Quiescence Entry, Maintenance, and Exit in Adult Stem Cells


Abstract

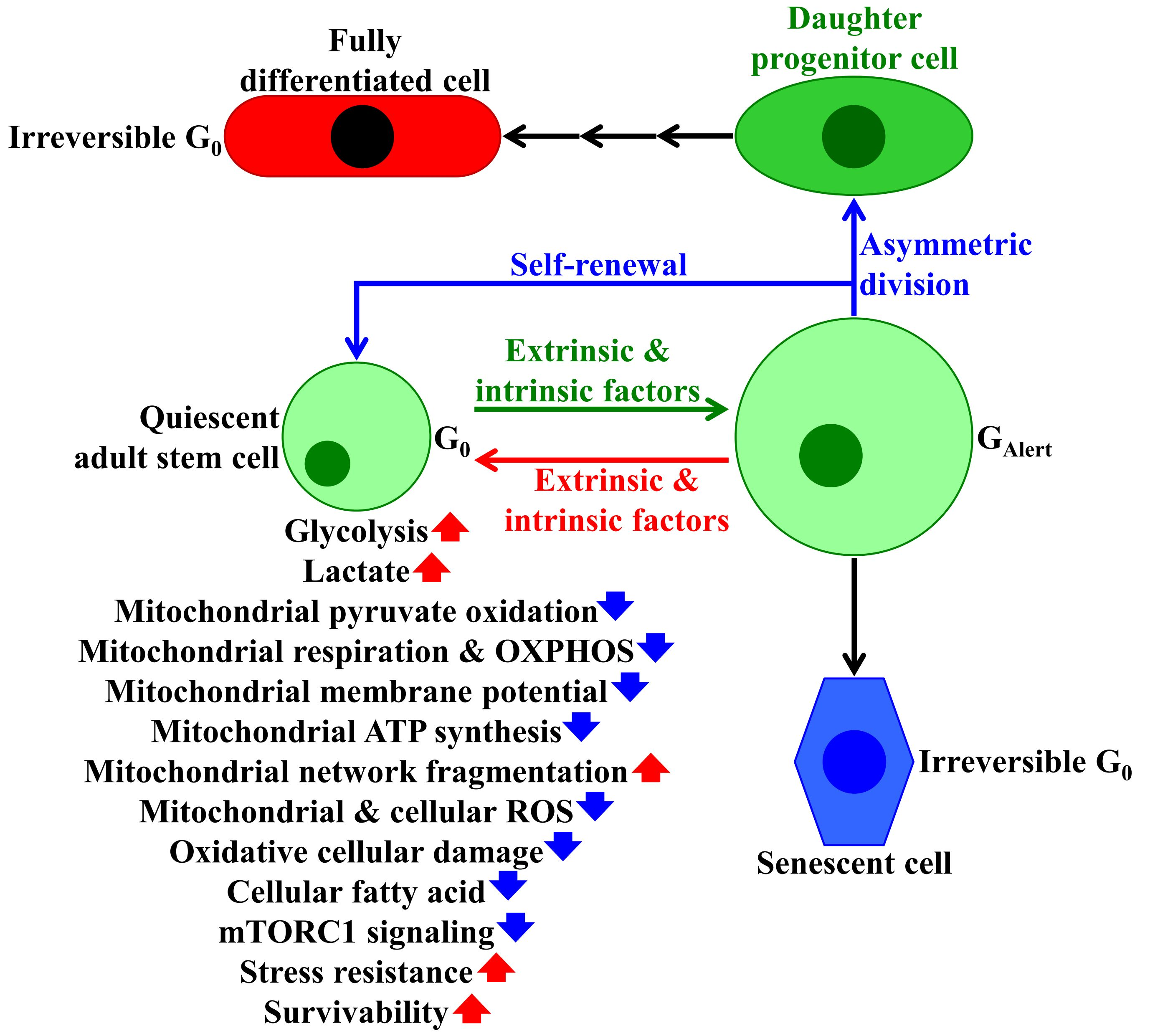{kind=link}
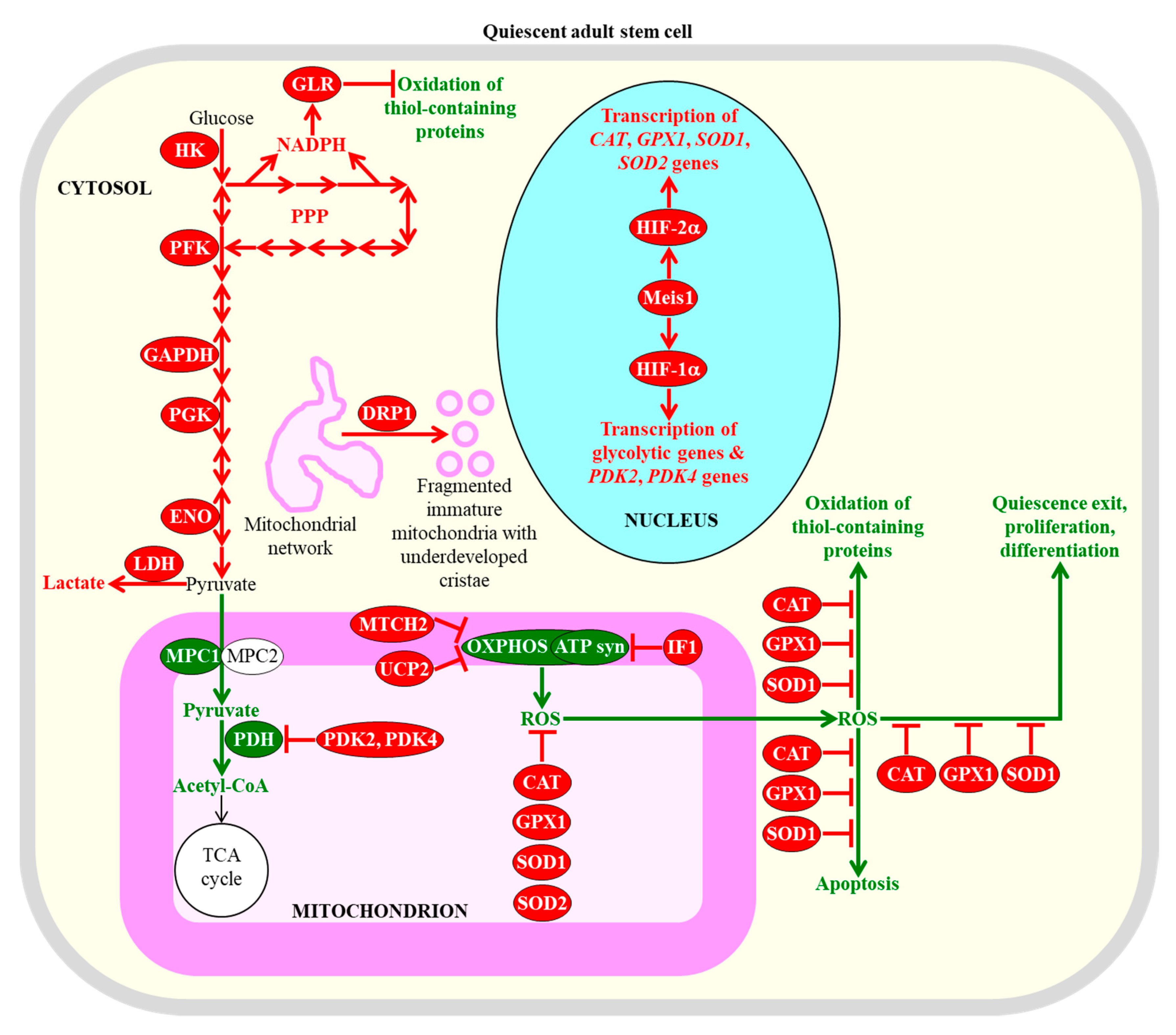{kind=link}
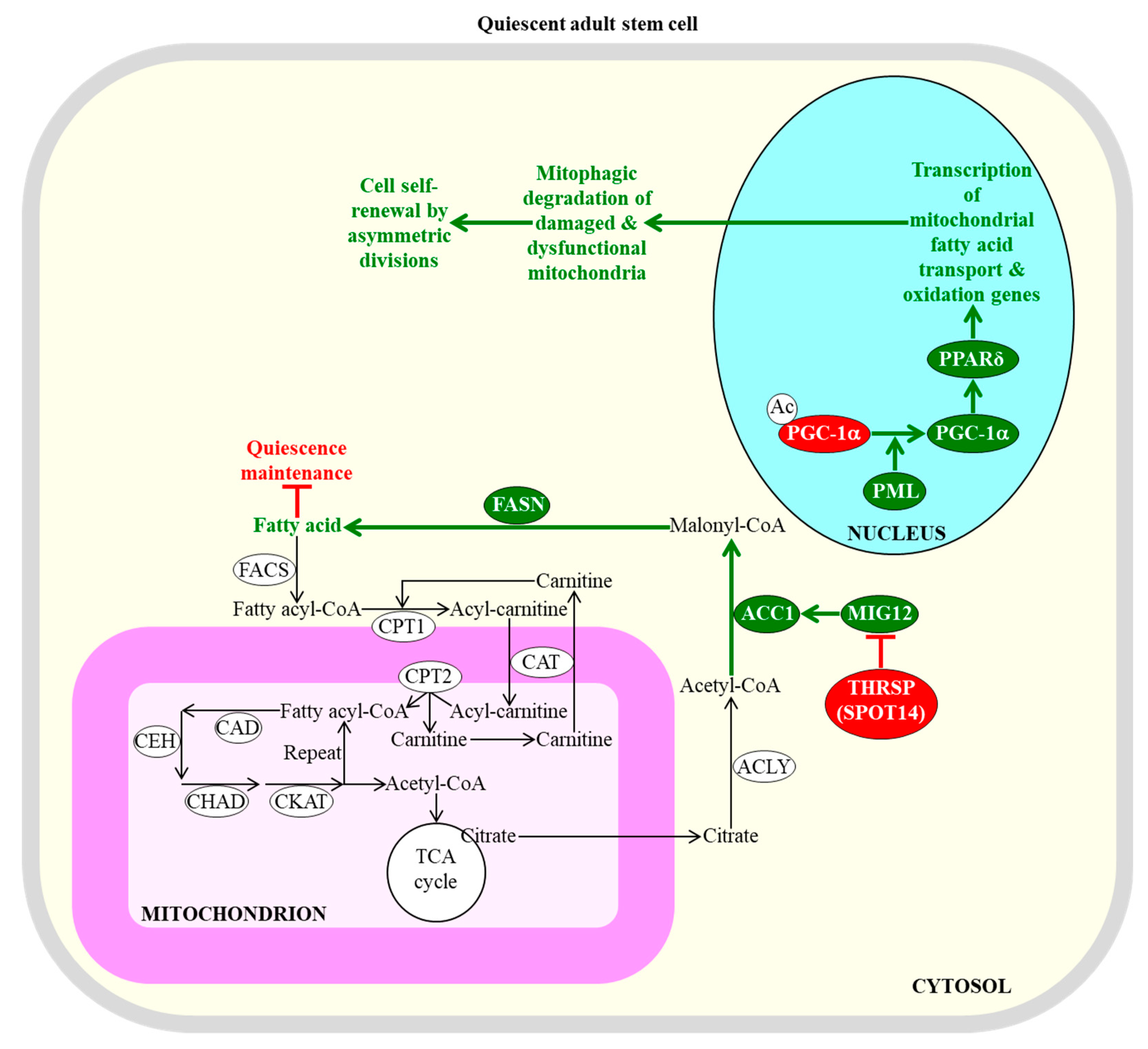{kind=link}
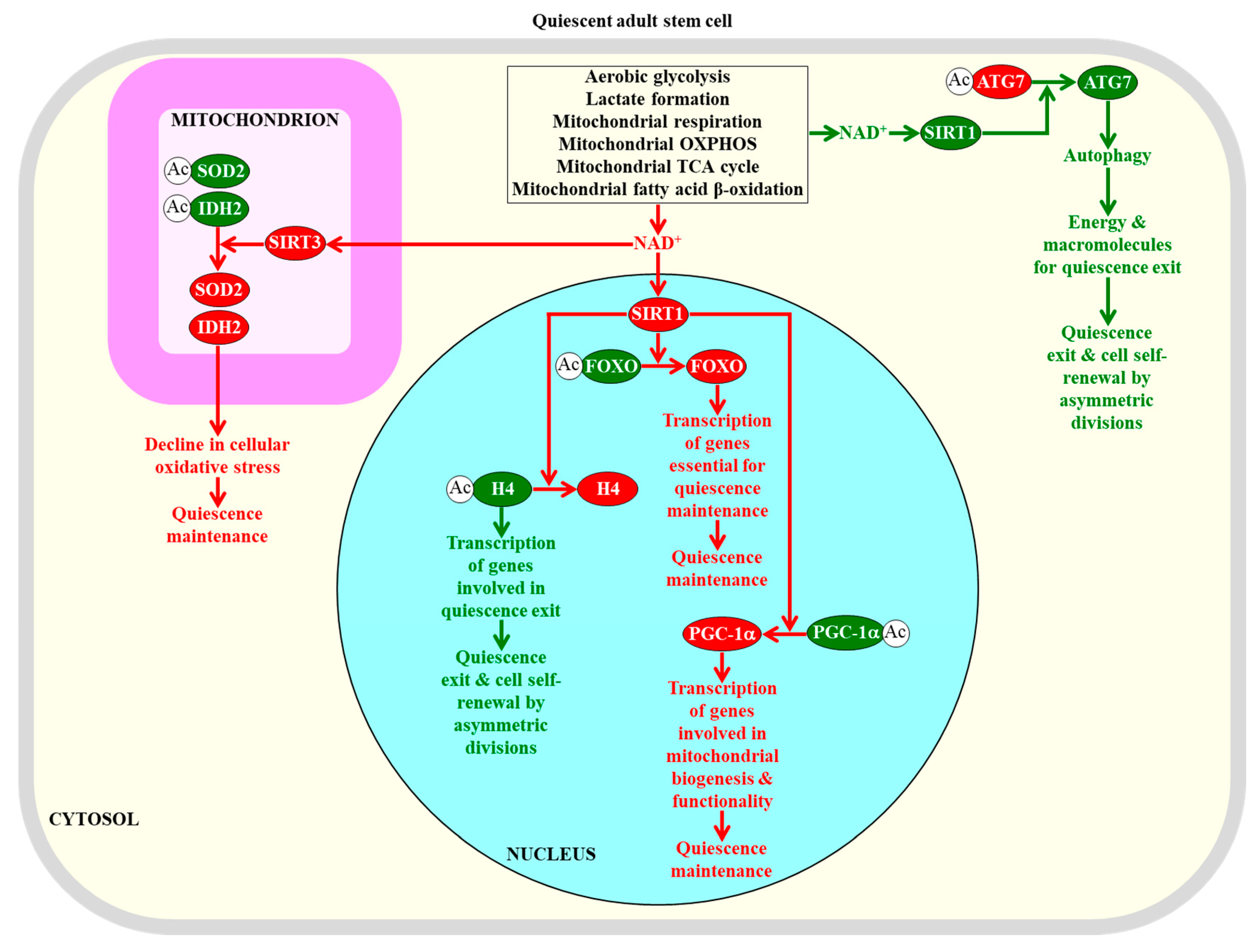{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Common Traits of Quiescent Adult Stem Cells and Cell-Intrinsic Mechanisms That in These Cells Control Quiescence Entry, Maintenance, and Exit
2.1. Common Metabolic Traits of Quiescent Adult Stem Cells Define Their Fate
2.2. Fatty Acid Oxidation and Synthesis Define the Fate of Quiescent Adult Stem Cells
2.3. A Dual Role of ROS in Defining the Homeostasis and Functionality of Quiescent Adult Stem Cells
2.4. Some Mitochondrial TCA Cycle Intermediates Contribute to Quiescence Maintenance by Adult Stem Cells
2.5. NAD+ Concentration within Adult Stem Cells Defines Their Fate
2.6. A Low-Energy Status of Adult Stem Cells Helps Sustain Their Quiescence
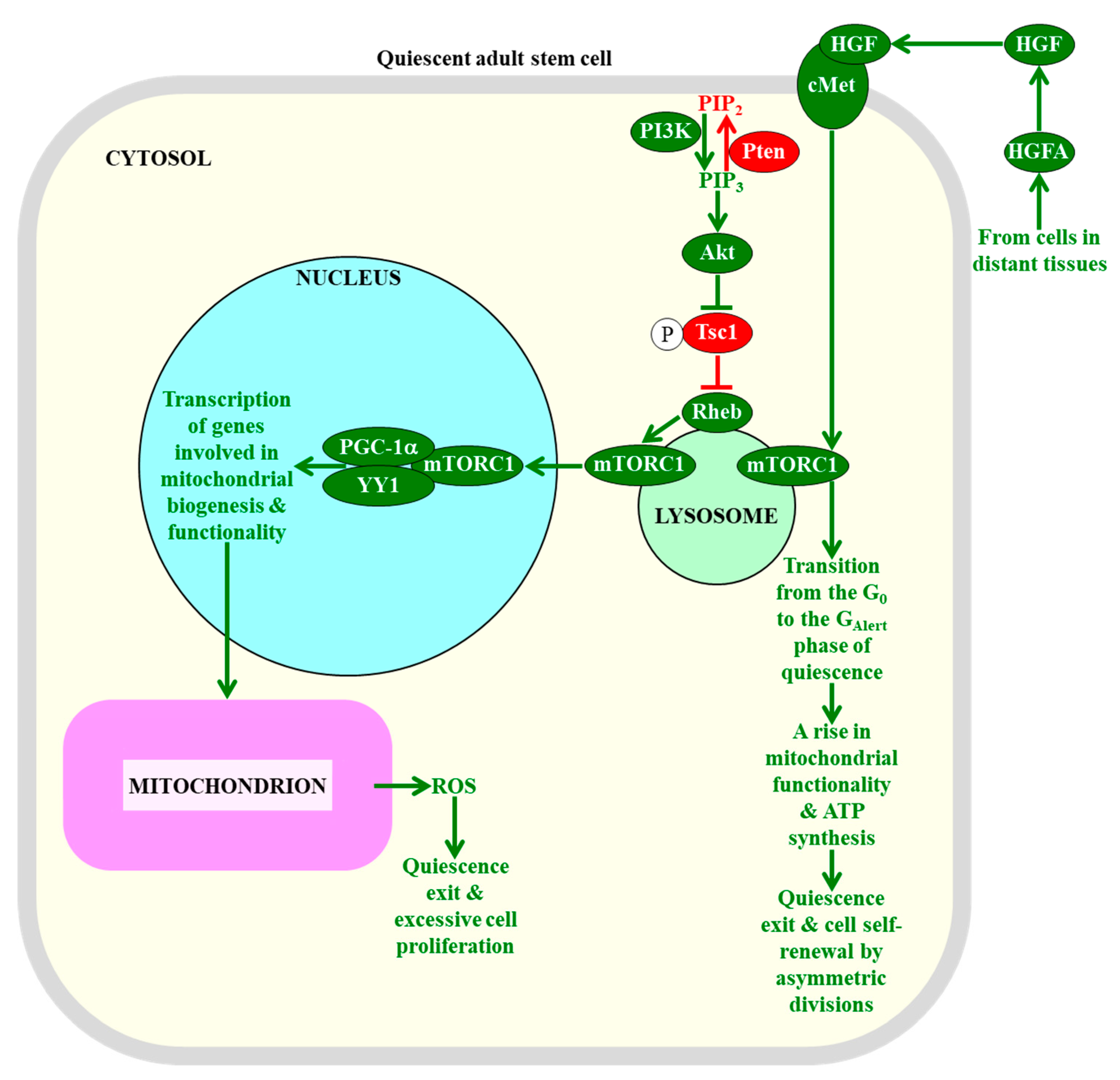
2.7. The Maintenance of mTORC1 Signaling at Low Intensity in Adult Stem Cells Is Essential for Sustaining Their Quiescence, Self-Renewal, and Functionality
2.8. Several Mechanisms of Proteostasis Maintenance in Quiescent Adult Stem Cells Define Their Fate
2.8.1. Protein Synthesis on Free Ribosomes in the Cytosol
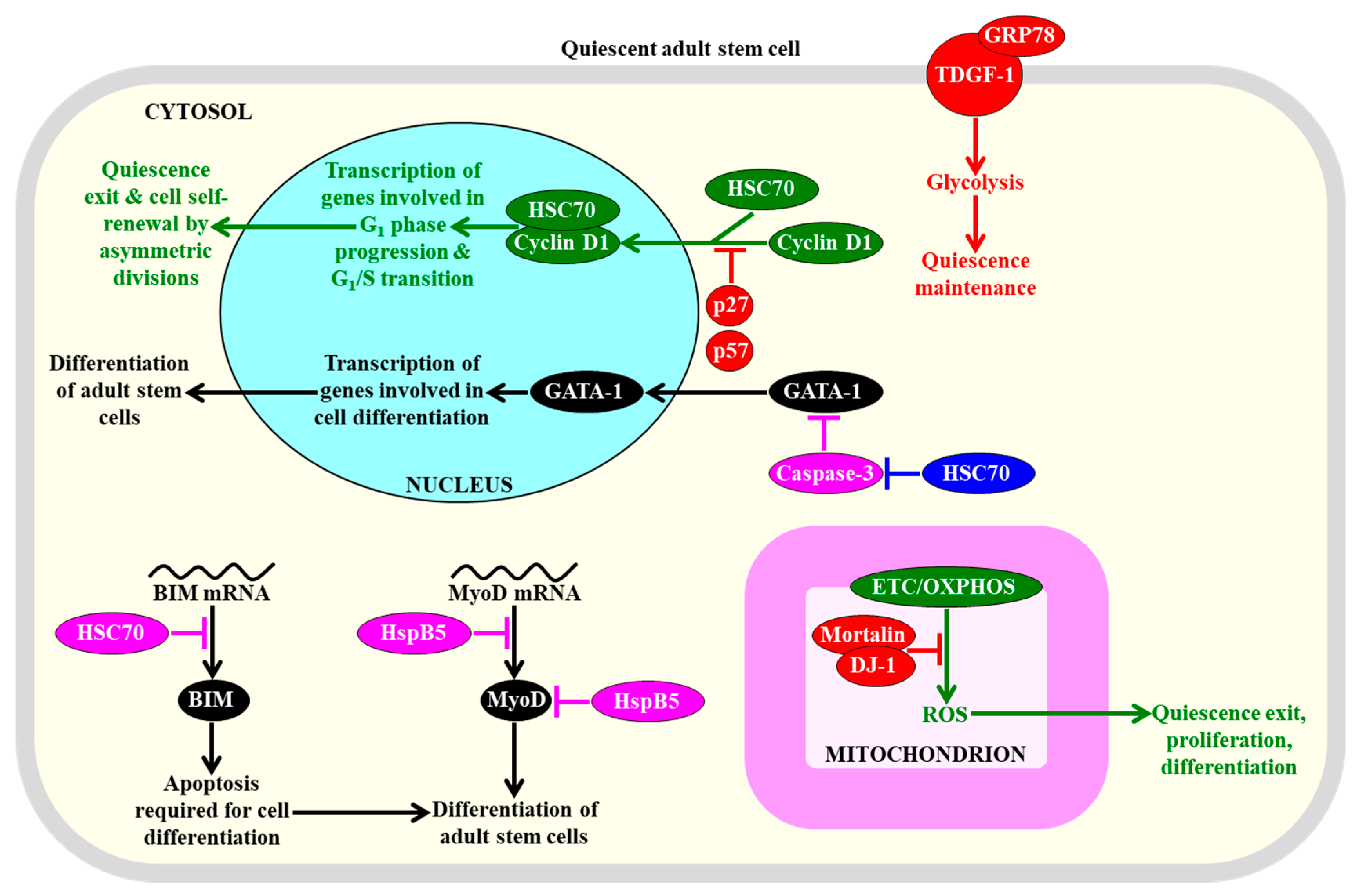
2.8.2. HSP Concentrations
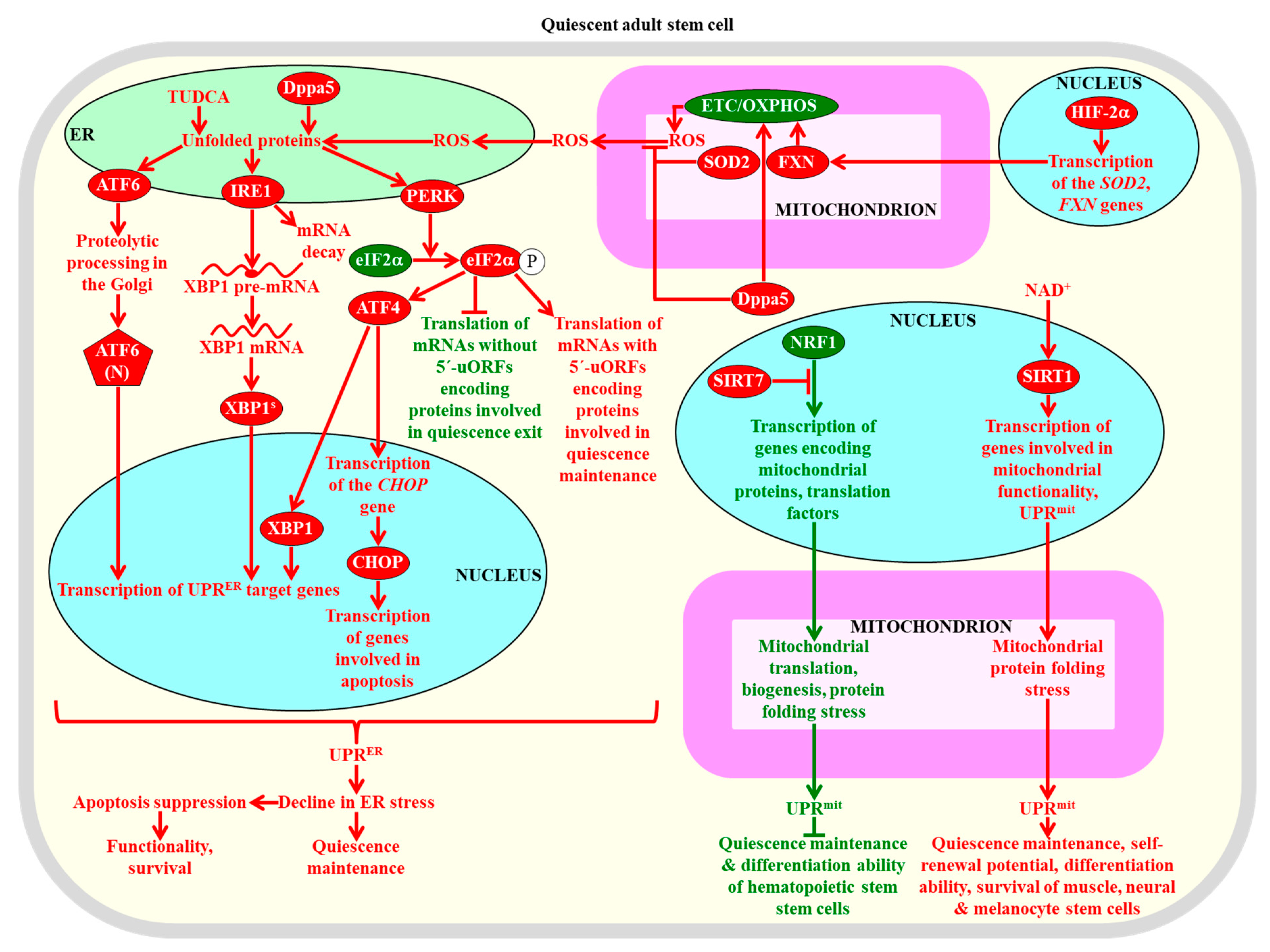
2.8.3. The UPRER and UPRmit Systems
2.8.4. The Ubiquitin System
2.8.5. Autophagy
2.9. Cell Cycle Regulatory Proteins Are Essential for Sustaining the Quiescence, Self-Renewal, Functionality, and Differentiation Potential of Adult Stem Cells
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, J.V.; Petsko, G.A.; Johnston, G.C.; Ringe, D.; Singer, R.A.; Werner-Washburne, M. “Sleeping beauty”: Quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004, 68, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell. Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, J.; Laxman, S. Decoding the stem cell quiescence cycle—Lessons from yeast for regenerative biology. J. Cell Sci. 2015, 128, 4467–4474. [Google Scholar] [CrossRef] [PubMed]
- Rumman, M.; Dhawan, J.; Kassem, M. Concise Review: Quiescence in Adult Stem Cells: Biological Significance and Relevance to Tissue Regeneration. Stem Cells 2015, 33, 2903–2912. [Google Scholar] [CrossRef]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A.; Yellen, P.; Xu, L.; Saqcena, M. Regulation of G1 Cell Cycle Progression: Distinguishing the Restriction Point from a Nutrient-Sensing Cell Growth Checkpoint(s). Genes Cancer 2010, 1, 1124–1131. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic control of the cell division cycle in yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef]
- Cameron, I.L.; Bols, N.C. Effect of cell population density on G2 arrest in Tetrahymena. J. Cell Biol. 1975, 67, 518–522. [Google Scholar] [CrossRef]
- Drewinko, B.; Yang, L.Y.; Barlogie, B.; Trujillo, J.M. Cultured human tumour cells may be arrested in all stages of the cycle during stationary phase: Demonstration of quiescent cells in G1, S and G2 phase. Cell Tissue Kinet. 1984, 17, 453–463. [Google Scholar] [CrossRef]
- Costello, G.; Rodgers, L.; Beach, D. Fission yeast enters the stationary phase G0 state from either mitotic G1 or G2. Curr. Genet. 1986, 11, 119–125. [Google Scholar] [CrossRef]
- Baisch, H. Different quiescence states of three culture cell lines detected by acridine orange staining of cellular RNA. Cytometry 1988, 9, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Nurse, P.; Broek, D. Yeast cells can enter a quiescent state through G1, S, G2, or M phase of the cell cycle. Cancer Res. 1993, 53, 1867–1870. [Google Scholar]
- Takeo, K.; Tanaka, R.; Miyaji, M.; Nishimura, K. Unbudded G2 as well as G1 arrest in the stationary phase of the basidiomycetous yeast Cryptococcus neoformans. FEMS Microbiol. Lett. 1995, 29, 231–235. [Google Scholar]
- Cooper, S. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. FASEB J. 2003, 17, 333–340. [Google Scholar] [CrossRef]
- Klosinska, M.M.; Crutchfield, C.A.; Bradley, P.H.; Rabinowitz, J.D.; Broach, J.R. Yeast cells can access distinct quiescent states. Genes Dev. 2011, 25, 336–349. [Google Scholar] [CrossRef]
- Daignan-Fornier, B.; Sagot, I. Proliferation/quiescence: The controversial “aller-retour”. Cell Div. 2011, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Laporte, D.; Lebaudy, A.; Sahin, A.; Pinson, B.; Ceschin, J.; Daignan-Fornier, B.; Sagot, I. Metabolic status rather than cell cycle signals control quiescence entry and exit. J. Cell Biol. 2011, 192, 949–957. [Google Scholar] [CrossRef]
- Roche, B.; Arcangioli, B.; Martienssen, R. Transcriptional reprogramming in cellular quiescence. RNA Biol. 2017, 14, 843–853. [Google Scholar] [CrossRef]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature 2014, 510, 393–396. [Google Scholar] [CrossRef]
- McGann, C.J.; Odelberg, S.J.; Keating, M.T. Mammalian myotube dedifferentiation induced by newt regeneration extract. Proc. Natl. Acad. Sci. USA 2001, 98, 13699–13704. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Corbel, S.Y.; Sage, J.; Pomerantz, J.H.; Blau, H.M. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010, 7, 198–213. [Google Scholar] [CrossRef]
- Clevers, H. Stem cells. What is an adult stem cell? Science 2015, 350, 1319–1320. [Google Scholar] [CrossRef]
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef]
- Ochocki, J.D.; Simon, M.C. Nutrient-sensing pathways and metabolic regulation in stem cells. J. Cell Biol. 2013, 203, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef]
- Kwon, J.S.; Everetts, N.J.; Wang, X.; Wang, W.; Della Croce, K.; Xing, J.; Yao, G. Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep. 2017, 20, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Ocampo, A.; Liu, G.H.; Izpisua Belmonte, J.C. Regulation of Stem Cell Aging by Metabolism and Epigenetics. Cell Metab. 2017, 26, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Wang, H.; Kuang, W. Stem cell rejuvenation and the role of autophagy in age retardation by caloric restriction: An update. Mech. Ageing Dev. 2018, 175, 46–54. [Google Scholar] [CrossRef]
- Lewis, D.B.; Gattie, G.T. The ecology of quiescent microbes. ASM News 1991, 57, 27–32. [Google Scholar]
