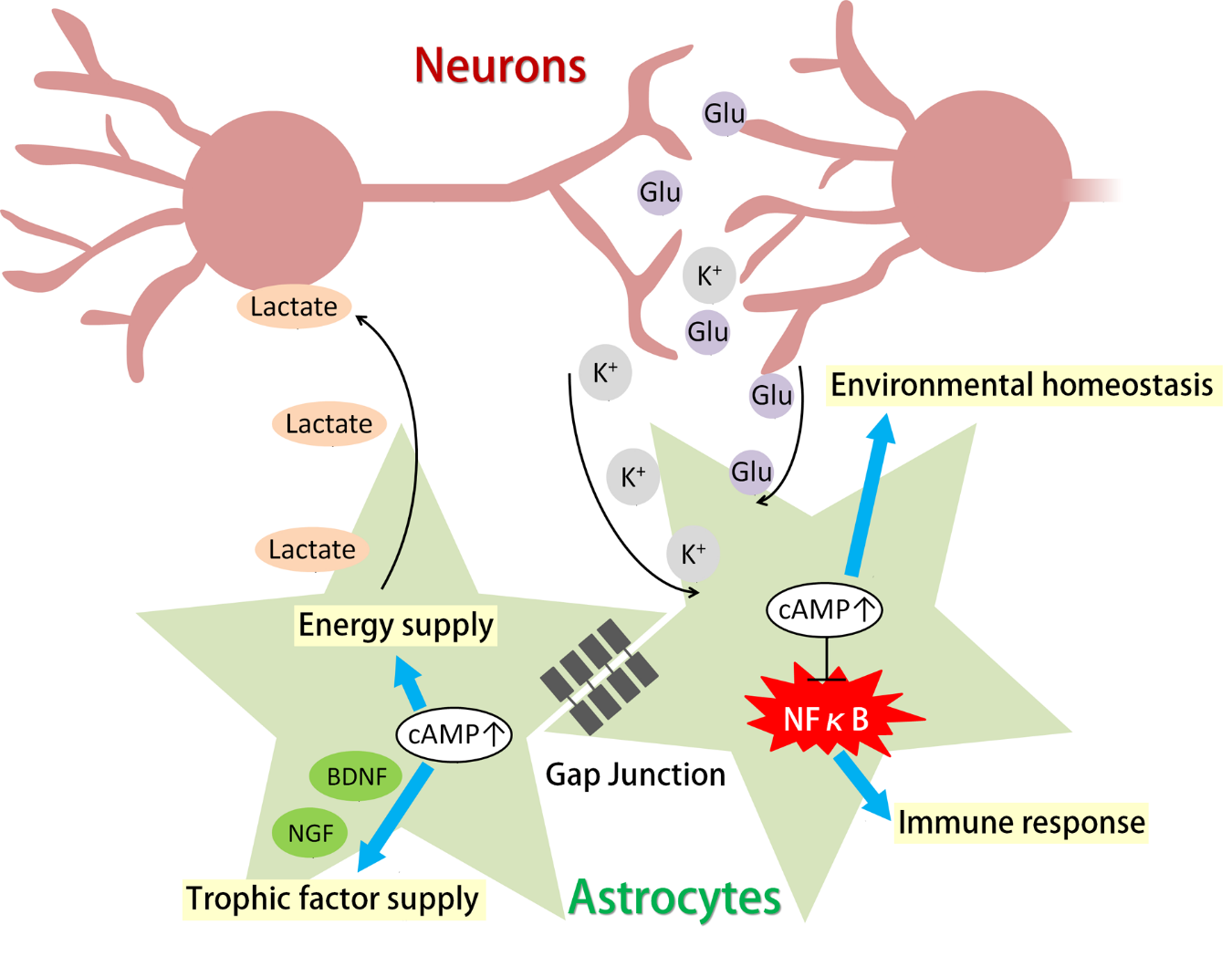{kind=link}
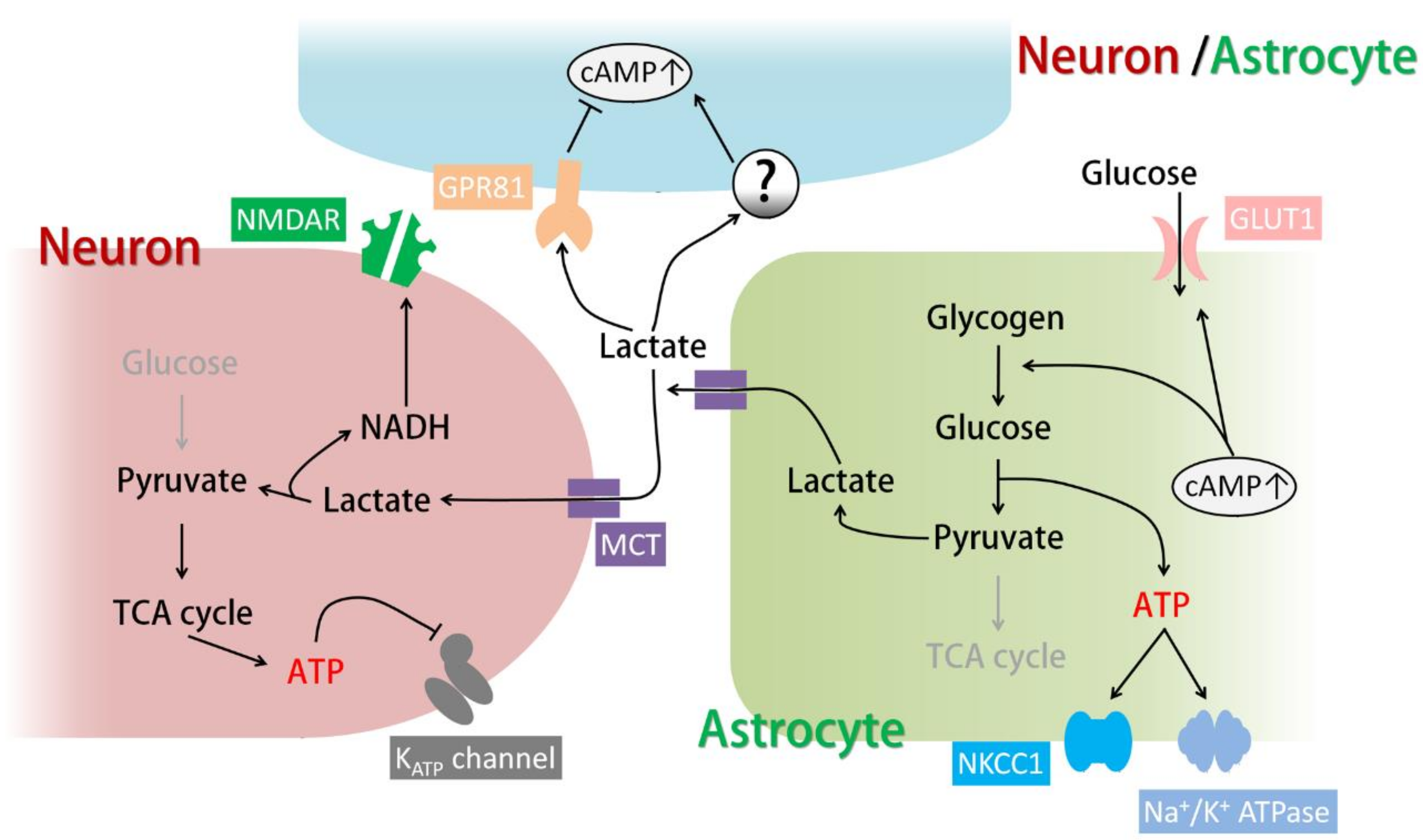{kind=link}
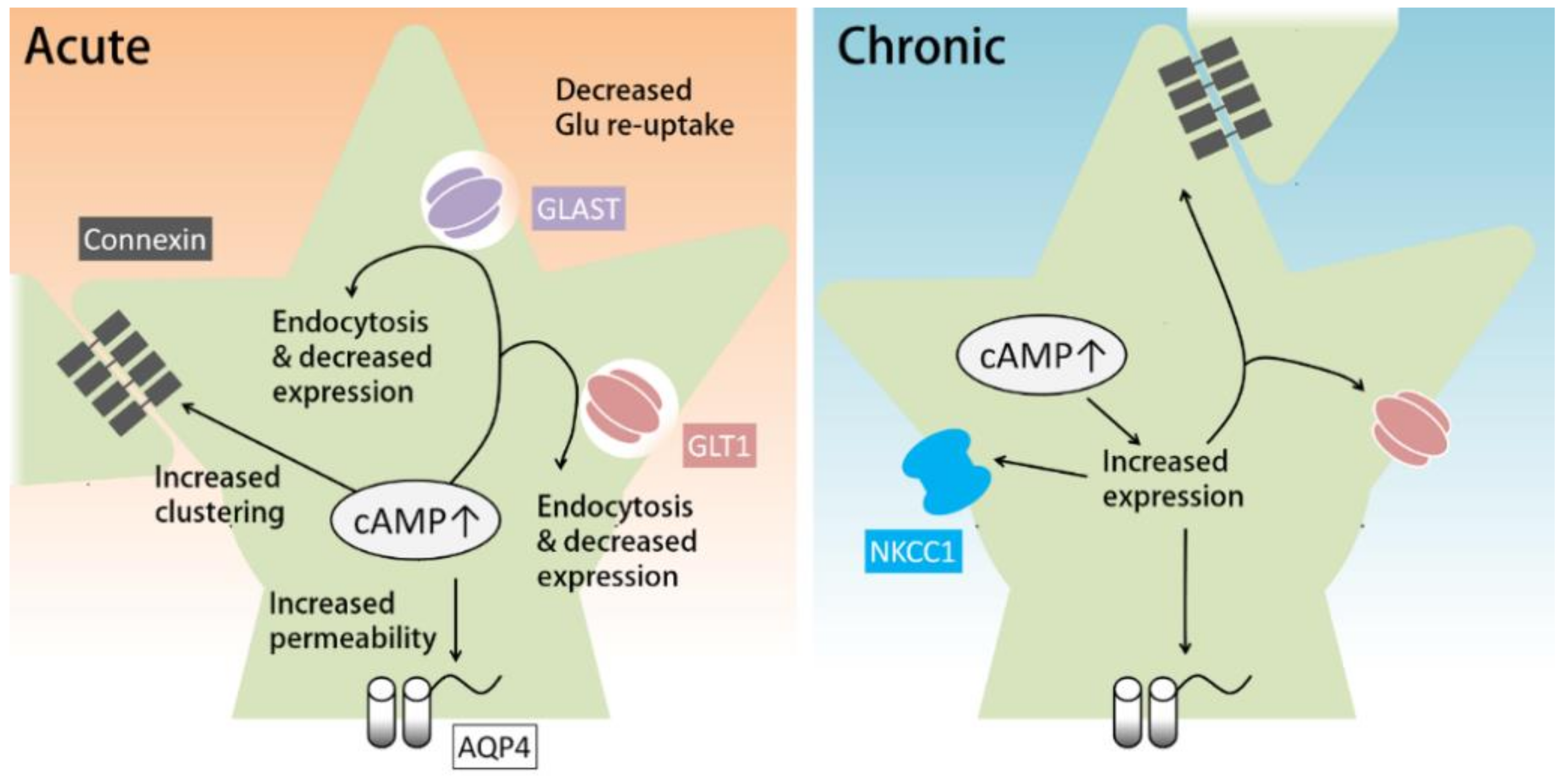{kind=link}
{kind=link}
1. Introduction
2. The cAMP Pathway in Astrocytes
2.1. cAMP Synthesis in Astrocytes
2.2. Pathways Downstream of Astrocytic cAMP
3. Glycogenolysis and Lactate Release from Astrocytes
3.1. Glycogenolysis in Astrocytes and Its Dependence on cAMP
3.2. Functions of Astrocytic Glycogenolysis and the Lactate Shuttle in Relation to cAMP Signals
3.3. Other Targets of Lactate
4. Astrocytes and Extracellular Maintenance
4.1. Astrocytic cAMP and Extracellular K+ Clearance
4.2. cAMP Regulates Glutamate Reuptake by Astrocytes
4.3. Astrocytic cAMP and Water Transport
5. Immune Response and Astrocytes
5.1. Astrocytic cAMP Regulates Activation of NF-κB
5.2. cAMP Pathways Regulate the Release of Cytokines and Inflammatory Factors
5.3. Astrocytes and Peripheral Immune Cell Infiltration
6. Neurotrophic Factors from Astrocytes
6.1. cAMP-Dependent Release of Trophic Factors from Astrocytes
6.2. Functions of Astrocyte-Derived Trophic Factors
6.3. Astrocytic cAMP and BDNF in Depression
7. Conclusions
Funding
Conflicts of Interest
References
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K+spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Royal, C.; Dupuis, J.; Groc, L.; Oliet, S.H.R. Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J. Neurosci. Res. 2017, 95, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Parpura, V. Central Role of Maladapted Astrocytic Plasticity in Ischemic Brain Edema Formation. Front. Cell. Neurosci. 2016, 10, 129. [Google Scholar] [CrossRef]
- Wang, Q.; Jie, W.; Liu, J.H.; Yang, J.M.; Gao, T.M. An astroglial basis of major depressive disorder? An overview. Glia 2017, 65, 1227–1250. [Google Scholar] [CrossRef]
- Martín, R.; Bajo-Grañeras, R.; Moratalla, R.; Perea, G.; Araque, A. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 2015, 349, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martin, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci. 2017, 20, 1540–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabst, M.; Braganza, O.; Dannenberg, H.; Hu, W.; Pothmann, L.; Rosen, J.; Mody, I.; van Loo, K.; Deisseroth, K.; Becker, A.J.; et al. Astrocyte Intermediaries of Septal Cholinergic Modulation in the Hippocampus. Neuron 2016, 90, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Dunphy, J.M.; Tolman, M.; Dineley, K.T.; Haydon, P.G. Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron 2017, 94, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Scharbarg, E.; Daenens, M.; Lemaître, F.; Geoffroy, H.; Guille-Collignon, M.; Gallopin, T.; Rancillac, A. Astrocyte-derived adenosine is central to the hypnogenic effect of glucose. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Vardjan, N. Astroglial cAMP signalling in space and time. Neurosci. Lett. 2019, 689, 5–10. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Tse, F.W.Y. Norepinephrine and cyclic adenosine 3′:5′-cyclic monophosphate enhance a nifedipine-sensitive calcium current in cultured rat astrocytes. Glia 1988, 1, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Chun, L.L.; Corey, D.P. Calcium current in cortical astrocytes: Induction by cAMP and neurotransmitters and permissive effect of serum factors. J. Neurosci. 1989, 9, 3169–3175. [Google Scholar] [CrossRef] [PubMed]
- Ujita, S.; Sasaki, T.; Asada, A.; Funayama, K.; Gao, M.; Mikoshiba, K.; Matsuki, N.; Ikegaya, Y. cAMP-Dependent Calcium Oscillations of Astrocytes: An Implication for Pathology. Cereb. Cortex 2017, 27, 1602–1614. [Google Scholar] [CrossRef]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial Excitability and Gliotransmission: An Appraisal of Ca 2+ as a Signalling Route. ASN Neuro 2012, 4, e00080. [Google Scholar] [CrossRef]
- Vardjan, N.; Parpura, V.; Zorec, R. Loose excitation–secretion coupling in astrocytes. Glia 2016, 64, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Zorec, R.; Vardjan, N. Adrenergic stimulation of single rat astrocytes results in distinct temporal changes in intracellular Ca2+ and cAMP-dependent PKA responses. Cell Calcium 2016, 59, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Catus, S.L.; Gibbs, M.E.; Sato, M.; Summers, R.J.; Hutchinson, D.S. Role of β-adrenoceptors in glucose uptake in astrocytes using β-adrenoceptor knockout mice. Br. J. Pharmacol. 2011, 162, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Sutin, J. Expression of adrenergic receptors in individual astrocytes and motor neurons isolated from the adult rat brain. Glia 1992, 6, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Gao, V.; Suzuki, A.; Magistretti, P.J.; Lengacher, S.; Pollonini, G.; Steinman, M.Q.; Alberini, C.M. Astrocytic β 2 -adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl. Acad. Sci. USA 2016, 113, 8526–8531. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, L.I.; Sims, R.E.; Dale, N.; Haydon, P.G. Wakefulness Affects Synaptic and Network Activity by Increasing Extracellular Astrocyte-Derived Adenosine. J. Neurosci. 2012, 32, 4417–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardjan, N.; Kreft, M.; Zorec, R. Dynamics of β-adrenergic/cAMP signaling and morphological changes in cultured astrocytes. Glia 2014, 62, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Zeinstra, E.M.; Wilczak, N.; Wilschut, J.C.; Glazenburg, L.; Chesik, D.; Kroese, F.G.M.; De Keyser, J. 5HT4agonists inhibit interferon-γ-induced MHC class II and B7 costimulatory molecules expression on cultured astrocytes. J. Neuroimmunol. 2006, 179, 191–195. [Google Scholar] [CrossRef]
