Comparison of Immune Responses to Different Versions of VLP Associated Stabilized RSV Pre-Fusion F Protein

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Plasmids, and Viruses
2.2. Antibodies
2.3. VLP Preparation, Purification, and Characterization
2.4. Preparation of Soluble F Proteins
2.5. Detection of Cell Surface Protein by Surface Biotinylation
2.6. Measures of Relative Binding of Mab to Purified VLPs
2.7. Determination of Stability of Pre-Fusion F Conformation
2.8. Determination of Total Anti-F Protein IgG in Sera
2.9. RSV Plaque Assays, Antibody Neutralization, and Antibody Blocking
2.10. Animals, Animal Immunization, and RSV Challenge
2.11. Statistical Analysis
3. Results
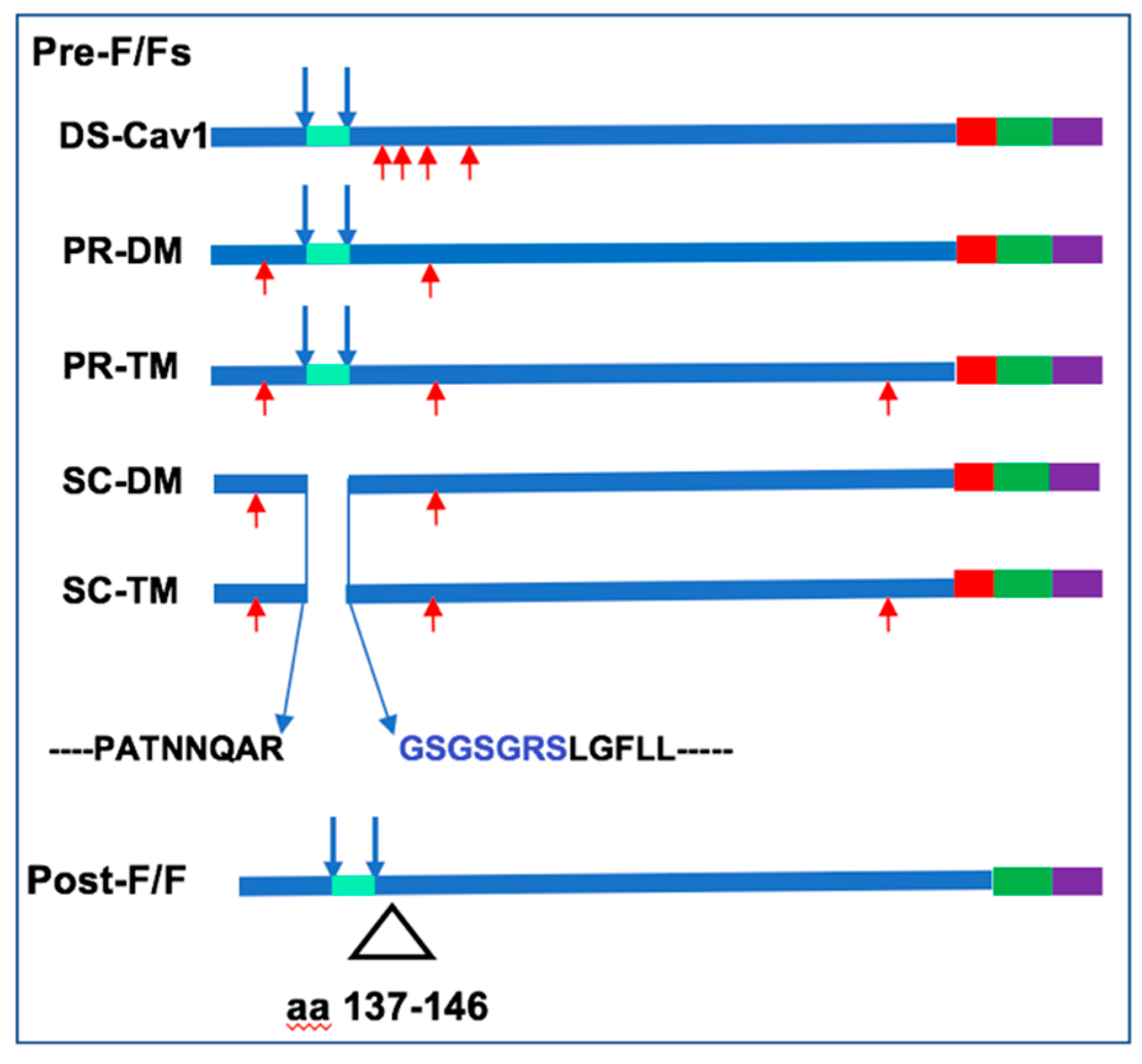
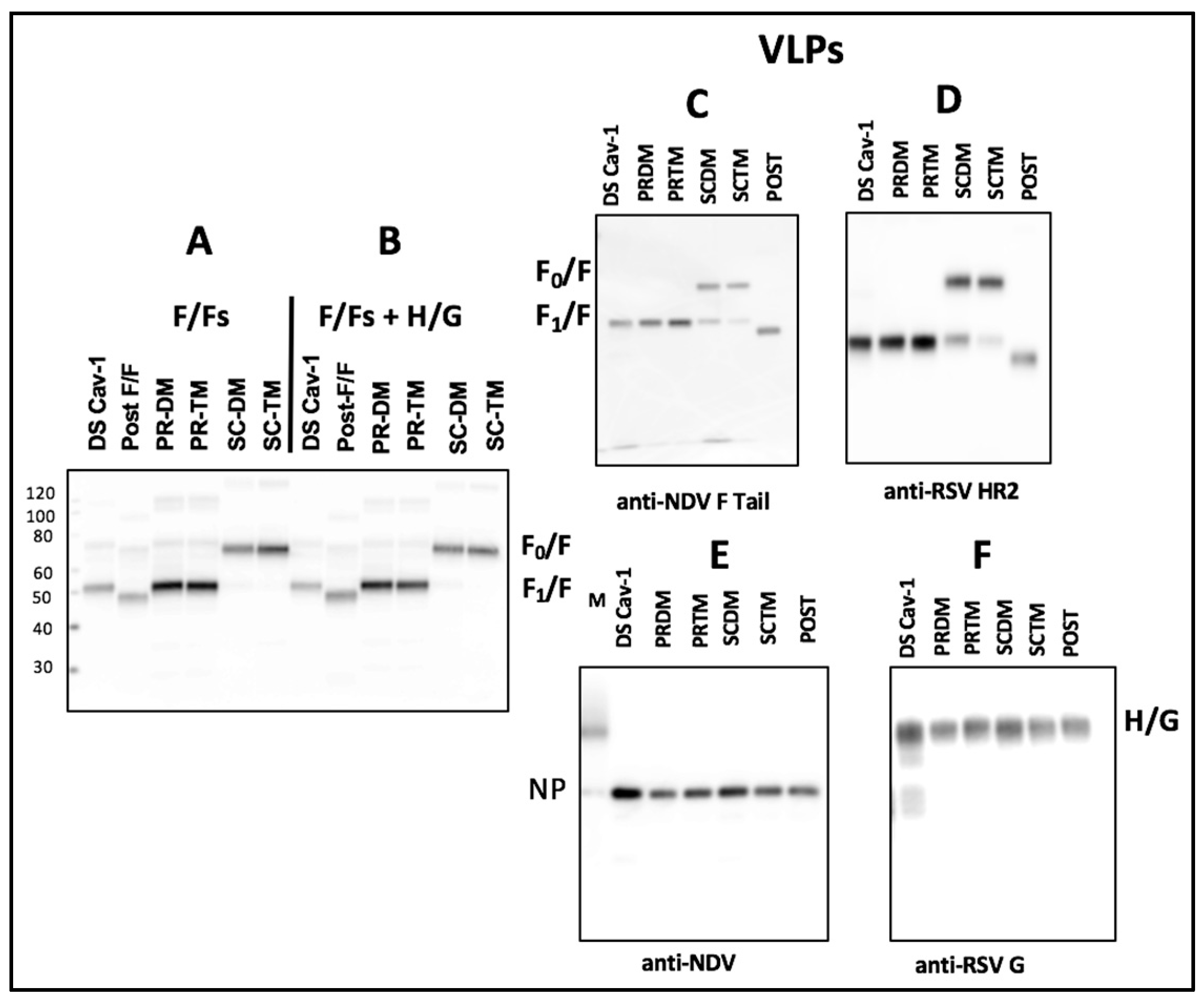
3.1. Incorporation of Pre-fusion F Proteins Into VLPs
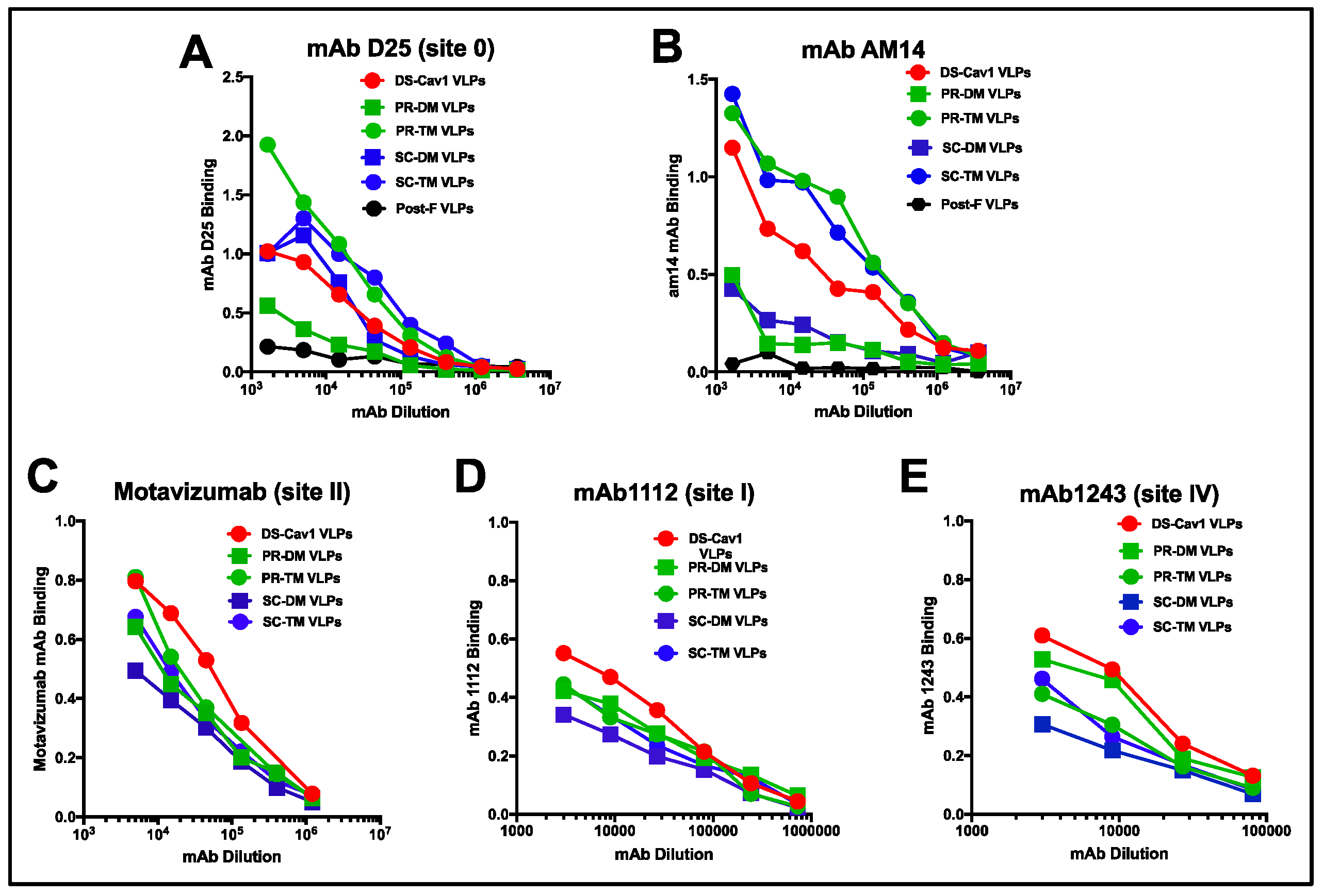
3.2. Relative Binding of Monoclonal Antibodies to Different VLPs
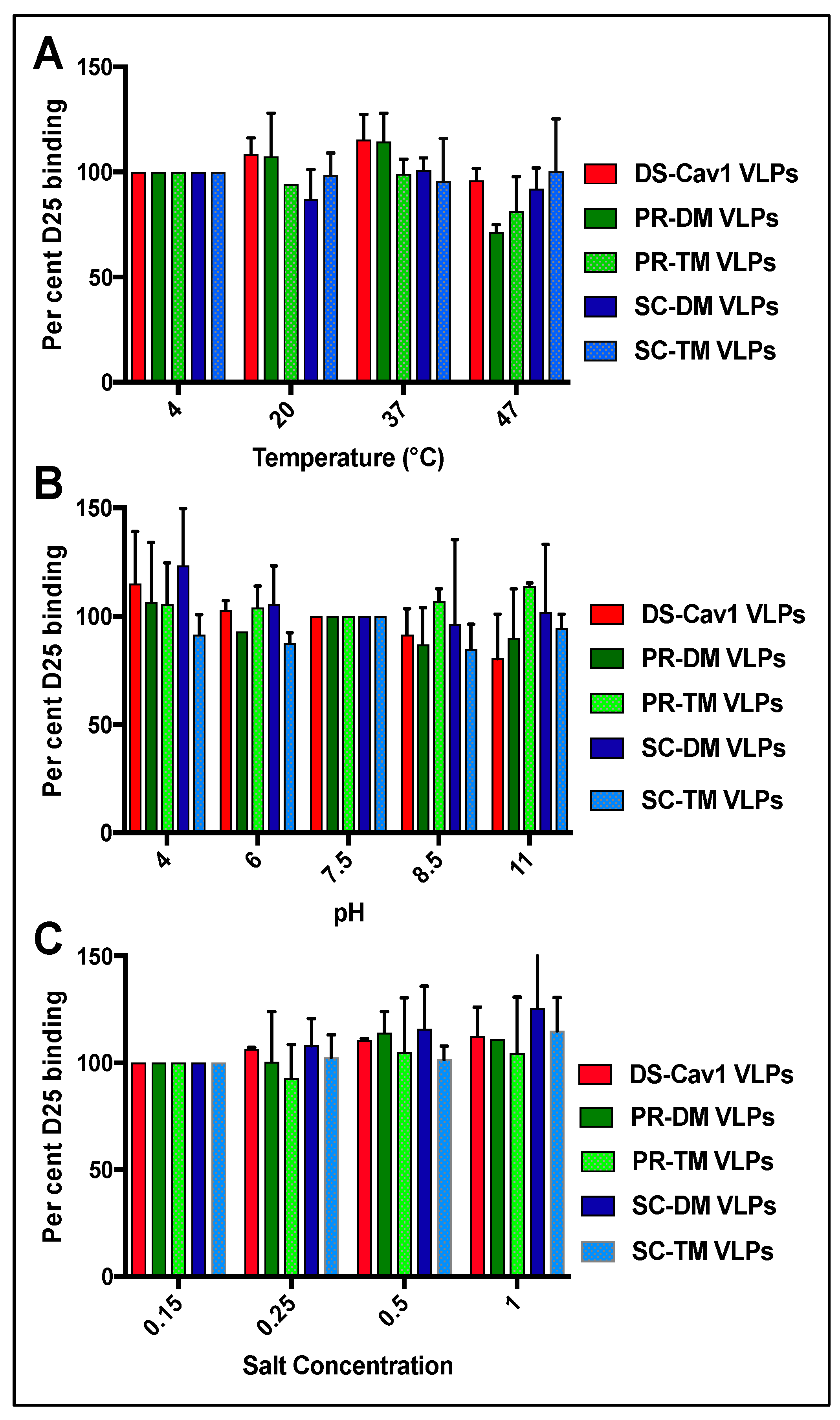
3.3. Stability of the Pre-Fusion F Proteins in the VLPs
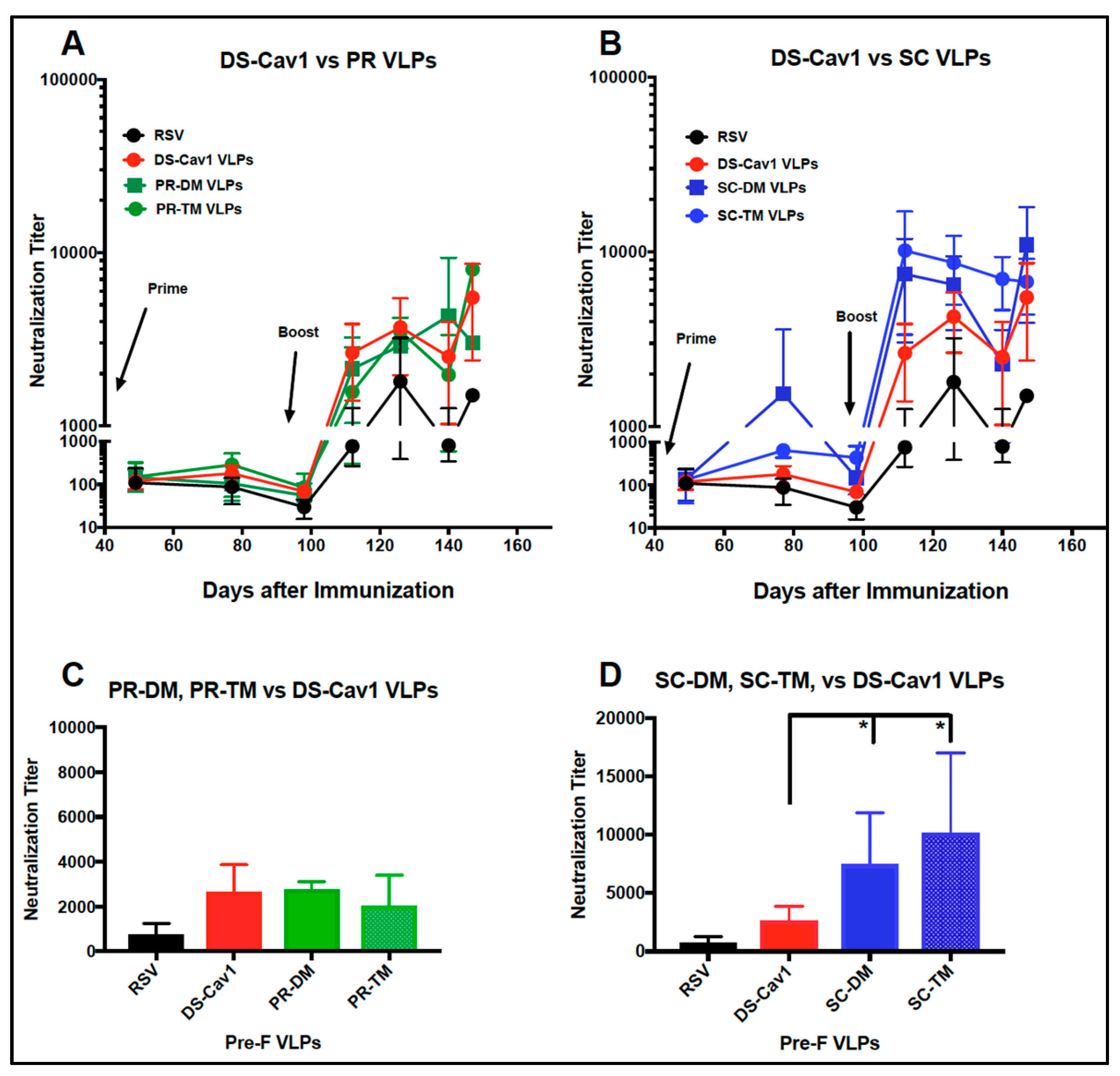
3.4. Induction of Neutralizing Antibodies in Mice
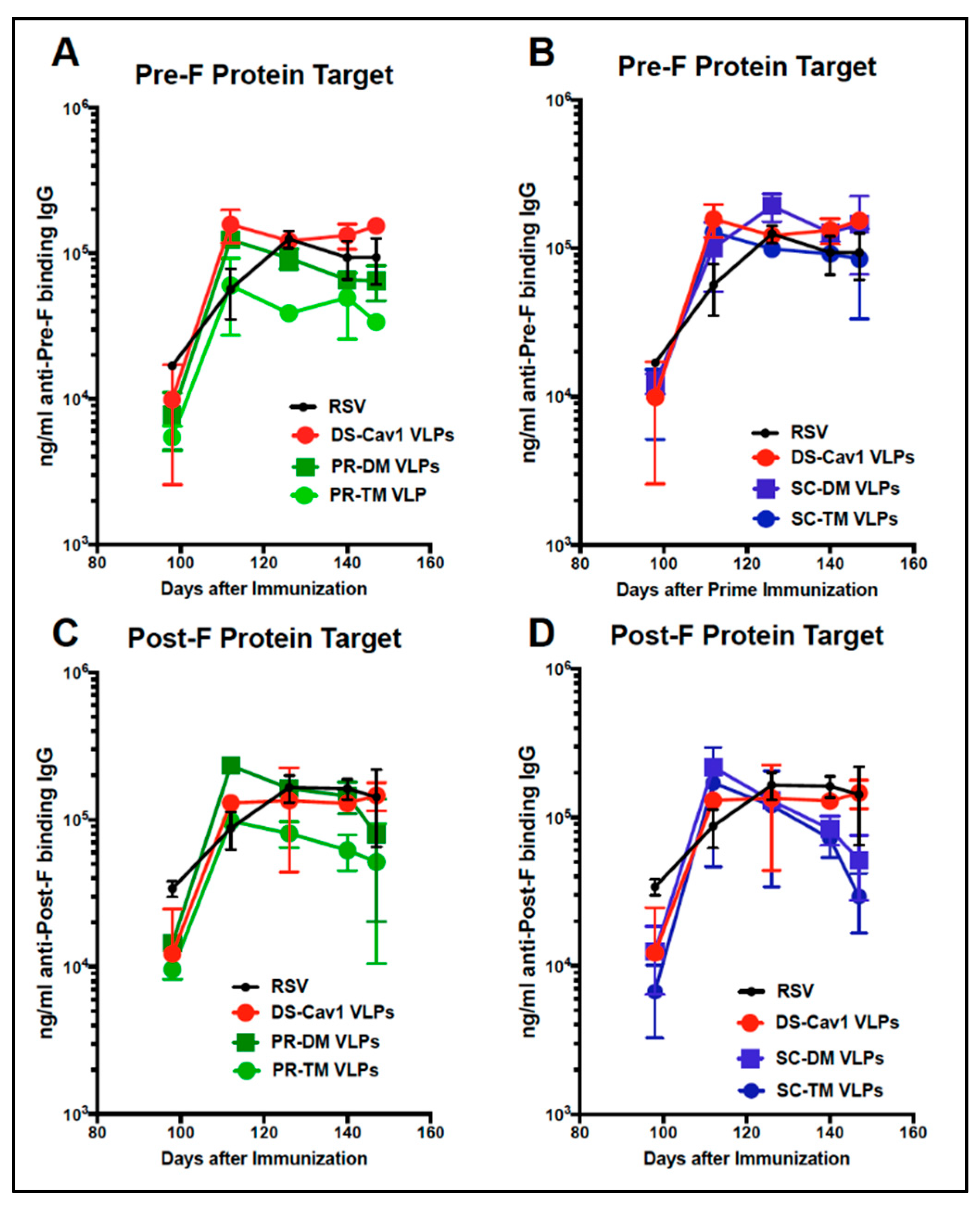
3.5. Total Anti-F Antibody Titers
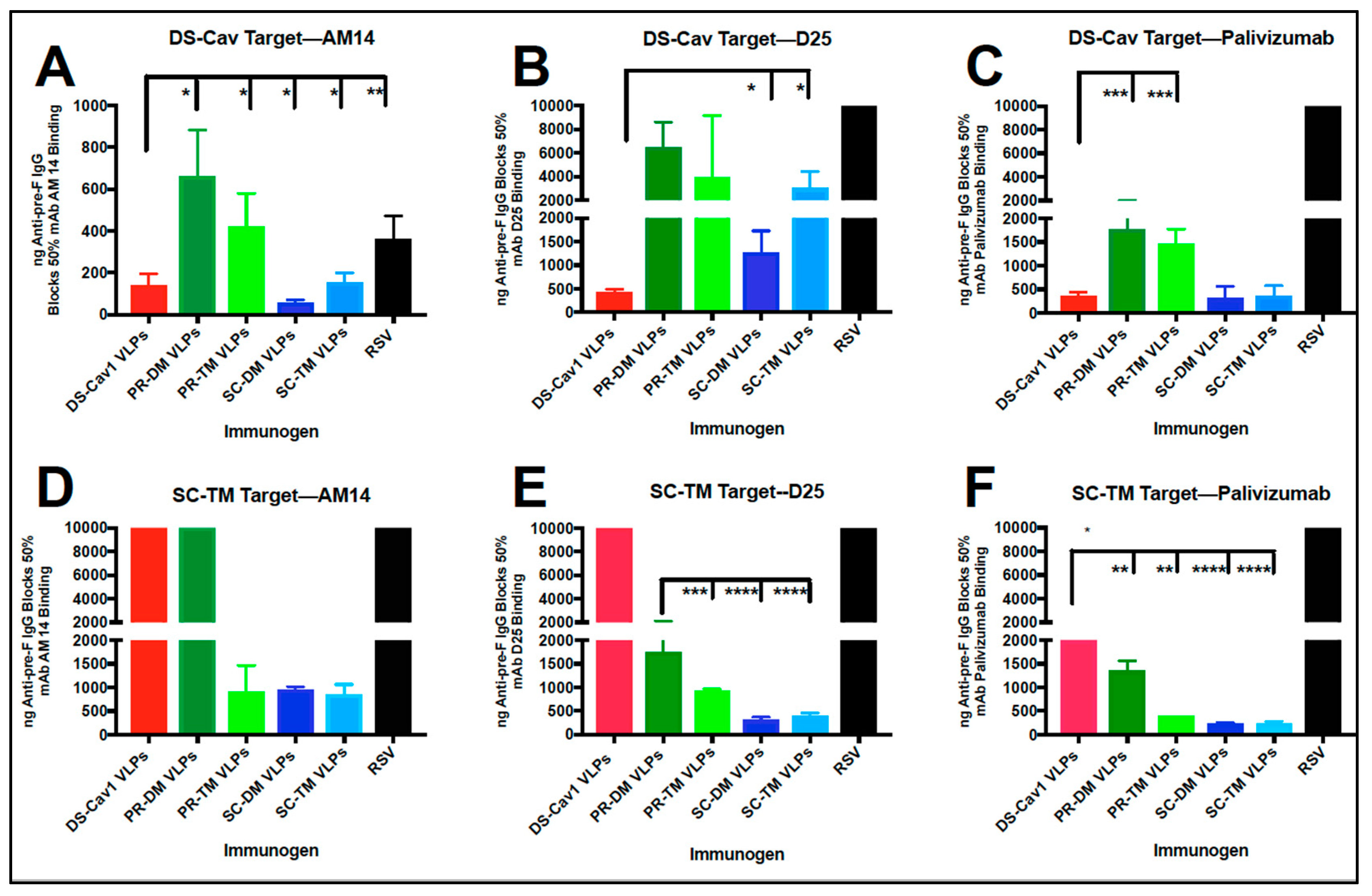
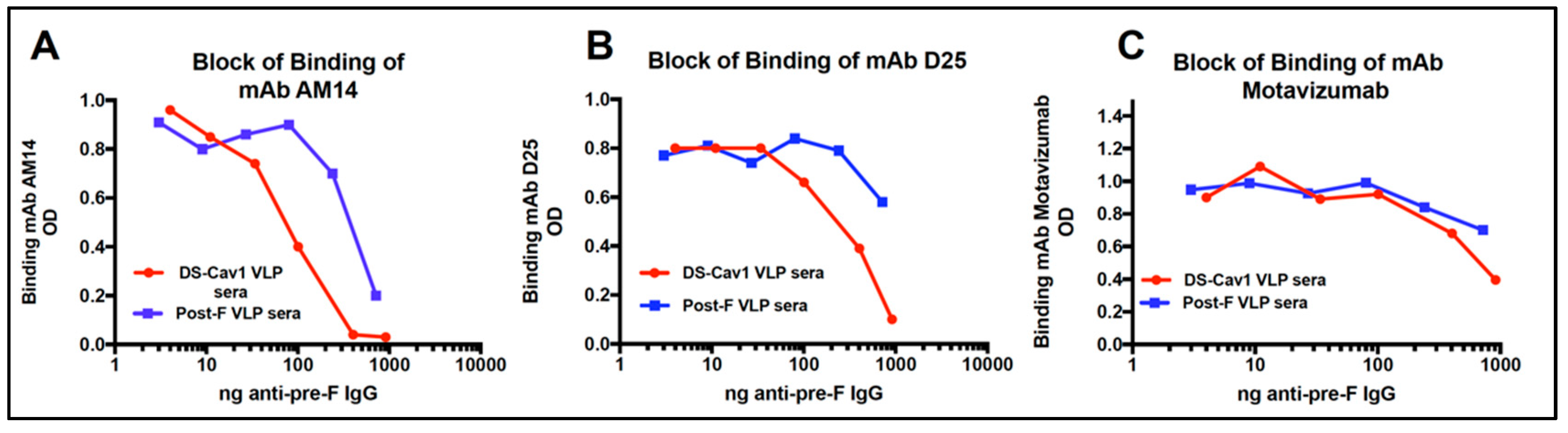
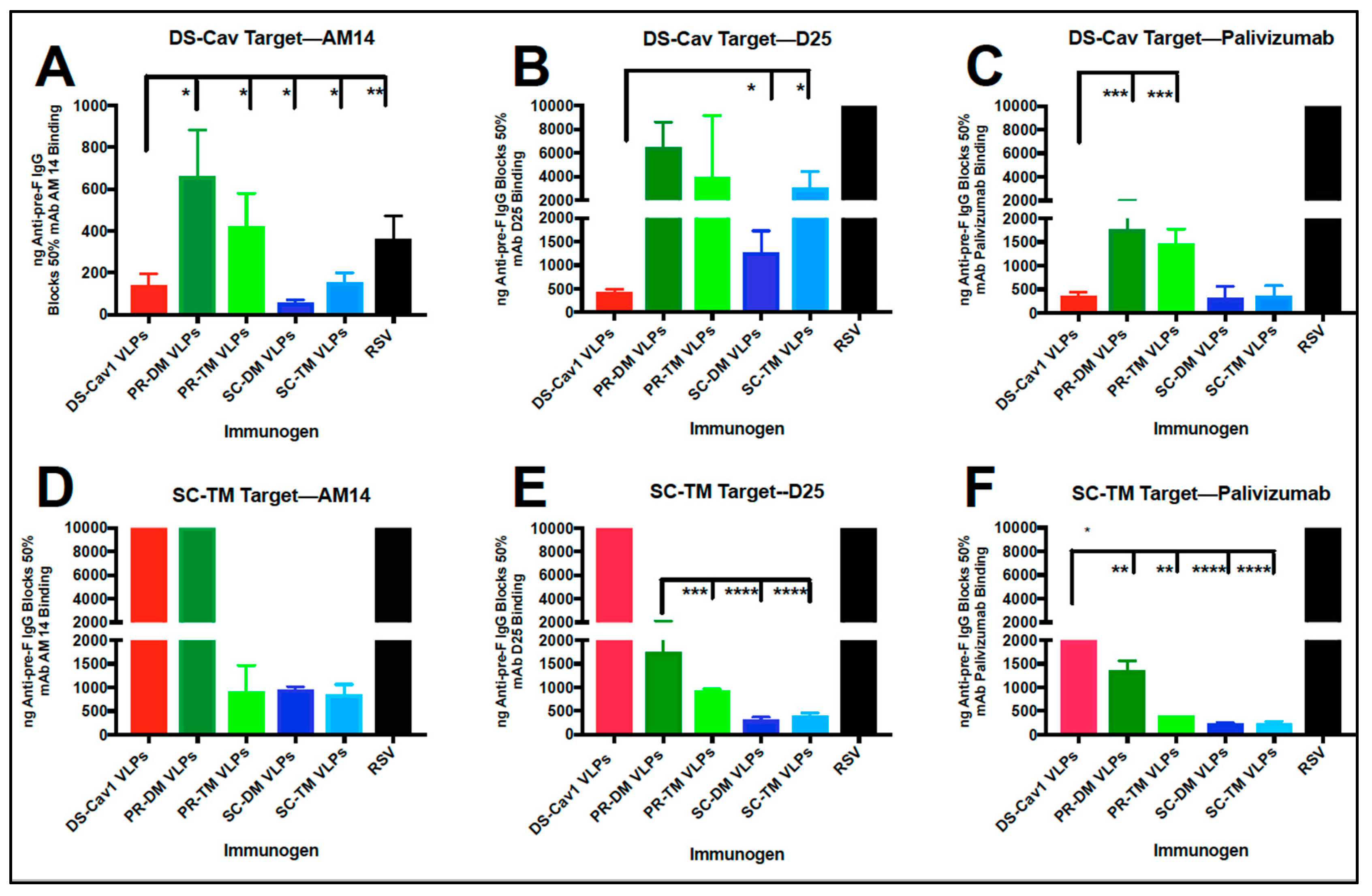
3.6. Relative Concentration of D25 and AM14 Blocking Antibodies in Sera