- Finkel, S.E. Long-term survival during stationary phase: Evolution and the GASP phenotype. Nat. Rev. Microbiol. 2006, 4, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Werner-Washburne, M.; Roy, S.; Davidson, G.S. Aging and the survival of quiescent and non-quiescent cells in yeast stationary-phase cultures. Subcell. Biochem. 2012, 57, 123–143. [Google Scholar] [PubMed]
- Rittershaus, E.S.; Baek, S.H.; Sassetti, C.M. The normalcy of dormancy: Common themes in microbial quiescence. Cell Host Microbe 2013, 13, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Scheres, B. Stem-cell niches: Nursery rhymes across kingdoms. Nat. Rev. Mol. Cell Biol. 2007, 8, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Mukherjee, T.; Banerjee, U. Direct sensing of systemic and nutritional signals by haematopoietic progenitors in Drosophila. Nat. Cell Biol. 2012, 14, 394–400. [Google Scholar] [CrossRef]
- Heyman, J.; Kumpf, R.P.; De Veylder, L. A quiescent path to plant longevity. Trends Cell Biol. 2014, 24, 443–448. [Google Scholar] [CrossRef]
- Seidel, H.S.; Kimble, J. Cell-cycle quiescence maintains Caenorhabditis elegans germline stem cells independent of GLP-1/Notch. eLife 2015, 4, e10832. [Google Scholar] [CrossRef]
- Narbonne, P.; Gerhold, A.R.; Maddox, P.S.; Labbé, J.C. The C. elegans GSCs: A Powerful Model for In Vivo Study of Adult Stem Cell Regulation. Int. J. Stem Cell Res. Ther. 2016, 3, 044. [Google Scholar] [CrossRef]
- Rovere, F.D.; Fattorini, L.; Ronzan, M.; Falasca, G.; Altamura, M.M. The quiescent center and the stem cell niche in the adventitious roots of Arabidopsis thaliana. Plant Signal. Behav. 2016, 11, e1176660. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Velappan, Y.; Signorelli, S.; Considine, M.J. Cell cycle arrest in plants: What distinguishes quiescence, dormancy and differentiated G1? Ann. Bot. 2017, 120, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.C.; Vaz, S.; Logarinho, E. Mitotic Dysfunction Associated with Aging Hallmarks. Adv. Exp. Med. Biol. 2017, 1002, 153–188. [Google Scholar]
- Ahlqvist, K.J.; Suomalainen, A.; Hämäläinen, R.H. Stem cells, mitochondria and aging. Biochim. Biophys. Acta 2015, 1847, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Eridani, S.; Sgaramella, V.; Cova, L. Stem cells: From embryology to cellular therapy? An appraisal of the present state of art. Cytotechnology 2004, 44, 125–141. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Sabatini, D.M.; Yilmaz, Ö.H. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell 2014, 14, 292–305. [Google Scholar] [CrossRef]
- Ito, K.; Ito, K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu. Rev. Cell Dev. Biol. 2016, 32, 399–409. [Google Scholar] [CrossRef]
- Santoro, A.; Vlachou, T.; Carminati, M.; Pelicci, P.G.; Mapelli, M. Molecular mechanisms of asymmetric divisions in mammary stem cells. EMBO Rep. 2016, 17, 1700–1720. [Google Scholar] [CrossRef]
- Moore, D.L.; Jessberger, S. Creating Age Asymmetry: Consequences of Inheriting Damaged Goods in Mammalian Cells. Trends Cell Biol. 2017, 27, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef]
- Clayton, E.; Doupé, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar] [CrossRef]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef]
- Zhang, Y.V.; Cheong, J.; Ciapurin, N.; McDermitt, D.J.; Tumbar, T. Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell 2009, 5, 267–278. [Google Scholar] [CrossRef]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- O’Brien, L.E.; Soliman, S.S.; Li, X.; Bilder, D. Altered modes of stem cell division drive adaptive intestinal growth. Cell 2011, 147, 603–614. [Google Scholar] [CrossRef]
- Shahriyari, L.; Komarova, N.L. Symmetric vs. asymmetric stem cell divisions: An adaptation against cancer? PLoS ONE 2013, 8, e76195. [Google Scholar] [CrossRef]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Izpisua Belmonte, J.C. Stem Cells: A Renaissance in Human Biology Research. Cell 2016, 165, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ocampo, A.; Izpisua Belmonte, J.C. Cellular metabolism and induced pluripotency. Cell 2016, 166, 1371–1385. [Google Scholar] [CrossRef]
- Ahmed, A.S.; Sheng, M.H.; Wasnik, S.; Baylink, D.J.; Lau, K.W. Effect of aging on stem cells. World, J. Exp. Med. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Functional dysregulation of stem cells during aging: A focus on skeletal muscle stem cells. FEBS J. 2013, 280, 4051–4062. [Google Scholar] [CrossRef]
- Li, M.; Izpisua Belmonte, J.C. Genetic rejuvenation of old muscle. Nature 2014, 506, 304–305. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Goodell, M.A.; Rando, T.A. Stem cells and healthy aging. Science 2015, 350, 1199–1204. [Google Scholar] [CrossRef]
- Akunuru, S.; Geiger, H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef]
- Chandel, N.S.; Jasper, H.; Ho, T.T.; Passegué, E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 2016, 18, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Izpisua Belmonte, J.C. Anti-Aging Strategies Based on Cellular Reprogramming. Trends Mol. Med. 2016, 22, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef]
- Soria-Valles, C.; López-Otín, C. iPSCs: On the Road to Reprogramming Aging. Trends Mol. Med. 2016, 22, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Artoni, F.; Kreipke, R.E.; Palmeira, O.; Dixon, C.; Goldberg, Z.; Ruohola-Baker, H. Loss of foxo rescues stem cell aging in Drosophila germ line. eLife 2017, 6, e27842. [Google Scholar] [CrossRef]
- Brunet, A.; Rando, T.A. Interaction between epigenetic and metabolism in aging stem cells. Curr. Opin. Cell Biol. 2017, 45, 1–7. [Google Scholar] [CrossRef]
- García-Prat, L.; Muñoz-Cánoves, P. Aging, metabolism and stem cells: Spotlight on muscle stem cells. Mol. Cell Endocrinol. 2017, 445, 109–117. [Google Scholar] [CrossRef]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Solanas, G.; Peixoto, F.O.; Perdiguero, E.; Jardí, M.; Ruiz-Bonilla, V.; Datta, D.; Symeonidi, A.; Castellanos, A.; Welz, P.S.; Caballero, J.M.; et al. Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 2017, 170, 678–692. [Google Scholar] [CrossRef]
- Keyes, B.E.; Fuchs, E. Stem cells: Aging and transcriptional fingerprints. J. Cell Biol. 2018, 217, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Brunet, A. Bursts of Reprogramming: A Path to Extend Lifespan? Cell 2016, 167, 1672–1674. [Google Scholar] [CrossRef]