- Kong, E.K.C.; Peng, L.; Chen, Y.; Yu, A.C.H.; Hertz, L. Up-regulation of 5-HT2B receptor density and receptor-mediated glycogenolysis in mouse astrocytes by long-term fluoxetine administration. Neurochem. Res. 2002, 27, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M.; Diaz-Corrales, F.J.; Miyoshi, K.; Ogawa, N. Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res. 2004, 1029, 120–123. [Google Scholar] [CrossRef]
- Koppel, I.; Jaanson, K.; Klasche, A.; Tuvikene, J.; Tiirik, T.; Pärn, A.; Timmusk, T. Dopamine cross-reacts with adrenoreceptors in cortical astrocytes to induce BDNF expression, CREB signaling and morphological transformation. Glia 2018, 66, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Hosli, E.; Hosli, L. Autoradiographic localization of binding sites for [3H]histamine and H1- and H2-antagonists on cultured neurones and glial cells. Neuroscience 1984, 13, 863–870. [Google Scholar] [CrossRef]
- Kubo, A.; Fukui, H.; Inagaki, N.; Kanamura, A.; Wada, H. Histamine-induced cyclic AMP accumulation in type-1 and type-2 astrocytes in primary culture. Eur. J. Pharmacol. Mol. Pharmacol. 1991, 208, 249–253. [Google Scholar] [CrossRef]
- Joo, K.M.; Chung, Y.H.; Kim, M.K.; Nam, R.H.; Lee, B.L.; Lee, K.H.; Cha, C.I. Distribution of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide receptors (VPAC1, VPAC2, and PAC1 receptor) in the rat brain. J. Comp. Neurol. 2004, 476, 388–413. [Google Scholar] [CrossRef]
- Masmoudi-Kouki, O.; Gandolfo, P.; Castel, H.; Leprince, J.; Fournier, A.; Dejda, A.; Vaudry, H.; Tonon, M.C. Role of PACAP and VIP in astroglial functions. Peptides 2007, 28, 1753–1760. [Google Scholar] [CrossRef]
- Schmid, A.; Meili, D.; Salathe, M. Soluble adenylyl cyclase in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2584–2592. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.B.; Gordon, G.R.J.; Zhou, N.; Tai, C.; Rungta, R.L.; Martinez, J.; Milner, T.A.; Ryu, J.K.; McLarnon, J.G.; Tresguerres, M.; et al. Metabolic Communication between Astrocytes and Neurons via Bicarbonate-Responsive Soluble Adenylyl Cyclase. Neuron 2012, 75, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Antonio, L.L.; Anderson, M.L.; Angamo, E.A.; Gabriel, S.; Klaft, Z.J.; Liotta, A.; Salar, S.; Sandow, N.; Heinemann, U. In vitro seizure like events and changes in ionic concentration. J. Neurosci. Methods 2016, 260, 33–44. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Chaverot, N.; Strosberg, A.D.; Couraud, P.O. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-alpha secretion. J. Immunol. 1999, 163, 668–674. [Google Scholar] [PubMed]
- Vardjan, N.; Chowdhury, H.H.; Horvat, A.; Velebit, J.; Malnar, M.; Muhič, M.; Kreft, M.; Krivec, Š.G.; Bobnar, S.T.; Miš, K.; et al. Enhancement of Astroglial Aerobic Glycolysis by Extracellular Lactate-Mediated Increase in cAMP. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Sa, M.; Hong, Y.R.; Lee, C.J.; Koo, J. Fatty Acid Increases cAMP-dependent Lactate and MAO-B-dependent GABA Production in Mouse Astrocytes by Activating a G αs Protein-coupled Receptor. Exp. Neurobiol. 2018, 27, 365. [Google Scholar] [CrossRef] [PubMed]
- Modi, K.K.; Sendtner, M.; Pahan, K. Up-regulation of ciliary neurotrophic factor in astrocytes by aspirin; Implications for remyelination in multiple sclerosis. J. Biol. Chem. 2013, 288, 18533–18545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.-K.; Philips, M.-A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1931. [Google Scholar] [CrossRef]
- Honsa, P.; Pivonkova, H.; Harantova, L.; Butenko, O.; Kriska, J.; Dzamba, D.; Rusnakova, V.; Valihrach, L.; Kubista, M.; Anderova, M. Increased expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in reactive astrocytes following ischemia. Glia 2014, 62, 2004–2021. [Google Scholar] [CrossRef]
- Seo, H.; Lee, K. Epac2 contributes to PACAP-induced astrocytic differentiation through calcium ion influx in neural precursor cells. BMB Rep. 2016, 49, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, S.H.; Selige, J.; Dunkern, T.; Rassov, A.; Leist, M. Combined anti-inflammatory effects of β2-adrenergic agonists and PDE4 inhibitors on astrocytes by upregulation of intracellular cAMP. Neurochem. Int. 2011, 59, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Voss, C.M.; Pajęcka, K.; Stridh, M.H.; Nissen, J.D.; Schousboe, A.; Waagepetersen, H.S. AMPK Activation Affects Glutamate Metabolism in Astrocytes. Neurochem. Res. 2015, 40, 2431–2442. [Google Scholar] [CrossRef]
- Rosenberg, P.A.; Dichter, M.A. Extracellular cAMP Accumulation and Degradation in Rat Cerebral Cortex in Dissociated Cell Culture. J. Neurosci. 1989, 9, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.A.; Knowles, R.; Knowles, K.P.; Li, Y. Beta-adrenergic receptor-mediated regulation of extracellular adenosine in cerebral cortex in culture. J Neurosci. 1994, 14, 2953–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Broadwell, R.D. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions: I. Neurons and glia. J. Electron Microsc. Tech. 1986, 3, 413–437. [Google Scholar] [CrossRef]
- Oe, Y.; Baba, O.; Ashida, H.; Nakamura, K.C.; Hirase, H. Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia 2016, 64, 1532–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasel, P.; Dando, O.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorg, O.; Magistretti, P.J. Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res. 1991, 563, 227–233. [Google Scholar] [CrossRef]
- Hertz, L.; Xu, J.; Song, D.; Du, T.; Li, B.; Yan, E.; Peng, L. Astrocytic glycogenolysis: Mechanisms and functions. Metab. Brain Dis. 2014, 30, 317–333. [Google Scholar] [CrossRef]
