Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
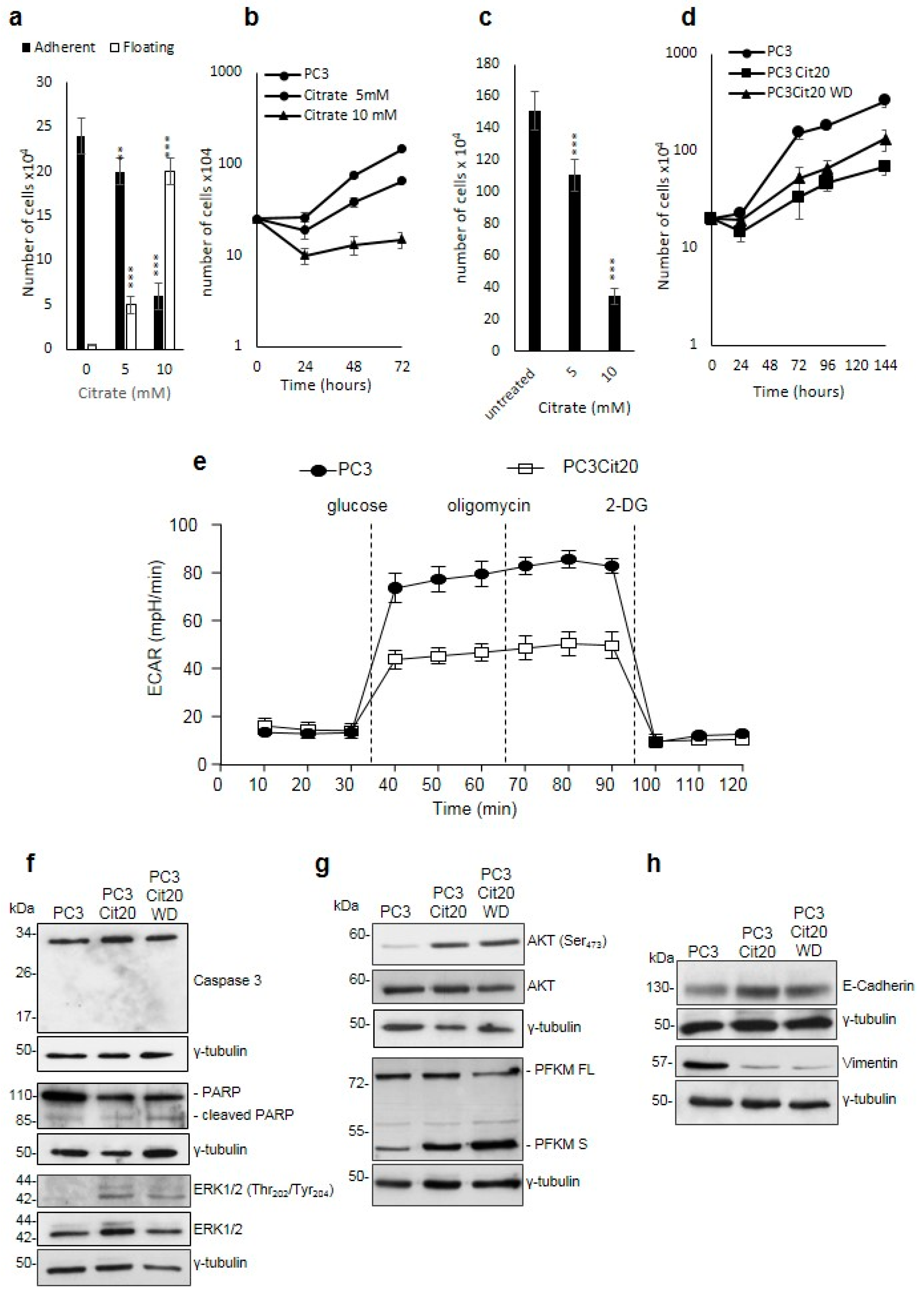
2.1. Citrate Treatment Affects PC3 Cell