- Meyer, K.; Yankner, B.A. Slowing Down Aging. Cell Metab. 2017, 26, 592–593. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Jones, K.M.; Basson, M.A.; Brack, A.S. The aged niche disrupts muscle stem cell quiescence. Nature 2012, 490, 355–360. [Google Scholar] [CrossRef]
- Conboy, I.M.; Rando, T.A. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle 2012, 11, 2260–2267. [Google Scholar] [CrossRef]
- Doles, J.; Storer, M.; Cozzuto, L.; Roma, G.; Keyes, W.M. Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 2012, 26, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Segal, J.P.; Heller, E.; Lien, W.H.; Chang, C.Y.; Guo, X.; Oristian, D.S.; Zheng, D.; Fuchs, E. Nfatc1 orchestrates aging in hair follicle stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E4950–E4959. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Murray, P.J.; Jiang, T.X.; Plikus, M.V.; Chang, Y.T.; Lee, O.K.; Widelitz, R.B.; Chuong, C.M. Regenerative hair waves in aging mice and extra-follicular modulators follistatin, dkk1, and sfrp4. J. Investig. Dermatol. 2014, 134, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- So, W.K.; Cheung, T.H. Molecular Regulation of Cellular Quiescence: A Perspective from Adult Stem Cells and Its Niches. Methods Mol. Biol. 2018, 1686, 1–25. [Google Scholar] [PubMed]
- Hsu, Y.C.; Fuchs, E. A family business: Stem cell progeny join the niche to regulate homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Scadden, D.T. Nice neighborhood: Emerging concepts of the stem cell niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Slack, R.S. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr Opin Cell Biol. 2017, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Ng, H.H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Wei, P.; Dove, K.K.; Bensard, C.; Schell, J.C.; Rutter, J. The Force Is Strong with This One: Metabolism (Over)powers Stem Cell Fate. Trends Cell Biol. 2018, 28, 551–559. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A.; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012, 2, 568. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.; Schell, J.; Krall, A.S.; Jelinek, D.; Miranda, M.; Grigorian, M.; Braas, D.; White, A.C.; Zhou, J.L.; Graham, N.A.; et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 2017, 19, 1017–1026. [Google Scholar] [CrossRef]
- St John, J.C.; Ramalho-Santos, J.; Gray, H.L.; Petrosko, P.; Rawe, V.Y.; Navara, C.S.; Simerly, C.R.; Schatten, G.P. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells 2005, 7, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef]
- Schuijers, J.; Clevers, H. Adult mammalian stem cells: The role of Wnt, Lgr5 and R-spondins. EMBO J. 2012, 31, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aragó, M.; García-Bermúdez, J.; Martínez-Reyes, I.; Santacatterina, F.; Cuezva, J.M. Degradation of IF1 controls energy metabolism during osteogenic differentiation of stem cells. EMBO Rep. 2013, 14, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Zaltsman, Y.; Ruggiero, A.; Goldman, A.; Shachnai, L.; Zaidman, S.L.; Porat, Z.; Golan, K.; Lapidot, T.; Gross, A. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 2015, 6, 7901. [Google Scholar] [CrossRef]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Schell, J.C.; Rutter, J. Mitochondria link metabolism and epigenetics in haematopoiesis. Nat. Cell Biol. 2017, 19, 589–591. [Google Scholar] [CrossRef]
- Eliasson, P.; Rehn, M.; Hammar, P.; Larsson, P.; Sirenko, O.; Flippin, L.A.; Cammenga, J.; Jönsson, J.I. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp. Hematol. 2010, 38, 301–310. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Klimmeck, D.; Hansson, J.; Raffel, S.; Vakhrushev, S.Y.; Trumpp, A.; Krijgsveld, J. Proteomic cornerstones of hematopoietic stem cell differentiation: Distinct signatures of multipotent progenitors and myeloid committed cells. Mol. Cell. Proteom. 2012, 11, 286–302. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runners, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Zhang, C.C.; Sadek, H.A. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid. Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Pattappa, G.; Thorpe, S.D.; Jegard, N.C.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. Continuous and uninterrupted oxygen tension influences the colony formation and oxidative metabolism of human mesenchymal stem cells. Tissue Eng. Part C Methods 2013, 19, 68–79. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Tsatmali, M.; Walcott, E.C.; Crossin, K.L. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005, 1040, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, K. Newly Identified Roles of PML in Stem Cell Biology. Front. Oncol. 2013, 3, 50. [Google Scholar] [CrossRef]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Stoll, E.A.; Makin, R.; Sweet, I.R.; Trevelyan, A.J.; Miwa, S.; Horner, P.J.; Turnbull, D.M. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem Cells 2015, 33, 2306–2319. [Google Scholar] [CrossRef]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, O.; Suter, U.; Machado, R.A.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Liang, R.; Ghaffari, S. Stem cells, redox signaling, and stem cell aging. Antioxid. Redox Signal. 2014, 20, 1902–1916. [Google Scholar] [CrossRef]
- Cervantes, R.B.; Stringer, J.R.; Shao, C.; Tischfield, J.A.; Stambrook, P.J. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. USA 2002, 99, 3586–3590. [Google Scholar] [CrossRef]
- Kirby, D.M.; Rennie, K.J.; Smulders-Srinivasan, T.K.; Acin-Perez, R.; Whittington, M.; Enriquez, J.A.; Trevelyan, A.J.; Turnbull, D.M.; Lightowlers, R.N. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell Prolif. 2009, 42, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hämäläinen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, M.; Ameur, A.; Moraghebi, R.; Norddahl, G.L.; Sten, G.; Woods, N.B.; Bryder, D. Somatic cells with a heavy mitochondrial DNA mutational load render induced pluripotent stem cells with distinct differentiation defects. Stem Cells 2014, 32, 1173–1182. [Google Scholar] [CrossRef]
- Ma, H.; Folmes, C.D.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef]
- Morimoto, H.; Iwata, K.; Ogonuki, N.; Inoue, K.; Atsuo, O.; Kanatsu-Shinohara, M.; Morimoto, T.; Yabe-Nishimura, C.; Shinohara, T. ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell 2013, 12, 774–786. [Google Scholar] [CrossRef]