- Dong, J.H.; Chen, X.; Cui, M.; Yu, X.; Pang, Q.; Sun, J.P. Beta2-adrenergic receptor and astrocyte glucose metabolism. J. Mol. Neurosci. 2012, 48, 456–463. [Google Scholar] [CrossRef]
- Hutchinson, D.S.; Summers, R.J.; Gibbs, M.E. β2- and β3-Adrenoceptors activate glucose uptake in chick astrocytes by distinct mechanisms: A mechanism for memory enhancement? J. Neurochem. 2007, 103, 997–1008. [Google Scholar] [CrossRef]
- Allaman, I.; Lengacher, S.; Magistretti, P.J.; Pellerin, L. A2B receptor activation promotes glycogen synthesis in astrocytes through modulation of gene expression. AJP Cell Physiol. 2003, 284, C696–C704. [Google Scholar] [CrossRef]
- Xu, J.; Song, D.; Xue, Z.; Gu, L.; Hertz, L.; Peng, L. Requirement of glycogenolysis for uptake of increased extracellular K+in astrocytes: Potential implications for K+homeostasis and glycogen usage in brain. Neurochem. Res. 2013, 38, 472–485. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; O’Dowd, B.S.; Ng, K.T.; Gibbs, M.E. Reciprocal changes in forebrain contents of glycogen and of glutamate/glutamine during early memory consolidation in the day-old chick. Brain Res. 2003, 994, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Morton, M.M.; Sagar, S.M.; Sharp, F.R. Sensory stimulation induces local cerebral glycogenolysis: Demonstration by autoradiography. Neuroscience 1992, 51, 451–461. [Google Scholar] [CrossRef]
- Gibbs, M.E.; Anderson, D.G.; Hertz, L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 2006, 54, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Boury-Jamot, B.; Carrard, A.; Martin, J.L.; Halfon, O.; Magistretti, P.J.; Boutrel, B. Disrupting astrocyte–neuron lactate transfer persistently reduces conditioned responses to cocaine. Mol Psychiatry. 2016, 21, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef] [PubMed]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef]
- Schousboe, A.; Bak, L.K.; Waagepetersen, H.S. Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front. Endocrinol. (Lausanne) 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Hutchinson, D.S.; Summers, R.J. Role of β-adrenoceptors in memory consolidation: β3- adrenoceptors act on glucose uptake and β2-adrenoceptors on glycogenolysis. Neuropsychopharmacology 2008, 33, 2384–2397. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Takahashi, Y.; Watabe, A.M.; Kubo, Y.; Kato, F. On-Site Energy Supply at Synapses through Monocarboxylate Transporters Maintains Excitatory Synaptic Transmission. J. Neurosci. 2014, 34, 2605–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ruchti, E.; Petit, J.-M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef] [PubMed]
- Bouzat, P.; Sala, N.; Suys, T.; Zerlauth, J.B.; Marques-Vidal, P.; Feihl, F.; Bloch, J.; Messerer, M.; Levivier, M.; Meuli, R.; et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, Y.; Meng, S.; Luo, Y.; Liang, J.; Li, J.; Ai, S.; Sun, C.; Shen, H.; Zhu, W.; et al. Inhibition of Lactate Transport Erases Drug Memory and Prevents Drug Relapse. Biol. Psychiatry 2016, 79, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Yokai, M.; Kurihara, T.; Miyata, A. Spinal astrocytic activation contributes to both induction and maintenance of pituitary adenylate cyclase-activating polypeptide type 1 receptor-induced long-lasting mechanical allodynia in mice. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Lane, S.; Korsak, A.; Paton, J.F.R.; Gourine, A.V.; Kasparov, S.; Teschemacher, A.G. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat. Commun. 2014, 5, 3284. [Google Scholar] [CrossRef]
- Larsen, B.R.; Stoica, A.; MacAulay, N. Managing brain extracellular K+during neuronal activity: The physiological role of the Na+/K+-ATPase subunit isoforms. Front. Physiol. 2016, 7, 141. [Google Scholar] [CrossRef]
- Su, S. Regulation of Na+-K+-Cl−cotransporter in primary astrocytes by dibutyryl cAMP and high [K+]o. Am. J. Physiol. Cell Physiol. 2000, 279, C1720–C1721. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Wallraff, A. The Impact of Astrocytic Gap Junctional Coupling on Potassium Buffering in the Hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafavi, H.; Khaksarian, M.; Joghataei, M.T.; Yoosefee, S.; Soleimannejad, M.; Gholamzadeh, R.; Bahnamiri, S.S.; Hadjighassem, M.R. cAMP-Epac Pathway Stimulation Modulate Connexin-43 and MicroRNA-21 Expression in Glioma Cells. Basic Clin Neurosci. 2015, 6, 52–57. [Google Scholar] [PubMed]
- Khaksarian, M.; Mostafavi, H.; Soleimani, M.; Karimian, S.M.; Ghahremani, M.H.; Joghataei, M.T.; Khorashadizadeh, M.; Aligholi, H.; Attari, F.; Hassanzadeh, G. Regulation of connexin 43 and microRNA expression via β2-adrenoceptor signaling in 1321N1 astrocytoma cells. Mol. Med. Rep. 2015, 12, 1941–1950. [Google Scholar] [CrossRef]
- Hanstein, R.; Trotter, J.; Behl, C.; Clement, A.B. Increased Connexin 43 Expression as a Potential Mediator of the Neuroprotective Activity of the Corticotropin-Releasing Hormone. Mol. Endocrinol. 2009, 23, 1479–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenbroek, E.M.; Lampe, P.D.; Solan, J.L.; Reynhout, J.K.; Johnson, R.G. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 2001, 155, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Petr, G.T.; Sun, Y.; Frederick, N.M.; Zhou, Y.; Dhamne, S.C.; Hameed, M.Q.; Miranda, C.; Bedoya, E.A.; Fischer, K.D.; Armsen, W.; et al. Conditional Deletion of the Glutamate Transporter GLT-1 Reveals That Astrocytic GLT-1 Protects against Fatal Epilepsy While Neuronal GLT-1 Contributes Significantly to Glutamate Uptake into Synaptosomes. J. Neurosci. 2015, 35, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Lehre, K.P.; Danbolt, N.C. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219–226. [Google Scholar] [CrossRef]