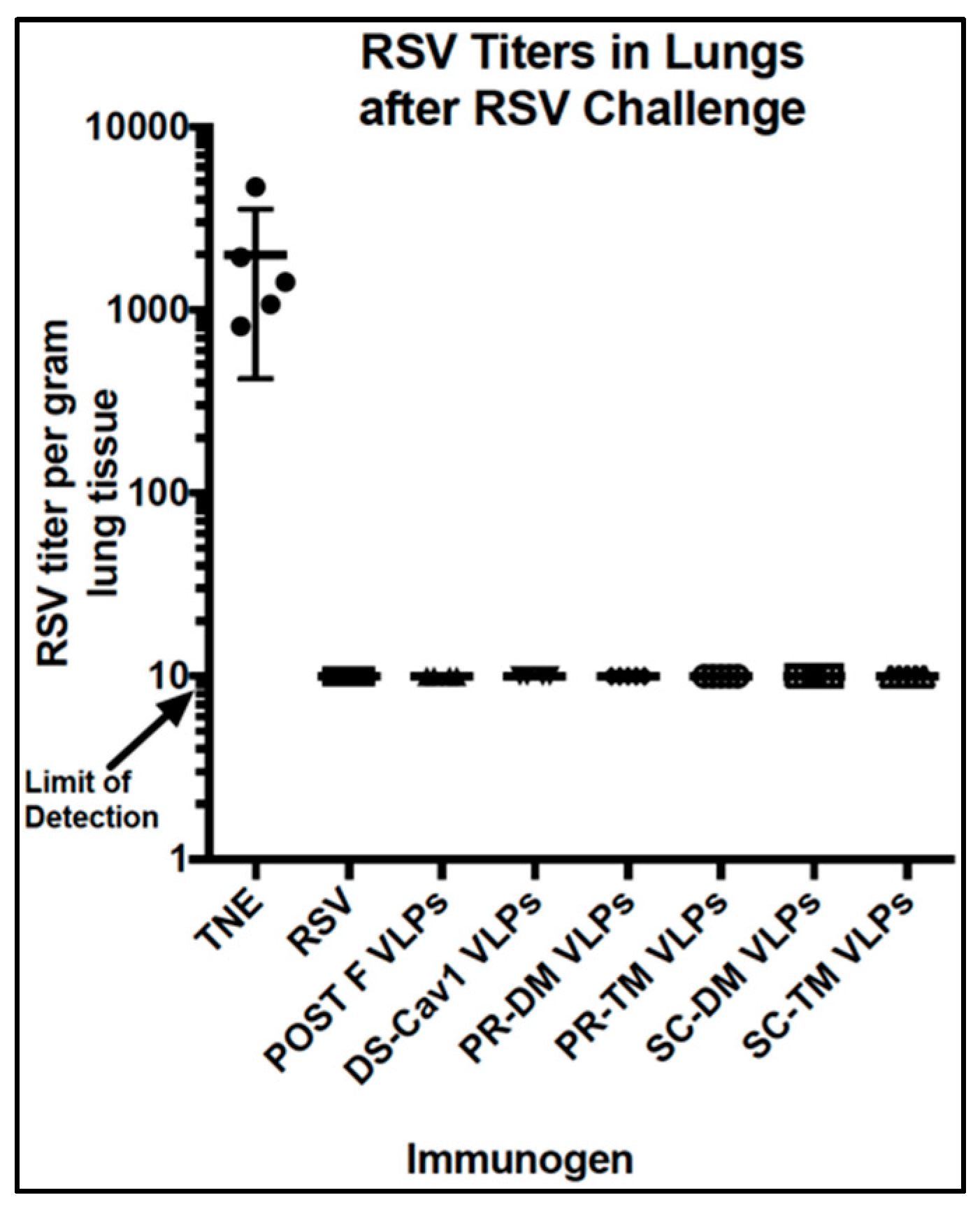
3.7. Protection From RSV Challenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Karron, R.A. Respiratory syncytial virus and parainfluenza virus vaccines. In Vaccines, 5th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P., Eds.; Saunders-Elsevier: Amsterdam, The Netherlands, 2008; Volume 6, p. 1146. [Google Scholar]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Han, L.L.; Alexander, J.P.; Anderson, L.J. Respiratory syncytial virus pneumonia among the elderly: An assessment of disease burden. J. Infect. Dis. 1999, 179, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Raboni, S.M.; Nogueira, M.B.; Tsuchiya, L.R.; Takahashi, G.A.; Pereira, L.A.; Pasquini, R. Respiratory tract viral infections in bone marrow transplant patients. Transplantation 2003, 76, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ison, M.G. Respiratory syncytial virus and other respiratory viruses in the setting of bone marrow transplantation. Curr. Opin. Oncol. 2009, 21, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.N.; Chemaly, R.F. Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 2011, 117, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Jardetsky, T.S.; Lamb, R.A. A class act. Nature 2004, 427, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The Viruses and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Strauss, S.E., Eds.; LippincottWilliams &Wilkins: Philadelphia, PA, USA, 2007; Volume 1, pp. 1450–1496. [Google Scholar]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat. Struct. Mol. Biol. 2011, 17, 248–250. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Modjarrad, K.; McLellan, J.S. Novel antigens for RSV vaccines. Curr. Opin. Immunol. 2015, 35, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuzil, K.M. Progress toward a Respiratory Syncytial Virus Vaccine. Clin. Vaccine Immunol. 2016, 23, 186–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falloon, J.; Yu, J.; Esser, M.T.; Villafana, T.; Yu, L.; Dubovsky, F.; Takas, T.; Levin, M.J.; Falsey, A.R. An adjuvanted, postfusion f protein–based vaccine did not prevent respiratory syncytial virus illness in older adults. J. Inf. Dis. 2017, 216, 1362–1370. [Google Scholar] [CrossRef]
- McGinnes, L.W.; Gravel, K.A.; Finberg, R.W.; Kurt-Jones, E.A.; Massare, M.J.; Smith, G. Assembly and immunological properties of Newcastle disease virus-like particles containing the respiratory syncytial virus F and G proteins. J. Virol. 2011, 85, 366–377. [Google Scholar] [CrossRef]
- Murawski, M.R.; McGinnes, L.W.; Finberg, R.W.; Kurt-Jones, E.A.; Massare, M.; Smith, G. Newcastle disease virus-like particles containing respiratory syncytial virus G protein induced protection in BALB/c mice with no evidence of immunopathology. J. Virol. 2010, 84, 1110–1123. [Google Scholar] [CrossRef]