- Kohli, L.; Passed, E. Surviving changes: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; Passegué, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923. [Google Scholar] [CrossRef]
- Adams, P.D.; Jasper, H.; Rudolph, K.L. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell 2015, 16, 601–612. [Google Scholar] [CrossRef]
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.T.; Da Cruz, A.B.; Rao, M.; de Souza-Pinto, N.C.; Zeng, X.; Bohr, V.A. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 2008, 26, 2266–2274. [Google Scholar] [CrossRef]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef]
- Dannenmann, B.; Lehle, S.; Hildebrand, D.G.; Kübler, A.; Grondona, P.; Schmid, V.; Holzer, K.; Fröschl, M.; Essmann, F.; Rothfuss, O.; et al. High glutathione and glutathione peroxidase-2 levels mediate cell-type-specific DNA damage protection in human induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 886–898. [Google Scholar] [CrossRef]
- Miyamoto, K.; Miyamoto, T.; Kato, R.; Yoshimura, A.; Motoyama, N.; Suda, T. FoxO3a regulates hematopoietic homeostasis through a negative feedback pathway in conditions of stress or aging. Blood 2008, 112, 4485–4493. [Google Scholar] [CrossRef]
- Renault, V.M.; Rafalski, V.A.; Morgan, A.A.; Salih, D.A.; Brett, J.O.; Webb, A.E.; Villeda, S.A.; Thekkat, P.U.; Guillerey, C.; Denko, N.C.; et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 2009, 5, 527–539. [Google Scholar] [CrossRef]
- Matsui, K.; Ezoe, S.; Oritani, K.; Shibata, M.; Tokunaga, M.; Fujita, N.; Tanimura, A.; Sudo, T.; Tanaka, H.; McBurney, M.W.; et al. NAD-dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2012, 418, 811–817. [Google Scholar] [CrossRef]
- Webb, A.E.; Pollina, E.A.; Vierbuchen, T.; Urbán, N.; Ucar, D.; Leeman, D.S.; Martynoga, B.; Sewak, M.; Rando, T.A.; Guillemot, F.; et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013, 4, 477–491. [Google Scholar] [CrossRef]
- Rimmelé, P.; Bigarella, C.L.; Liang, R.; Izac, B.; Dieguez-Gonzalez, R.; Barbet, G.; Donovan, M.; Brugnara, C.; Blander, J.M.; Sinclair, D.A.; et al. Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. 2014, 3, 44–59. [Google Scholar] [CrossRef]
- Mehta, A.; Zhao, J.L.; Sinha, N.; Marinov, G.K.; Mann, M.; Kowalczyk, M.S.; Galimidi, R.P.; Du, X.; Erikci, E.; Regev, A.; et al. The MicroRNA-132 and MicroRNA-212 Cluster Regulates Hematopoietic Stem Cell Maintenance and Survival with Age by Buffering FOXO3 Expression. Immunity 2015, 42, 1021–1032. [Google Scholar] [CrossRef]
- Rimmelé, P.; Liang, R.; Bigarella, C.L.; Kocabas, F.; Xie, J.; Serasinghe, M.N.; Chipuk, J.; Sadek, H.; Zhang, C.C.; Ghaffari, S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015, 16, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, C.E.; Biteau, B.; Bohmann, D.; Jasper, H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell 2011, 8, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegué, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegué, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Lyublinskaya, O.G.; Borisov, Y.G.; Pugovkina, N.A.; Smirnova, I.S.; Obidina, J.V.; Ivanova, J.S.; Zenin, V.V.; Shatrova, A.N.; Borodkina, A.V.; Aksenov, N.D.; et al. Reactive Oxygen Species Are Required for Human Mesenchymal Stem Cells to Initiate Proliferation after the Quiescence Exit. Oxid. Med. Cell. Longev. 2015, 2015, 502105. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef]
- Biteau, B.; Jasper, H. EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 2011, 138, 1045–1055. [Google Scholar] [CrossRef]
- Malinska, D.; Kudin, A.P.; Bejtka, M.; Kunz, W.S. Changes in mitochondrial reactive oxygen species synthesis during differentiation of skeletal muscle cells. Mitochondrion 2012, 12, 144–148. [Google Scholar] [CrossRef]
- Ueda, T.; Nagamachi, A.; Takubo, K.; Yamasaki, N.; Matsui, H.; Kanai, A.; Nakata, Y.; Ikeda, K.; Konuma, T.; Oda, H.; et al. Fbxl10 overexpression in murine hematopoietic stem cells induces leukemia involving metabolic activation and upregulation of Nsg2. Blood 2015, 125, 3437–3446. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Karigane, D.; Kobayashi, H.; Morikawa, T.; Ootomo, Y.; Sakai, M.; Nagamatsu, G.; Kubota, Y.; Goda, N.; Matsumoto, M.; Nishimura, E.K.; et al. p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 2016, 19, 192–204. [Google Scholar] [CrossRef]
- Kwon, B. p38α-mediated purine metabolism is linked to exit from quiescence of hematopoietic stem cells. Stem Cell Investig. 2016, 3, 69. [Google Scholar] [CrossRef][Green Version]
- Essers, M.A.G. Stressed-Out HSCs Turn Up p38α and Purine to Proliferate. Cell Stem Cell 2016, 19, 143–144. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Sørensen, A.L.; Timoskainen, S.; West, F.D.; Vekterud, K.; Boquest, A.C.; Ahrlund-Richter, L.; Stice, S.L.; Collas, P. Lineage-specific promoter DNA methylation patterns segregate adult progenitor cell types. Stem Cells Dev. 2010, 19, 1257–1266. [Google Scholar] [CrossRef]
- Yannarelli, G.; Pacienza, N.; Cuniberti, L.; Medin, J.; Davies, J.; Keating, A. Brief report: The potential role of epigenetics on multipotent cell differentiation capacity of mesenchymal stromal cells. Stem Cells 2013, 31, 215–220. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Hwang, I.Y.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H.; Jin, X.; et al. Psat1-Dependent Fluctuations in α-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab. 2016, 24, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Gao, Y.; Guo, H.; Xia, B.; Song, J.; Wu, X.; Zeng, H.; Kee, K.; Tang, F.; Yi, C. Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution. Cell Stem Cell 2017, 20, 720–731. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Noer, A.; Lindeman, L.C.; Collas, P. Histone H3 modifications associated with differentiation and long-term culture of mesenchymal adipose stem cells. Stem Cells Dev. 2009, 18, 725–736. [Google Scholar] [CrossRef]
- Weishaupt, H.; Sigvardsson, M.; Attema, J.L. Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood 2010, 115, 247–256. [Google Scholar] [CrossRef]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef]