- Paco, S.; Hummel, M.; Plá, V.; Sumoy, L.; Aguado, F. Cyclic AMP signaling restricts activation and promotes maturation and antioxidant defenses in astrocytes. BMC Genom. 2016, 17, 304. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Maguire, J.L.; McMinn, M.T.; Scholz, R.E.; Sutherland, M.L. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Mol. Brain Res. 2004, 124, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Schlag, B.D.; Vondrasek, J.R.; Munir, M.; Kalandadze, A.; Zelenaia, O.A.; Rothstein, J.D.; Robinson, M.B. Regulation of the Glial Na + -Dependent Glutamate Transporters by Cyclic AMP Analogs and Neurons. Mol. Pharmacol. 1998, 53, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo, H.; Sayed, M.D.S.; Lu, Y.; Yang, T.; Zhou, D.; Chen, Z.; Wang, H.; Wang, C.; Xu, J. cAMP/PKA/CREB/GLT1 signaling involved in the antidepressant-like effects of phosphodiesterase 4D inhibitor (GEBR-7b) in rats. Neuropsychiatr. Dis. Treat. 2016, 12, 219–227. [Google Scholar] [PubMed] [Green Version]
- Nishizaki, T.; Nagai, K.; Nomura, T.; Tada, H.; Kanno, T.; Tozaki, H.; Li, X.X.; Kondoh, T.; Kodama, N.; Takahashi, E.; et al. A new neuromodulatory pathway with a glial contribution mediated via A2a adenosine receptors. Glia 2002, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Agostinho, P. Adenosine A2Areceptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Adolph, O.; Köster, S.; Räth, M.; Georgieff, M.; Weigt, H.U.; Engele, J.; Senftleben, U.; Föhr, K.J. Rapid increase of glial glutamate uptake via blockade of the protein kinase A pathway. Glia 2007, 55, 1699–1707. [Google Scholar] [CrossRef]
- Matos, M.; Shen, H.Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.F. Deletion of Adenosine A2A Receptors from Astrocytes Disrupts Glutamate Homeostasis Leading to Psychomotor and Cognitive Impairment: Relevance to Schizophrenia. Biol. Psychiatry 2015, 78, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, I.E.; Ghorpade, A. Methamphetamine and HIV-1-induced neurotoxicity: Role of trace amine associated receptor 1 cAMP signaling in astrocytes. Neuropharmacology 2014, 85, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Räth, M.; Föhr, K.J.; Weigt, H.U.; Gauss, A.; Engele, J.; Georgieff, M.; Köster, S.; Adolph, O. Etomidate reduces glutamate uptake in rat cultured glial cells: Involvement of PKA. Br. J. Pharmacol. 2008, 155, 925–933. [Google Scholar] [CrossRef]
- Lim, G.; Wang, S.; Mao, J. cAMP and protein kinase A contribute to the downregulation of spinal glutamate transporters after chronic morphine. Neurosci. Lett. 2005, 376, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.H.; Wang, Y.H.; Tsai, R.Y.; Wang, J.J.; Tao, P.L.; Liu, T.M.; Wang, Y.C.; Wong, C.S. Amitriptyline preserves morphine’s antinociceptive effect by regulating the glutamate transporter GLAST and GLT-1 trafficking and excitatory amino acids concentration in morphine-tolerant rats. Pain 2007, 129, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Yoshitoshi, T.; Nagai, Y.; Kitahara, K. Increased glutamate uptake and GLAST expression by cyclic AMP in retinal glial cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hérault, K.; Zylbersztejn, K.; Lauterbach, M.A.; Guillon, M.; Oheim, M.; Ropert, N. Astrocyte VAMP3 vesicles undergo Ca2+-independent cycling and modulate glutamate transporter trafficking. J. Physiol. 2015, 593, 2807–2832. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Shanker, G.; Aschner, M. Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Res. 2001, 894, 131–140. [Google Scholar] [CrossRef]
- Nasca, C.; Bigio, B.; Zelli, D.; de Angelis, P.; Lau, T.; Okamoto, M.; Soya, H.; Ni, J.; Brichta, L.; Greengard, P.; et al. Role of the Astroglial Glutamate Exchanger xCT in Ventral Hippocampus in Resilience to Stress. Neuron 2017, 96, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Gochenauer, G.E.; Robinson, M.B. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc(-) subunits. J. Neurochem. 2001, 78, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system x(C)- cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Ottersen, O.P. Physiological Roles of Aquaporin-4 in Brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.; Kim, J.E.; Im, J.J.; Lee, J.; Jeong, H.S.; Park, S.; Jung, S.Y.; An, H.; Yoon, S.; Lim, S.M.; et al. Astrocytic water channel aquaporin-4 modulates brain plasticity in both mice and humans: A potential gliogenetic mechanism underlying language-associated learning. Mol. Psychiatry 2018, 23, 1021–1030. [Google Scholar] [CrossRef]
- Scharfman, H.E.; Binder, D.K. Aquaporin-4 water channels and synaptic plasticity in the hippocampus. Neurochem. Int. 2013, 63, 702–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of Select Forms of Spatial Memory and Neurotrophin-Dependent Synaptic Plasticity by Deletion of Glial Aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lackland, D.T.; Roccella, E.J.; Deutsch, A.F.; Fornage, M.; George, M.G.; Howard, G.; Kissela, B.M.; Kittner, S.J.; Lichtman, J.H.; Lisabeth, L.D.; et al. Factors influencing the decline in stroke mortality a statement from the american heart association/american stroke association. Stroke 2014, 45, 315–353. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Kahle, K.T.; Walcott, B.P.; Gerzanich, V.; Simard, J.M. Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl. Stroke Res. 2014, 5, 3–16. [Google Scholar] [CrossRef]