- Cullen, L.M.; Blanco, J.C.G.; Morrison, T.G. Cotton rat immune responses to virus-like particles containing the pre-fusion form of respiratory syncytial virus fusion protein. J. Transl. Med. 2015, 13, 1–13. [Google Scholar] [CrossRef]
- McGinnes, L.; Schmidt, M.R.; Kenward, S.A.; Woodland, R.T.; Morrison, T.G. Murine immune responses to virus-like particle-associated pre- and postfusion forms of the respiratory syncytial virus f protein. J. Virol. 2015, 89, 6835–6847. [Google Scholar] [CrossRef] [PubMed]
- Cullen, L.M.; Schmidt, M.R.; Morrison, T.G. The importance of RSV F protein conformation in VLPs in stimulation of neutralizing antibody titers in mice previously infected with RSV. Hum. Vaccines Immunother. 2017, 1–10. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.C.G.; Pletneva, L.M.; McGinnes-Cullen, L.; Otoa, R.O.; Patel, M.C.; Fernando, L.R.; Boukhvalova, M.S.; Morrison, T.G. Efficacy of a respiratory syncytial virus vaccine candidate in a maternal immunization model. Nat. Commun. 2018, 9, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Krarup, A.; Truan, D.; Furmanova-Hollenstein, P.; Bogaert, L.; Bouchier, P.; Bisschop, I.J.M.; Widjojoatmodjo, M.N.; Zahn, R.; Schuitemaker, H.; McLellan, J.S.; et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat. Commun. 2015, 6, 8143–8155. [Google Scholar] [CrossRef]