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.B.; Mancini, E.; et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014, 158, 673–688. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Beerman, I.; Rossi, D.J. Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease. Cell Stem Cell 2015, 16, 613–625. [Google Scholar] [CrossRef]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef]
- Ugarte, F.; Sousae, R.; Cinquin, B.; Martin, E.W.; Krietsch, J.; Sanchez, G.; Inman, M.; Tsang, H.; Warr, M.; Passegué, E.; et al. Progressive Chromatin Condensation and H3K9 Methylation Regulate the Differentiation of Embryonic and Hematopoietic Stem Cells. Stem Cell Rep. 2015, 5, 728–740. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Faralli, H.; Wang, C.; Nakka, K.; Benyoucef, A.; Sebastian, S.; Zhuang, L.; Chu, A.; Palii, C.G.; Liu, C.; Camellato, B.; et al. UTX demethylase activity is required for satellite cell-mediated muscle regeneration. J. Clin. Investig. 2016, 126, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef]
- Fukada, S.; Uezumi, A.; Ikemoto, M.; Masuda, S.; Segawa, M.; Tanimura, N.; Yamamoto, H.; Miyagoe-Suzuki, Y.; Takeda, S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 2007, 25, 2448–2459. [Google Scholar] [CrossRef]
- Forsberg, E.C.; Passegué, E.; Prohaska, S.S.; Wagers, A.J.; Koeva, M.; Stuart, J.M.; Weissman, I.L. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS ONE 2010, 5, e8785. [Google Scholar] [CrossRef]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Lien, W.H.; Stokes, N.; Pasolli, H.A.; Silva, J.M.; Fuchs, E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011, 25, 485–498. [Google Scholar] [CrossRef]
- Juan, A.H.; Derfoul, A.; Feng, X.; Ryall, J.G.; Dell’Orso, S.; Pasut, A.; Zare, H.; Simone, J.M.; Rudnicki, M.A.; Sartorelli, V. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011, 25, 789–794. [Google Scholar] [CrossRef]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nuñez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 2012, 11, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Libert, S.; Guarente, L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu. Rev. Physiol. 2013, 75, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Yu, A.; Dang, W. Regulation of stem cell aging by SIRT1—Linking metabolic signaling to epigenetic modifications. Mol. Cell. Endocrinol. 2017, 455, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, V.A.; Ho, P.P.; Brett, J.O.; Ucar, D.; Dugas, J.C.; Pollina, E.A.; Chow, L.M.; Ibrahim, A.; Baker, S.J.; Barres, B.A.; et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat. Cell Biol. 2013, 15, 614–624. [Google Scholar] [CrossRef]
- Saharan, S.; Jhaveri, D.J.; Bartlett, P.F. SIRT1 regulates the neurogenic potential of neural precursors in the adult subventricular zone and hippocampus. J. Neurosci. Res. 2013, 91, 642–659. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, E.; Arlotta, P. The quest for myelin in the adult brain. Nat. Cell Biol. 2013, 15, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, Y.; Chen, C.; Li, Q.; Niu, X.; Guo, S.; Zhang, A.; Wang, Y.; Deng, Z. Repression of SIRT1 promotes the differentiation of mouse induced pluripotent stem cells into neural stem cells. Cell Mol. Neurobiol. 2014, 34, 905–912. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Sirtuins in Neuroendocrine Regulation and Neurological Diseases. Front. Neurosci. 2018, 12, 778. [Google Scholar] [CrossRef]
- Hisahara, S.; Chiba, S.; Matsumoto, H.; Tanno, M.; Yagi, H.; Shimohama, S.; Sato, M.; Horio, Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 15599–15604. [Google Scholar] [CrossRef]
- Ma, C.Y.; Yao, M.J.; Zhai, Q.W.; Jiao, J.W.; Yuan, X.B.; Poo, M.M. SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development 2014, 141, 4697–4709. [Google Scholar] [CrossRef] [PubMed]
- Fawal, M.A.; Davy, A. Impact of Metabolic Pathways and Epigenetics on Neural Stem Cells. Epigenet. Insights 2018, 11, 2516865718820946. [Google Scholar] [CrossRef]
- Diaz-Ruiz, A.; Gonzalez-Freire, M.; Ferrucci, L.; Bernier, M.; Cabo, R. SIRT1 synchs satellite cell metabolism with stem cell fate. Cell Stem Cell 2015, 16, 103–104. [Google Scholar] [CrossRef]
- Yun, J.; Johnson, J.L.; Hanigan, C.L.; Locasale, J.W. Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2012, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Diecke, S.; Zhang, W.Y.; Lan, F.; He, C.; Mordwinkin, N.M.; Chua, K.F.; Wu, J.C. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 18439–18447. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P. Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. The resurgence of NAD⁺. Science 2016, 352, 1396–1397. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef] [PubMed]
- Wagers, A.J. How stem cells get “turned on”. EMBO J. 2014, 33, 2743–2744. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link—Ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Gan, B.; Hu, J.; Jiang, S.; Liu, Y.; Sahin, E.; Zhuang, L.; Fletcher-Sananikone, E.; Colla, S.; Wang, Y.A.; Chin, L.; et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010, 468, 701–704. [Google Scholar] [CrossRef]
- Gurumurthy, S.; Xie, S.Z.; Alagesan, B.; Kim, J.; Yusuf, R.Z.; Saez, B.; Tzatsos, A.; Ozsolak, F.; Milos, P.; Ferrari, F.; et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 2010, 468, 659–663. [Google Scholar] [CrossRef]
- Nakada, D.; Saunders, T.L.; Morrison, S.J. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 2010, 468, 653–658. [Google Scholar] [CrossRef]
- Durand, E.M.; Zon, L.I. Stem cells: The blood balance. Nature 2010, 468, 644–645. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.; Skuli, N.; Simon, M.C. The tumor suppressor LKB1 emerges as a critical factor in hematopoietic stem cell biology. Cell Metab. 2011, 13, 8–10. [Google Scholar] [CrossRef][Green Version]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Gan, B.; Sahin, E.; Jiang, S.; Sanchez-Aguilera, A.; Scott, K.L.; Chin, L.; Williams, D.A.; Kwiatkowski, D.J.; DePinho, R.A. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc. Natl. Acad. Sci. USA 2008, 105, 19384–19389. [Google Scholar] [CrossRef] [PubMed]
- Kharas, M.G.; Gritsman, K. Akt: A double-edged sword for hematopoietic stem cells. Cell Cycle 2010, 9, 1223–1224. [Google Scholar] [CrossRef][Green Version]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Zabkiewicz, J.; McCarthy, A.; Pearson, H.B.; Winton, D.J.; Sansom, O.J.; Ashworth, A.; Clarke, A.R. Lkb1 deficiency alters goblet and paneth cell differentiation in the small intestine. PLoS ONE 2009, 4, e4264. [Google Scholar] [CrossRef]