- Vardjan, N.; Horvat, A.; Anderson, J.E.; Yu, D.; Croom, D.; Zeng, X.; Lužnik, Z.; Kreft, M.; Teng, Y.D.; Kirov, S.A.; et al. Adrenergic activation attenuates astrocyte swelling induced by hypotonicity and neurotrauma. Glia 2016, 64, 1034–1049. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Yao, X.; Derugin, N.; Manley, G.T.; Verkman, A.S. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci. Lett. 2015, 584, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Gunnarson, E. Potassium dependent regulation of astrocyte water permeability is mediated by camp signaling. PLoS ONE 2012, 7, e34936. [Google Scholar] [CrossRef]
- Su, G.; Kintner, D.B.; Flagella, M.; Shull, G.E.; Sun, D. Astrocytes from Na+-K+-Cl−cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 2002, 282, C1147–C1160. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Liu, M.; Moriyama, M.; Ramakrishnan, R.; Forbush, B.; Reddy, P.V.B.; Norenberg, M.D. Na-K-Cl cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem. 2008, 283, 33874–33882. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Okabe, K.; Shibasaki, K.; Funatsu, T.; Matsuki, N.; Ikegaya, Y.; Koyama, R. Ischemic Brain Injury Leads to Brain Edema via Hyperthermia-Induced TRPV4 Activation. J. Neurosci. 2018, 38, 5700–5709. [Google Scholar] [CrossRef] [PubMed]
- Wurm, A.; Lipp, S.; Pannicke, T.; Linnertz, R.; Krügel, U.; Schulz, A.; Färber, K.; Zahn, D.; Grosse, J.; Wiedemann, P.; et al. Endogenous purinergic signaling is required for osmotic volume regulation of retinal glial cells. J. Neurochem. 2010, 112, 1261–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faff, L.; Reichenbach, A.; Albrecht, J. Ammonia-induced taurine release from cultured rabbit Muller cells is an osmoresistant process mediated by intracellular accumulation of cyclic AMP. J. Neurosci. Res. 1996, 46, 231–238. [Google Scholar] [CrossRef]
- Hertz, L.; Xu, J.; Chen, Y.; Gibbs, M.; Du, T. Antagonists of the Vasopressin V1 Receptor and of the β1-Adrenoceptor Inhibit Cytotoxic Brain Edema in Stroke by Effects on Astrocytes—But the Mechanisms Differ. Curr. Neuropharmacol. 2014, 12, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Xu, J.; Hertz, L.; Peng, L. Regulatory volume increase in astrocytes exposed to hypertonic medium requires β1-adrenergic Na+/K+-ATPase stimulation and glycogenolysis. J. Neurosci. Res. 2015, 93, 130–139. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroin fl ammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Syed, S.A.; Beurel, E.; Loewenstein, D.A.; Lowell, J.A.; Craighead, W.E.; Dunlop, B.W.; Mayberg, H.S.; Dhabhar, F.; Dietrich, W.D.; Keane, R.W.; et al. Defective Inflammatory Pathways in Never-Treated Depressed Patients Are Associated with Poor Treatment Response. Neuron 2018, 99, 914–924. [Google Scholar] [CrossRef]
- Brambilla, R.; Morton, P.D.; Ashbaugh, J.J.; Karmally, S.; Lambertsen, K.L.; Bethea, J.R. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia 2014, 62, 452–467. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron 2015, 85, 101–116. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggu, R.; Schumacher, T.; Gerich, F.; Rakers, C.; Tai, K.; Delekate, A.; Petzold, G.C. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 2016, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Zhuang, K.; Liu, Z.; Huang, C.; Gao, Y.; Chen, G.; Lin, H.; Hu, Y.; Wu, D.; Shi, M.; et al. Menin Deficiency Leads to Depressive-like Behaviors in Mice by Modulating Astrocyte-Mediated Neuroinflammation. Neuron 2018, 100, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Gavrilyuk, V.; Dello Russo, C.; Heneka, M.T.; Pelligrino, D.; Weinberg, G.; Feinstein, D.L. Norepinephrine increases I kappa B alpha expression in astrocytes. J. Biol. Chem. 2002, 277, 29662–29668. [Google Scholar] [CrossRef] [PubMed]
- Laureys, G.; Gerlo, S.; Spooren, A.; Demol, F.; De Keyser, J.; Aerts, J.L. β2-adrenergic agonists modulate TNFα induced astrocytic inflammatory gene expression and brain inflammatory cell populations. J. Neuroinflam. 2014, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; Van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Walle, L.V.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef]
- Guedes, R.P.; Csizmadia, E.; Moll, H.P.; Ma, A.; Ferran, C.; da Silva, C.G. A20 deficiency causes spontaneous neuroinflammation in mice. J. Neuroinflam. 2014, 11, 1–16. [Google Scholar] [CrossRef]
- Wang, X.; Deckert, M.; Xuan, N.T.; Nishanth, G.; Just, S.; Waisman, A.; Naumann, M.; Schlüter, D. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-κB- and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol. 2013, 126, 711–724. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type i interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesch-Bartlomowicz, B.; Huelster, A.; Wiss, O.; Antoniou-Lipfert, P.; Dietrich, C.; Arand, M.; Weiss, C.; Bockamp, E.; Oesch, F. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: Divergent signaling pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 9218–9223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spooren, A.; Kooijman, R.; Lintermans, B.; Van Craenenbroeck, K.; Vermeulen, L.; Haegeman, G.; Gerlo, S. Cooperation of NFκB and CREB to induce synergistic IL-6 expression in astrocytes. Cell Signal. 2010, 22, 871–881. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Ryan, K.M.; Kilroy, D.; Connor, T.J. Noradrenaline induces IL-1ra and IL-1 type II receptor expression in primary glial cells and protects against IL-1β-induced neurotoxicity. Eur. J. Pharmacol. 2010, 626, 219–228. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezzani, A.; Moneta, D.; Conti, M.; Richichi, C.; Ravizza, T.; De Luigi, A.; De Simoni, M.G.; Sperk, G.; Andell-Jonsson, S.; Lundkvist, J.; et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11534–11539. [Google Scholar] [CrossRef] [Green Version]