- Flynn, J.A.; Durr, E.; Swoyer, R.; Cejas, P.J.; Horton, M.S.; Galli, J.D.; Cosmi, S.A.; Espeseth, A.S.; Bett, A.J.; Zhang, L. Stability characterization of a vaccine antigen based on the respiratory syncytial virus fusion glycoprotein. PLoS ONE 2016, 11, e0164789. [Google Scholar] [CrossRef]
- Russell, C.J.; Simões, E.A.F.; Hurwitz, J.L. Vaccines for the Paramyxoviruses and Pneumoviruses: Successes, candidates, and hurdles. Viral Immunol. 2018, 31, 133–141. [Google Scholar] [CrossRef]
- McGinnes, L.W.; Morrison, T.G. Newcastle Disease Virus-Like Particles: Preparation, Purification, Quantification, and Incorporation of Foreign Glycoproteins. In Current Protocols in Microbiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Frank, S.; Kammerer, R.A.; Mechling, D.; Schulthess, T.; Landwehr, R.; Bann, J. Stabilization of short collagen-like triple helices by protein engineering. J. Mol. Biol. 2001, 308, 1081–1089. [Google Scholar] [CrossRef]
- Gilman, M.S.A.; Moin, S.M.; Mas, V.; Chen, M.; Patel, N.K.; Kramer, K.; Zhu, Q.; Kabeche, S.C.; Kumar, A.; Palomo, C.; et al. Characterization of a prefusion-specific antibody that recognizes a quaternary, cleavage-dependent epitope on the rsv fusion glycoprotein. PLoS Pathog. 2015, 11, e1005035. [Google Scholar] [CrossRef]
- Smith, G.; Raghunandan, R.; Wu, Y.; Liu, Y.; Massare, M.; Nathan, M.; Zhou, B.; Lu, H.; Boddapati, S.; Li, J.; et al. Respiratory syncytial virus fusion glycoprotein expressed in insect cells form protein nanoparticles that induce protective immunity in cotton rats. PLoS ONE 2012, 7, e50852. [Google Scholar] [CrossRef]