- Yeung, T.M.; Chia, L.A.; Kosinski, C.M.; Kuo, C.J. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell. Mol. Life Sci. 2011, 68, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Richmond, C.A.; Shah, M.S.; Carlone, D.L.; Breault, D.T. Factors regulating quiescent stem cells: Insights from the intestine and other self-renewing tissues. J. Physiol. 2016, 594, 4805–4813. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Rosario, F.J.; Gupta, M.B.; Myatt, L.; Powell, T.L.; Glenn, J.P.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 2019, 9, 246. [Google Scholar] [CrossRef]
- Zhou, J.; Shrikhande, G.; Xu, J.; McKay, R.M.; Burns, D.K.; Johnson, J.E.; Parada, L.F. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev. 2011, 25, 1595–1600. [Google Scholar] [CrossRef]
- Yilmaz, Ö.H.; Katajisto, P.; Lamming, D.W.; Gültekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef]
- Malam, Z.; Cohn, R.D. Stem cells on alert: Priming quiescent stem cells after remote injury. Cell Stem Cell 2014, 15, 7–8. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Schroeder, M.D.; Ma, C.; Rando, T.A. HGFA Is an Injury-Regulated Systemic Factor that Induces the Transition of Stem Cells into G(Alert). Cell Rep. 2017, 19, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Signer, R.A.; Morrison, S.J. Cellular differences in protein synthesis regulate tissue homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Vilchez, D.; Simic, M.S.; Dillin, A. Proteostasis and aging of stem cells. Trends Cell Biol. 2014, 24, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Brombin, A.; Joly, J.S.; Jamen, F. New tricks for an old dog: Ribosome biogenesis contributes to stem cell homeostasis. Curr. Opin. Genet. Dev. 2015, 34, 61–70. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.B.; Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012, 33, 357–363. [Google Scholar] [CrossRef][Green Version]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef]
- Strikoudis, A.; Guillamot, M.; Aifantis, I. Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 2014, 15, 365–382. [Google Scholar] [CrossRef]
- Werner, A.; Manford, A.G.; Rape, M. Ubiquitin-Dependent Regulation of Stem Cell Biology. Trends Cell Biol. 2017, 27, 568–579. [Google Scholar] [CrossRef]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Duc, A.C.; Rao, S.; Sun, X.L.; Bilbee, A.N.; Rhodes, M.; Li, Q.; Kappes, D.J.; Rhodes, J.; Wiest, D.L. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell 2013, 24, 411–425. [Google Scholar] [CrossRef]
- Cai, X.; Gao, L.; Teng, L.; Ge, J.; Oo, Z.M.; Kumar, A.R.; Gilliland, D.G.; Mason, P.J.; Tan, K.; Speck, N.A. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 17, 165–177. [Google Scholar] [CrossRef]
- Le Bouteiller, M.; Souilhol, C.; Beck-Cormier, S.; Stedman, A.; Burlen-Defranoux, O.; Vandormael-Pournin, S.; Bernex, F.; Cumano, A.; Cohen-Tannoudji, M. Notchless-dependent ribosome synthesis is required for the maintenance of adult hematopoietic stem cells. J. Exp. Med. 2013, 210, 2351–2369. [Google Scholar] [CrossRef]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef]
- Liakath-Ali, K.; Mills, E.W.; Sequeira, I.; Lichtenberger, B.M.; Pisco, A.O.; Sipilä, K.H.; Mishra, A.; Yoshikawa, H.; Wu, C.C.; Ly, T.; et al. An evolutionarily conserved ribosome-rescue pathway maintains epidermal homeostasis. Nature 2018, 556, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Zismanov, V.; Chichkov, V.; Colangelo, V.; Jamet, S.; Wang, S.; Syme, A.; Koromilas, A.E.; Crist, C. Phosphorylation of eIF2α Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 2016, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef]
- Crist, C.G.; Montarras, D.; Buckingham, M. Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell 2012, 11, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C. Role of heat shock proteins in stem cell behavior. Prog. Mol. Biol. Transl. Sci. 2012, 111, 305–322. [Google Scholar] [PubMed]
- Baharvand, H.; Fathi, A.; van Hoof, D.; Salekdeh, G.H. Concise review: Trends in stem cell proteomics. Stem Cells 2007, 25, 1888–1903. [Google Scholar] [CrossRef]
- DeLany, J.P.; Floyd, Z.E.; Zvonic, S.; Smith, A.; Gravois, A.; Reiners, E.; Wu, X.; Kilroy, G.; Lefevre, M.; Gimble, J.M. Proteomic analysis of primary cultures of human adipose-derived stem cells: Modulation by adipogenesis. Mol. Cell. Proteom. 2005, 4, 731–740. [Google Scholar] [CrossRef]
- Matsui, H.; Asou, H.; Inaba, T. Cytokines direct the regulation of Bim mRNA stability by heat-shock cognate protein 70. Mol. Cell 2007, 25, 99–112. [Google Scholar] [CrossRef]
- Zou, P.; Yoshihara, H.; Hosokawa, K.; Tai, I.; Shinmyozu, K.; Tsukahara, F.; Maru, Y.; Nakayama, K.; Nakayama, K.I.; Suda, T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell 2011, 9, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Miharada, K.; Karlsson, G.; Rehn, M.; Rörby, E.; Siva, K.; Cammenga, J.; Karlsson, S. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell 2011, 9, 330–344. [Google Scholar] [CrossRef]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Tai-Nagara, I.; Matsuoka, S.; Ariga, H.; Suda, T. Mortalin and DJ-1 coordinately regulate hematopoietic stem cell function through the control of oxidative stress. Blood 2014, 123, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tomé, F.; Dupret, J.M.; et al. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Sanbe, A.; Osinska, H.; Saffitz, J.E.; Glabe, C.G.; Kayed, R.; Maloyan, A.; Robbins, J. Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10132–10136. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Rao, K.S.; Rao, C.M. Ubiquitin-proteasome-mediated degradation and synthesis of MyoD is modulated by alphaB-crystallin, a small heat shock protein, during muscle differentiation. Biochim. Biophys. Acta 2010, 1803, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shi, Z.D.; Ji, X.; Morales, J.; Zhang, J.; Kaur, N.; Wang, S. Enhanced osteogenesis of human mesenchymal stem cells by periodic heat shock in self-assembling peptide hydrogel. Tissue Eng. Part A 2013, 19, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sunderic, K.; Nicoll, S.B.; Wang, S. Downregulation of Heat Shock Protein 70 Impairs Osteogenic and Chondrogenic Differentiation in Human Mesenchymal Stem Cells. Sci. Rep. 2018, 8, 553. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007559. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; de Verneuil, H.; Gribben, J.; et al. HIF-2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell 2013, 13, 549–563. [Google Scholar] [CrossRef]