- Auvin, S.; Shin, D.; Mazarati, A.; Sankar, R. Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia 2010, 51, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, B.D.; O’Brien, T.J.; Gimlin, K.; Wright, D.K.; Eun Kim, S.; Casillas-Espinosa, P.M.; Webster, K.M.; Petrou, S.; Noble-Haeusslein, L.J. Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J. Neurosci. 2017, 37, 7864–7877. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Brady, R.D.; Wright, D.K.; Kim, H.A.; Zhang, S.R.; Sobey, C.G.; Johnstone, M.R.; O’Brien, T.J.; Semple, B.D.; McDonald, S.J.; et al. Treatment with an interleukin-1 receptor antagonist mitigates neuroinflammation and brain damage after polytrauma. Brain Behav. Immun. 2017, 66, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Garber, C.; Vasek, M.J.; Vollmer, L.L.; Sun, T.; Jiang, X.; Klein, R.S. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1 article. Nat. Immunol. 2018, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Lebonville, C.L.; Paniccia, J.E.; Balentine, M.E.; Reissner, K.J.; Lysle, D.T. Hippocampal interleukin-1 mediates stress-enhanced fear learning: A potential role for astrocyte-derived interleukin-1β. Brain Behav. Immun. 2018, 67, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006, 12, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Toft-Hansen, H.; Füchtbauer, L.; Owens, T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 2011, 59, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Haroon, F.; Drögemüller, K.; Händel, U.; Brunn, A.; Reinhold, D.; Nishanth, G.; Mueller, W.; Trautwein, C.; Ernst, M.; Deckert, M.; et al. Gp130-Dependent Astrocytic Survival Is Critical for the Control of Autoimmune Central Nervous System Inflammation. J. Immunol. 2011, 186, 6521–6531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petković, F.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Reduced cuprizone-induced cerebellar demyelination in mice with astrocyte-targeted production of IL-6. J. Neuroimmunol. 2017, 310, 97–102. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Reis, D.J. Norepinephrine Suppresses Inducible Nitric Oxide Synthase Activity in Rat Astroglial Cultures. J. Neurochem. 1993, 60, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Pahan, K.; Namboodiri, A.M.S.; Sheikh, F.G.; Smith, B.T.; Singh, I. Increasing cAMP attenuates induction of inducible nitric-oxide synthase in rat primary astrocytes. J. Biol. Chem. 1997, 272, 7786–7791. [Google Scholar] [CrossRef]
- Gavrilyuk, V.; Horvath, P.; Weinberg, G.; Feinstein, D.L. A 27-bp region of the inducible nitric oxide synthase promoter regulates expression in glial cells. J. Neurochem. 2001, 78, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Bannerman, P.; Ma, J.; Guo, F.; Miers, L.; Soulika, A.M.; Pleasure, D. Conditional Ablation of Astroglial CCL2 Suppresses CNS Accumulation of M1 Macrophages and Preserves Axons in Mice with MOG Peptide EAE. J. Neurosci. 2014, 34, 8175–8185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.Y.; Hoffman, A.S.; Itoh, N.; Ao, Y.; Spence, R.; Sofroniew, M.V.; Voskuhl, R.R. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal, J.L.M.; Leza, J.C.; Polak, P.; Kalinin, S.; Feinstein, D.L. Astrocyte-Derived MCP-1 Mediates Neuroprotective Effects of Noradrenaline. J. Neurosci. 2009, 29, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.-S.; Peng, J.; Murugan, M.; Feng, L.-J.; Liu, J.-L.; Eyo, U.B.; Zhou, L.-J.; Mogilevsky, R.; Wang, W.; Wu, L.-J. Chemokine CCL2–CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1β Production after Status Epilepticus. J. Neurosci. 2017, 37, 7878–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Zhang, X.D.; Miao, W.Y.; Sun, Y.J.; Xiong, G.; Wu, Q.; Li, G.; Yang, P.; Yu, H.; Li, H.; et al. PDGFRβ Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 2018, 100, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; Leclair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Bullard, D.C.; Hu, X.; Schoeb, T.R.; Collins, R.G.; Beaudet, A.L.; Barnum, S.R. Intercellular Adhesion Molecule-1 Expression Is Required on Multiple Cell Types for the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2007, 178, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, M.A.T.; Sim, J.E.; Russell, J.H. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J. Neuroimmunol. 2004, 151, 116–125. [Google Scholar] [CrossRef]
- Ballestas, M.E.; Benveniste, E.N. Elevation of cyclic AMP levels in astrocytes antagonizes cytokine-induced adhesion molecule expression. J. Neurochem. 1997, 69, 1438–1448. [Google Scholar] [CrossRef]
- Frohman, E.M.; Vayuvegula, B.; Gupta, S.; van den Noort, S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc. Natl. Acad. Sci. USA 1988, 85, 1292–1296. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Von Bohlen und Halbach, O.; von Bohlen und Halbach, V. BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res. 2018, 373, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Jurič, D.M.; Miklič, Š.; Čarman-Kržan, M. Monoaminergic neuronal activity up-regulates BDNF synthesis in cultured neonatal rat astrocytes. Brain Res. 2006, 1108, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Tanaka, T.; Sakai, Y.; Fukuchi, T.; Abe, H.; Nawa, H.; Takei, N. Müller cells as a source of brain-derived neurotrophic factor in the retina: Noradrenaline upregulates brain-derived neurotrophic factor levels in cultured rat Müller cells. Neurochem. Res. 2005, 30, 1163–1170. [Google Scholar] [CrossRef]
- Krzan, M.; Wu, V.W.; Schwartz, J.P. Serotonin regulation of nerve growth factor synthesis in neonatal and adult astrocytes: Comparison to the β-adrenergic agonist isoproterenol. J. Neurosci. Res. 2001, 64, 261–267. [Google Scholar] [CrossRef]