- Magro, M.; Mas, V.; Chappell, K.; Vazquez, M.; Cano, O.; Luque, D.; Terron, M.C.; Melero, J.A.; Palomo, C. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc. Natl. Acad. Sci. USA 2012, 109, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Blais, N.; Gagné, M.; Hamuro, Y.; Rheault, P.; Boyer, M.; Steff, A.-M.; Baudoux, G.; Dewar, V.; Demers, J.; Ruelle, J.-L.; et al. Characterization of Pre-F-GCN4t, a modified human respiratory syncytial virus fusion protein stabilized in a noncleaved prefusion conformation. J. Virol. 2017, 91, e02437-16. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Boigard, H.; Bhatia, B.; Fallon, J.T.; Alimova, A.; Gottlieb, P.; Galarza, J.M. Novel respiratory syncytial virus-like particle vaccine composed of the postfusion and prefusion conformations of the f glycoprotein. Clin. Vaccine Immunol. 2016, 23, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Ngwuta, J.O.; Surman, S.; Kabatova, B.; Liu, X.; Lingemann, M.; Liu, X.; Yang, L.; Herbert, R.; Swerczek, J.; et al. Improved prefusion stability, optimized codon usage, and augmented virion packaging enhance the immunogenicity of respiratory syncytial virus fusion protein in a vectored-vaccine candidate. J. Virol. 2017, 91, e00189-17. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Mas, V.; Thom, M.; Vázquez, M.; Cano, O.; Terrón, M.C.; Luque, D.; Taylor, G.; Melero, J.A. Influence of respiratory syncytial virus f glycoprotein conformation on induction of protective immune responses. J. Virol. 2016, 90, 5485–5498. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.A.; Balabanis, K.; Xie, Y.; Aggarwal, Y.; Palomo, C.; Mas, V.; Metrick, C.; Yang, H.; Shaw, C.A.; Melero, J.A.; et al. A Monomeric uncleaved respiratory syncytial virus f antigen retains prefusion-specific neutralizing epitopes. J. Virol. 2014, 88, 11802–11810. [Google Scholar] [CrossRef]
- Wu, H.; Pfarr, D.S.; Tang, Y.; An, L.-L.; Patel, N.K.; Watkins, J.D.; Huse, W.D.; Kiener, P.A.; Young, J.F. Ultra-potent antibodies against respiratory syncytial virus: Effects of binding kinetics and binding valence on viral neutralization. J. Mol. Biol. 2005, 350, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Mousa, J.J.; Sauer, M.F.; Sevy, A.M.; Finn, J.A.; Bates, J.T.; Alvarado, G.; King, H.G.; Loerinc, L.B.; Fong, R.H.; Doranz, B.J.; et al. Structural basis for nonneutralizing antibody competition at antigenic site II of the respiratory syncytial virus fusion protein. Proc. Natl. Acad. Sci. USA 2016, 113, E6849–E6858. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monoclonal Antibody | VLP Immunogen | ||
|---|---|---|---|
| Post-F | DS-Cav1 F | ||