- Miharada, K.; Sigurdsson, V.; Karlsson, S. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 2014, 7, 1381–1392. [Google Scholar] [CrossRef]
- Sigurdsson, V.; Miharada, K. Regulation of unfolded protein response in hematopoietic stem cells. Int. J. Hematol. 2018, 107, 627–633. [Google Scholar] [CrossRef]
- van Galen, P.; Kreso, A.; Mbong, N.; Kent, D.G.; Fitzmaurice, T.; Chambers, J.E.; Xie, S.; Laurenti, E.; Hermans, K.; Eppert, K.; et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 2014, 510, 268–272. [Google Scholar] [CrossRef]
- Cui, K.; Coutts, M.; Stahl, J.; Sytkowski, A.J. Novel interaction between the transcription factor CHOP (GADD153) and the ribosomal protein FTE/S3a modulates erythropoiesis. J. Biol. Chem. 2000, 275, 7591–7596. [Google Scholar] [CrossRef]
- Skalet, A.H.; Isler, J.A.; King, L.B.; Harding, H.P.; Ron, D.; Monroe, J.G. Rapid B cell receptor-induced unfolded protein response in non-secretory B cells correlates with pro- versus antiapoptotic cell fate. J. Biol. Chem. 2005, 280, 39762–39771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef]
- Sigurdsson, V.; Takei, H.; Soboleva, S.; Radulovic, V.; Galeev, R.; Siva, K.; Leeb-Lundberg, L.M.; Iida, T.; Nittono, H.; Miharada, K. Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 2016, 18, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Hindi, S.M.; Mann, A.K.; Gallot, Y.S.; Bohnert, K.R.; Cavener, D.R.; Whittemore, S.R.; Kumar, A. The PERK arm of the unfolded protein response regulates satellite cell-mediated skeletal muscle regeneration. eLife 2017, 6, e22871. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Gupta-Rossi, N.; Le Bail, O.; Gonen, H.; Brou, C.; Logeat, F.; Six, E.; Ciechanover, A.; Israël, A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J. Biol. Chem. 2001, 276, 34371–34378. [Google Scholar] [CrossRef] [PubMed]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef]
- Goh, E.L.; Zhu, T.; Leong, W.Y.; Lobie, P.E. c-Cbl is a negative regulator of GH-stimulated STAT5-mediated transcription. Endocrinology 2002, 143, 3590–3603. [Google Scholar] [CrossRef][Green Version]
- Jehn, B.M.; Dittert, I.; Beyer, S.; von der Mark, K.; Bielke, W. c-Cbl binding and ubiquitin-dependent lysosomal degradation of membrane-associated Notch1. J. Biol. Chem. 2002, 277, 8033–8040. [Google Scholar] [CrossRef]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef]
- Tsunematsu, R.; Nakayama, K.; Oike, Y.; Nishiyama, M.; Ishida, N.; Hatakeyama, S.; Bessho, Y.; Kageyama, R.; Suda, T.; Nakayama, K.I. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J. Biol. Chem. 2004, 279, 9417–9423. [Google Scholar] [CrossRef]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef]
- Schmidt, M.H.H.; Dikic, I. The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 907–918. [Google Scholar] [CrossRef]
- Zeng, S.; Xu, Z.; Lipkowitz, S.; Longley, J.B. Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl). Blood 2005, 105, 226–232. [Google Scholar] [CrossRef]
- Naujokat, C.; Sarić, T. Concise review: Role and function of the ubiquitin-proteasome system in mammalian stem and progenitor cells. Stem Cells 2007, 25, 2408–2418. [Google Scholar] [CrossRef]
- Reavie, L.; Della Gatta, G.; Crusio, K.; Aranda-Orgilles, B.; Buckley, S.M.; Thompson, B.; Lee, E.; Gao, J.; Bredemeyer, A.L.; Helmink, B.A.; et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat. Immunol. 2010, 11, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, C.; Matesic, L.E.; Flavell, R.A. The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat. Immunol. 2011, 12, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Takeishi, S.; Matsumoto, A.; Onoyama, I.; Naka, K.; Hirao, A.; Nakayama, K.I. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell 2013, 23, 347–361. [Google Scholar] [CrossRef] [PubMed]
- King, B.; Boccalatte, F.; Moran-Crusio, K.; Wolf, E.; Wang, J.; Kayembe, C.; Lazaris, C.; Yu, X.; Aranda-Orgilles, B.; Lasorella, A.; et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat. Immunol. 2016, 17, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Oike, Y.; Onoyama, I.; Iwama, A.; Arai, F.; Takubo, K.; Mashimo, Y.; Oguro, H.; Nitta, E.; Ito, K.; et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008, 22, 986–991. [Google Scholar] [CrossRef]
- Rathinam, C.; Thien, C.B.; Langdon, W.Y.; Gu, H.; Flavell, R.A. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008, 22, 992–997. [Google Scholar] [CrossRef]
- Thompson, B.J.; Jankovic, V.; Gao, J.; Buonamici, S.; Vest, A.; Lee, J.M.; Zavadil, J.; Nimer, S.D.; Aifantis, I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 2008, 205, 1395–1408. [Google Scholar] [CrossRef]
- Rodriguez, S.; Wang, L.; Mumaw, C.; Srour, E.F.; Lo Celso, C.; Nakayama, K.; Carlesso, N. The SKP2 E3 ligase regulates basal homeostasis and stress-induced regeneration of HSCs. Blood 2011, 117, 6509–6519. [Google Scholar] [CrossRef]
- Wang, J.; Han, F.; Wu, J.; Lee, S.W.; Chan, C.H.; Wu, C.Y.; Yang, W.L.; Gao, Y.; Zhang, X.; Jeong, Y.S.; et al. The role of Skp2 in hematopoietic stem cell quiescence, pool size, and self-renewal. Blood 2011, 118, 5429–5438. [Google Scholar] [CrossRef]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Stegmüller, J.; Matsuda, T.; Bonni, S.; Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 2004, 303, 1026–1030. [Google Scholar] [CrossRef]
- Lasorella, A.; Stegmüller, J.; Guardavaccaro, D.; Liu, G.; Carro, M.S.; Rothschild, G.; de la Torre-Ubieta, L.; Pagano, M.; Bonni, A.; Iavarone, A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006, 442, 471–474. [Google Scholar] [CrossRef]
- Stegmüller, J.; Konishi, Y.; Huynh, M.A.; Yuan, Z.; Dibacco, S.; Bonni, A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron 2006, 50, 389–400. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, A.H.; Yamada, T.; Wu, B.; Bilimoria, P.M.; Ikeuchi, Y.; de la Iglesia, N.; Shen, J.; Bonni, A. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 2009, 326, 575–578. [Google Scholar] [CrossRef]