- Mele, T.; Čarman-Kržan, M.; Jurič, D.M. Regulatory role of monoamine neurotransmitters in astrocytic NT-3 synthesis. Int. J. Dev. Neurosci. 2010, 28, 13–19. [Google Scholar] [CrossRef]
- Jurič, D.M.; Lončar, D.; Čarman-Kržan, M. Noradrenergic stimulation of BDNF synthesis in astrocytes: Mediation via α1- and β1/β2-adrenergic receptors. Neurochem. Int. 2008, 52, 297–306. [Google Scholar] [CrossRef]
- Schwartz, J.P.; Nishiyama, N. Neurotrophic factor gene expression in astrocytes during development and following injury. Brain Res. Bull. 1994, 35, 403–407. [Google Scholar] [CrossRef]
- Qian, C.; Tan, D.; Wang, X.; Li, L.; Wen, J.; Pan, M.; Li, Y.; Wu, W.; Guo, J. Peripheral nerve injury-induced astrocyte activation in spinal ventral horn contributes to nerve regeneration. Neural Plast. 2018, 2018, 8561704. [Google Scholar] [CrossRef]
- Fulmer, C.G.; VonDran, M.W.; Stillman, A.A.; Huang, Y.; Hempstead, B.L.; Dreyfus, C.F. Astrocyte-Derived BDNF Supports Myelin Protein Synthesis after Cuprizone-Induced Demyelination. J. Neurosci. 2014, 34, 8186–8196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Huang, Y.; Jia, B.; Zhang, X.; Mo, D.; Ma, N.; Gao, F.; Song, L.; Wang, B.; Miao, Z. Quetiapine prevents Aβ25-35-induced cell death in cultured neuron by enhancing brain-derived neurotrophic factor release from astrocyte. Neuroreport 2018, 29, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, Y.; Inagaki, Y.; Logan, S.; Bosnjak, Z.J.; Bai, X. Insufficient astrocyte-derived brain-derived neurotrophic factor contributes to propofol-induced neuron death through Akt/Glycogen Synthase Kinase 3β/Mitochondrial Fission Pathway. Anesth. Analg. 2017, 125, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Turati, J.; Ramírez, D.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J. Neurochem. 2018, 146, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef]
- Chun, H.; An, H.; Lim, J.; Woo, J.; Lee, J.; Ryu, H.; Lee, C.J. Astrocytic proBDNF and Tonic GABA Distinguish Active versus Reactive Astrocytes in Hippocampus. Exp. Neurobiol. 2018, 27, 155. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef]
- Quesseveur, G.; David, D.J.; Gaillard, M.C.; Pla, P.; Wu, M.V.; Nguyen, H.T.; Nicolas, V.; Auregan, G.; David, I.; Dranovsky, A.; et al. BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl. Psychiatry 2013, 3, e253. [Google Scholar] [CrossRef]
- Shirayama, Y.; Chen, A.C.-H.; Nakagawa, S.; Russell, D.S.; Duman, R.S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002, 22, 3251–3261. [Google Scholar] [CrossRef]
- Björkholm, C.; Monteggia, L.M. BDNF e a key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Hisaoka-Nakashima, K.; Morioka, N.; Okada-Tsuchioka, M.; Kaneko, M.; Kasai, M.; Shibasaki, C.; Nakata, Y.; Takebayashi, M. Antidepressant Acts on Astrocytes Leading to an Increase in the Expression of Neurotrophic/Growth Factors: Differential Regulation of FGF-2 by Noradrenaline. PLoS ONE 2012, 7, e51197. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka-Nakashima, K.; Kajitani, N.; Kaneko, M.; Shigetou, T.; Kasai, M.; Matsumoto, C.; Yokoe, T.; Azuma, H.; Takebayashi, M.; Morioka, N.; et al. Amitriptyline induces brain-derived neurotrophic factor (BDNF) mRNA expression through ERK-dependent modulation of multiple BDNF mRNA variants in primary cultured rat cortical astrocytes and microglia. Brain Res. 2016, 1634, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Boku, S.; Hisaoka-Nakashima, K.; Nakagawa, S.; Kato, A.; Kajitani, N.; Inoue, T.; Kusumi, I.; Takebayashi, M. Tricyclic antidepressant amitriptyline indirectly increases the proliferation of adult dentate gyrus-derived neural precursors: An involvement of astrocytes. PLoS ONE 2013, 8, e79371. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Fiumelli, H.; Magistretti, P.J.; Martin, J.L. Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology (Berl.) 2011, 216, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kittel-Schneider, S.; Kenis, G.; Schek, J.; van den Hove, D.; Prickaerts, J.; Lesch, K.P.; Steinbusch, H.; Reif, A. Expression of monoamine transporters, nitric oxide synthase 3, and neurotrophin genes in antidepressant-stimulated astrocytes. Front. Psychiatry 2012, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.; Sakai, N.; Shin, K.-H.; Steffen, C.; Zhang, Y.-J.; Impey, S.; Storm, D.; Duman, R.S. cAMP Response Element-Mediated Gene Transcription Is Upregulated by Chronic Antidepressant Treatment. J. Neurosci. 2000, 20, 4030–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, K.; Yamasaki, H.; Kawabe, K.; Moriyama, M.; Nakamura, Y. Imipramine Induces Brain-Derived Neurotrophic Factor mRNA Expression in Cultured Astrocytes. J. Pharmacol. Sci. 2012, 120, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramontina, A.C.; Tramontina, F.; Bobermin, L.D.; Zanotto, C.; Souza, D.F.; Leite, M.C.; Nardin, P.; Gottfried, C.; Gonçalves, C.A. Secretion of S100B, an astrocyte-derived neurotrophic protein, is stimulated by fluoxetine via a mechanism independent of serotonin. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1580–1583. [Google Scholar] [CrossRef]
- Yang, H.; Xu, W.; Zhao, W.; Gu, M.; Wang, W. 1,3,7-Trihydroxyxanthone, derived from Polygalae Radix, a herbal medicine, stimulates the expression of neurotrophic factors in rat astrocyte primary cultures via cAMP- and ERK-dependent pathways. Biomed. Pharmacother. 2018, 98, 762–768. [Google Scholar] [CrossRef]