| Am14 | Exp 1 | 950 +/− 70 | 65 +/− 5 |
| Exp 2 | 675 +/− 170 | 95 +/− 7 | |
| Exp 3 | 1500 +/− 100 | 138 +/− 56 | |
| D25 | Exp 1 | 7500 +/− 4000 | 550 +/− 50 |
| Exp 2 | 2333 +/− 577 | 425 +/− 50 | |
| Exp 3 | 8333 +/− 2100 | 505 +/− 130 | |
| Motavizamab | Exp 1 | 2000 +/− 100 | 825 +/− 25 |
| Exp 2 | 1000 +/− 50 | 1000 +/− 100 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cullen, L.M.; Schmidt, M.R.; Torres, G.M.; Capoferri, A.A.; Morrison, T.G. Comparison of Immune Responses to Different Versions of VLP Associated Stabilized RSV Pre-Fusion F Protein. Vaccines 2019, 7, 21. https://doi.org/10.3390/vaccines7010021
Cullen LM, Schmidt MR, Torres GM, Capoferri AA, Morrison TG. Comparison of Immune Responses to Different Versions of VLP Associated Stabilized RSV Pre-Fusion F Protein. Vaccines. 2019; 7(1):21. https://doi.org/10.3390/vaccines7010021
Chicago/Turabian StyleCullen, Lori M., Madelyn R. Schmidt, Gretel M. Torres, Adam A. Capoferri, and Trudy G. Morrison. 2019. "Comparison of Immune Responses to Different Versions of VLP Associated Stabilized RSV Pre-Fusion F Protein" Vaccines 7, no. 1: 21. https://doi.org/10.3390/vaccines7010021
APA StyleCullen, L. M., Schmidt, M. R., Torres, G. M., Capoferri, A. A., & Morrison, T. G. (2019). Comparison of Immune Responses to Different Versions of VLP Associated Stabilized RSV Pre-Fusion F Protein. Vaccines, 7(1), 21. https://doi.org/10.3390/vaccines7010021