- Huang, J.; Ikeuchi, Y.; Malumbres, M.; Bonni, A. A Cdh1-APC/FMRP Ubiquitin Signaling Link Drives mGluR-Dependent Synaptic Plasticity in the Mammalian Brain. Neuron 2015, 86, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Liu, J.; Yan, G.; Zhang, J.; Han, Y.; Guo, J.; Xu, Z.; Yuan, Z.; Liu, J.; Malumbres, M.; et al. Cdh1 regulates craniofacial development via APC-dependent ubiquitination and activation of Goosecoid. Cell Res. 2016, 26, 699–712. [Google Scholar] [CrossRef][Green Version]
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 2005, 123, 409–421. [Google Scholar] [CrossRef]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef]
- Zhao, X.; Heng, J.I.; Guardavaccaro, D.; Jiang, R.; Pagano, M.; Guillemot, F.; Iavarone, A.; Lasorella, A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 2008, 10, 643–653. [Google Scholar] [CrossRef]
- Forget, A.; Bihannic, L.; Cigna, S.M.; Lefevre, C.; Remke, M.; Barnat, M.; Dodier, S.; Shirvani, H.; Mercier, A.; Mensah, A.; et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev. Cell 2014, 29, 649–661. [Google Scholar] [CrossRef]
- Urbán, N.; van den Berg, D.L.; Forget, A.; Andersen, J.; Demmers, J.A.; Hunt, C.; Ayrault, O.; Guillemot, F. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science 2016, 353, 292–295. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Muñoz-Cánoves, P. Autophagy: A decisive process for stemness. Oncotarget 2016, 7, 12286–12288. [Google Scholar] [CrossRef]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22, 432–435. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.J.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, P.; Arroba, A.I.; Cecconi, F.; de la Rosa, E.J.; Boya, P.; de Pablo, F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012, 8, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Huang, Y.; Chang, J.Y.; Liu, L.; McKeehan, W.L.; Martin, J.F.; Wang, F. FRS2α-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ. Res. 2012, 110, e29–e39. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Liu, L.; McKeehan, W.L.; Wang, F. The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy 2012, 8, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liang, C.C.; Bian, Z.C.; Zhu, Y.; Guan, J.L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef]
- Fiacco, E.; Castagnetti, F.; Bianconi, V.; Madaro, L.; De Bardi, M.; Nazio, F.; D’Amico, A.; Bertini, E.; Cecconi, F.; Puri, P.L.; et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 2016, 23, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Iorio, E.; Cagnin, M.; Garribba, L.; Pietraforte, D.; Falchi, M.; Pascucci, B.; Baccarini, S.; Morani, F.; et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell Death Dis. 2016, 7, e2168. [Google Scholar] [CrossRef]
- Pietras, E.M.; Warr, M.R.; Passegué, E. Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 2011, 195, 709–720. [Google Scholar] [CrossRef]
- Sage, J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012, 26, 1409–1420. [Google Scholar] [CrossRef]
- Matsumoto, A.; Nakayama, K.I. Role of key regulators of the cell cycle in maintenance of hematopoietic stem cells. Biochim. Biophys. Acta 2013, 1830, 2335–2344. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takizawa, H.; Suda, T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 2014, 141, 4656–4666. [Google Scholar] [CrossRef] [PubMed]
- Popov, B.; Petrov, N. pRb-E2F signaling in life of mesenchymal stem cells: Cell cycle, cell fate, and cell differentiation. Genes Dis. 2014, 1, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- TeKippe, M.; Harrison, D.E.; Chen, J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp. Hematol. 2003, 31, 521–527. [Google Scholar] [CrossRef]
- Chen, J.; Ellison, F.M.; Keyvanfar, K.; Omokaro, S.O.; Desierto, M.J.; Eckhaus, M.A.; Young, N.S. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp. Hematol. 2008, 36, 1236–1243. [Google Scholar] [CrossRef]
- Akala, O.O.; Park, I.K.; Qian, D.; Pihalja, M.; Becker, M.W.; Clarke, M.F. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature 2008, 453, 228–232. [Google Scholar] [CrossRef]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef]
- Asai, T.; Liu, Y.; Di Giandomenico, S.; Bae, N.; Ndiaye-Lobry, D.; Deblasio, A.; Menendez, S.; Antipin, Y.; Reva, B.; Wevrick, R.; et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood 2012, 120, 1601–1612. [Google Scholar] [CrossRef]
- Yamashita, M.; Nitta, E.; Suda, T. Regulation of hematopoietic stem cell integrity through p53 and its related factors. Ann. N. Y. Acad. Sci. 2016, 1370, 45–54. [Google Scholar] [CrossRef]
- Meletis, K.; Wirta, V.; Hede, S.M.; Nistér, M.; Lundeberg, J.; Frisén, J. p53 suppresses the self-renewal of adult neural stem cells. Development 2006, 133, 363–369. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar]
- Viatour, P.; Somervaille, T.C.; Venkatasubrahmanyam, S.; Kogan, S.; McLaughlin, M.E.; Weissman, I.L.; Butte, A.J.; Passegué, E.; Sage, J. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 2008, 3, 416–428. [Google Scholar] [PubMed]
- Hosoyama, T.; Nishijo, K.; Prajapati, S.I.; Li, G.; Keller, C. Rb1 gene inactivation expands satellite cell and postnatal myoblast pools. J. Biol. Chem. 2011, 286, 19556–19564. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef]
- Kippin, T.E.; Martens, D.J.; van der Kooy, D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005, 19, 756–767. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, K.; Dakik, P.; Medkour, Y.; Mitrofanova, D.; Titorenko, V.I. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. Int. J. Mol. Sci. 2019, 20, 2158. https://doi.org/10.3390/ijms20092158
Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. International Journal of Molecular Sciences. 2019; 20(9):2158. https://doi.org/10.3390/ijms20092158
Chicago/Turabian StyleMohammad, Karamat, Paméla Dakik, Younes Medkour, Darya Mitrofanova, and Vladimir I. Titorenko. 2019. "Quiescence Entry, Maintenance, and Exit in Adult Stem Cells" International Journal of Molecular Sciences 20, no. 9: 2158. https://doi.org/10.3390/ijms20092158
APA StyleMohammad, K., Dakik, P., Medkour, Y., Mitrofanova, D., & Titorenko, V. I. (2019). Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. International Journal of Molecular Sciences, 20(9), 2158. https://doi.org/10.3390/ijms20092158