- Kinoshita, M.; Hirayama, Y.; Fujishita, K.; Shibata, K.; Shinozaki, Y.; Shigetomi, E.; Takeda, A.; Le, H.P.N.; Hayashi, H.; Hiasa, M.; et al. Anti-Depressant Fluoxetine Reveals its Therapeutic Effect Via Astrocytes. EBioMedicine 2018, 32, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.H.; Schappi, J.M.; Singh, H.; Senese, N.B.; Rasenick, M.M. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rasenick, M.M. Chronic treatment with escitalopram but not R-citalopram translocates Galpha(s) from lipid raft domains and potentiates adenylyl cyclase: A 5-hydroxytryptamine transporter-independent action of this antidepressant compound. J. Pharmacol. Exp. Ther. 2010, 332, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Donati, R.J.; Schappi, J.; Czysz, A.H.; Jackson, A.; Rasenick, M.M. Differential effects of antidepressants escitalopram versus lithium on Gs alpha membrane relocalization. BMC Neurosci. 2015, 16, 40. [Google Scholar] [CrossRef]
- Allen, J.A.; Yu, J.Z.; Dave, R.H.; Bhatnagar, A.; Roth, B.L.; Rasenick, M.M. Caveolin-1 and lipid microdomains regulate Gs trafficking and attenuate Gs/adenylyl cyclase signaling. Molecular 2009, 76, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Mukai, T.; Nagao, Y.; Matsuba, Y.; Tsuji, Y.; Tanaka, S.; Tokudome, K.; Shimizu, S.; Ito, H.; Ikeda, A.; et al. Inhibition of Inwardly Rectifying Potassium (Kir) 4.1 Channels Facilitates Brain-Derived Neurotrophic Factor (BDNF) Expression in Astrocytes. Front. Mol. Neurosci. 2017, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- John, C.S.; Smith, K.L.; Van’T Veer, A.; Gompf, H.S.; Carlezon, W.A.; Cohen, B.M.; Ngür, D.; Bechtholt-Gompf, A.J. Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology 2012, 37, 2467–2475. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, L.; Gao, C.; Zhai, L.; Zhang, N.; Guo, L. Astrocytes activation contributes to the antidepressant-like effect of ketamine but not scopolamine. Pharmacol. Biochem. Behav. 2018, 170, 1–8. [Google Scholar] [CrossRef]
- Braun, D.; Madrigal, J.; Feinstein, D. Noradrenergic Regulation of Glial Activation: Molecular Mechanisms and Therapeutic Implications. Curr. Neuropharmacol. 2014, 12, 342–352. [Google Scholar] [CrossRef]
- Nicol, X.; Voyatzis, S.; Muzerelle, A.; Narboux-Nême, N.; Südhof, T.C.; Miles, R.; Gaspar, P. cAMP oscillations and retinal activity are permissive for ephrin signaling during the establishment of the retinotopic map. Nat. Neurosci. 2007, 10, 340–347. [Google Scholar] [CrossRef]
- Nicol, X.; Hong, K.P.; Spitzer, N.C. Spatial and temporal second messenger codes for growth cone turning. Proc. Natl. Acad. Sci. USA 2011, 108, 13776–13781. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Tanaka, K.F.; Matsunaga, S.; Iseki, M.; Watanabe, M.; Matsuki, N.; Ikegaya, Y.; Koyama, R. Photoactivated adenylyl cyclase (PAC) reveals novel mechanisms underlying cAMP-dependent axonal morphogenesis. Sci. Rep. 2016, 5, 19679. [Google Scholar] [CrossRef]
- Harada, K.; Ito, M.; Wang, X.; Tanaka, M.; Wongso, D.; Konno, A.; Hirai, H.; Hirase, H.; Tsuboi, T.; Kitaguchi, T. Red fluorescent protein-based cAMP indicator applicable to optogenetics and in vivo imaging. Sci. Rep. 2017, 7, 7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Jongbloets, B.C.; Xiong, W.H.; Melander, J.B.; Qin, M.; Lameyer, T.J.; Harrison, M.F.; Zemelman, B.V.; Mao, T.; Zhong, H. A Highly Sensitive A-Kinase Activity Reporter for Imaging Neuromodulatory Events in Awake Mice. Neuron 2018, 99, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Iseki, M.; Matsunaga, S.; Murakami, A.; Ohno, K.; Shiga, K.; Yoshida, K.; Sugai, M.; Takahashi, T.; Hori, T.; Watanabe, M. A blue-light-activated adenylyl cyclase mediates photoavoidance in Euglena gracilis. Nature 2002, 415, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Stierl, M.; Stumpf, P.; Udwari, D.; Gueta, R.; Hagedorn, R.; Losi, A.; Gärtner, W.; Petereit, L.; Efetova, M.; Schwarzel, M.; et al. Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J. Biol. Chem. 2011, 286, 1181–1188. [Google Scholar] [CrossRef]
- Ohki, M.; Sugiyama, K.; Kawai, F.; Tanaka, H.; Nihei, Y.; Unzai, S.; Takebe, M.; Matsunaga, S.; Adachi, S.; Shibayama, N.; et al. Structural insight into photoactivation of an adenylate cyclase from a photosynthetic cyanobacterium. Proc. Natl. Acad. Sci. USA 2016, 113, 6659–6664. [Google Scholar] [CrossRef] [Green Version]
- Jansen, V.; Alvarez, L.; Balbach, M.; Strünker, T.; Hegemann, P.; Kaupp, U.B.; Wachten, D. Controlling fertilization and cAMP signaling in sperm by optogenetics. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Schröder-Lang, S.; Schwärzel, M.; Seifert, R.; Strünker, T.; Kateriya, S.; Looser, J.; Watanabe, M.; Kaupp, U.B.; Hegemann, P.; Nagel, G. Fast manipulation of cellular cAMP level by light in vivo. Nat. Methods 2007, 4, 39–42. [Google Scholar] [CrossRef]
- Blain-Hartung, M.; Rockwell, N.C.; Moreno, M.V.; Martin, S.S.; Gan, F.; Bryant, D.A.; Lagarias, J.C. Cyanobacteriochrome-based photoswitchable adenylyl cyclases (cPACs) for broad spectrum light regulation of cAMP levels in cells. J. Biol. Chem. 2018, 293, 8473–8483. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Ikegaya, Y.; Koyama, R. The Astrocytic cAMP Pathway in Health and Disease. Int. J. Mol. Sci. 2019, 20, 779. https://doi.org/10.3390/ijms20030779
Zhou Z, Ikegaya Y, Koyama R. The Astrocytic cAMP Pathway in Health and Disease. International Journal of Molecular Sciences. 2019; 20(3):779. https://doi.org/10.3390/ijms20030779
Chicago/Turabian StyleZhou, Zhiwen, Yuji Ikegaya, and Ryuta Koyama. 2019. "The Astrocytic cAMP Pathway in Health and Disease" International Journal of Molecular Sciences 20, no. 3: 779. https://doi.org/10.3390/ijms20030779
APA StyleZhou, Z., Ikegaya, Y., & Koyama, R. (2019). The Astrocytic cAMP Pathway in Health and Disease. International Journal of Molecular Sciences, 20(3), 779. https://doi.org/10.3390/ijms20030779