Proliferation, Survival, and Metabolism
2.2. Citrate Alters Signaling Pathways Governing the Proliferation, Differentiation, and Survival of PC3 Cells
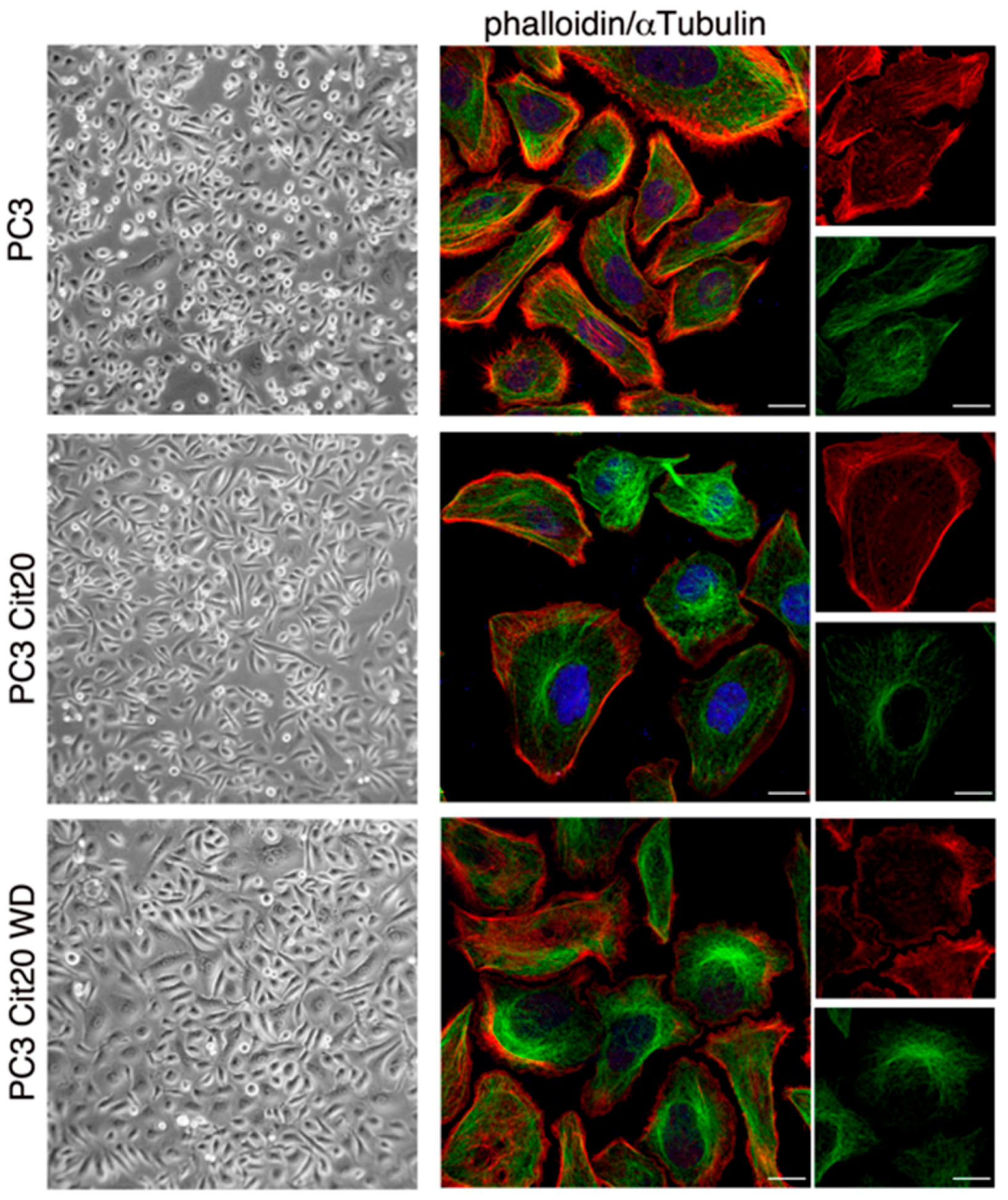
2.3. Cytoskeleton Dynamics is Altered in Citrate-Resistant PC3 Cells
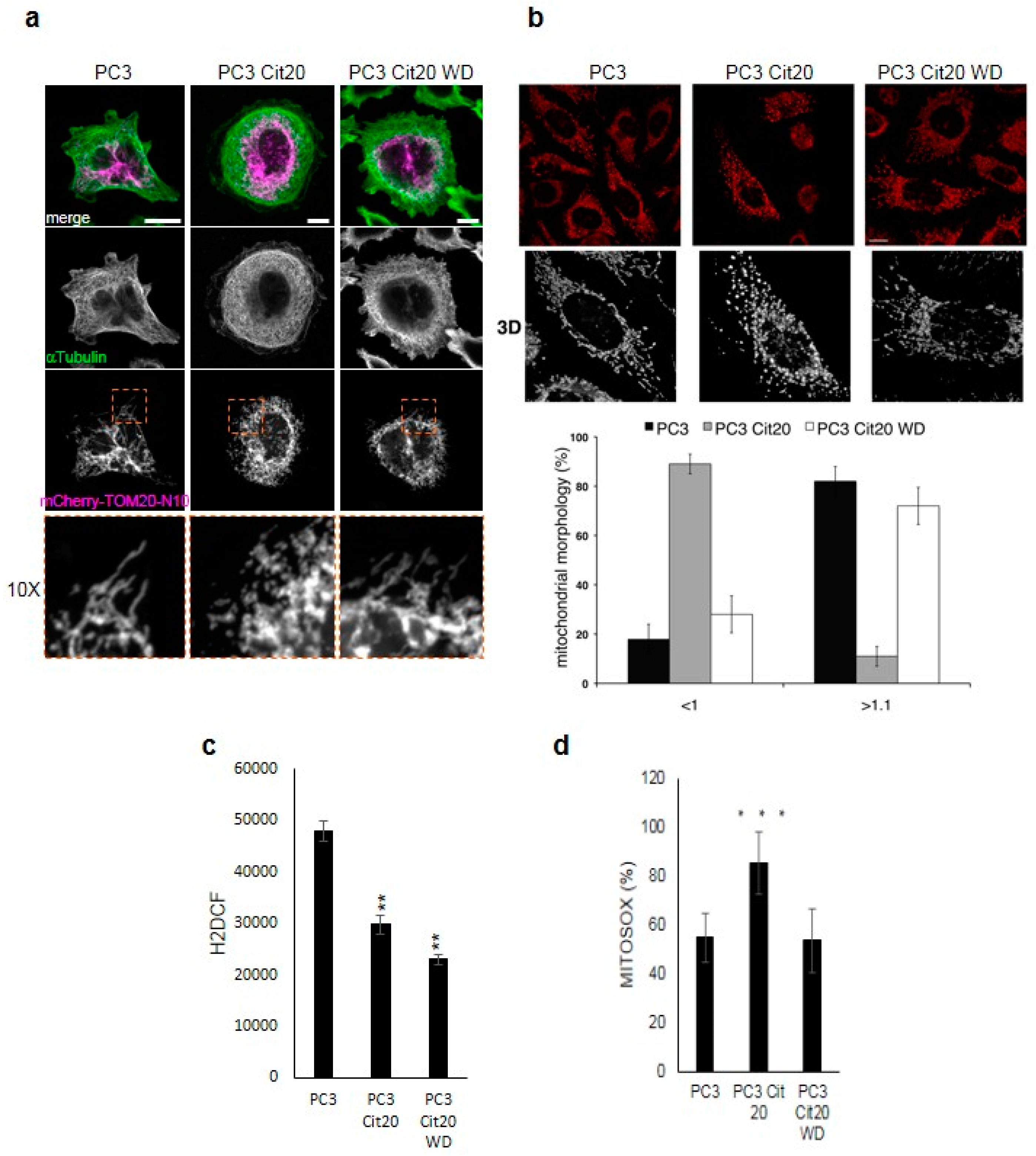
2.4. The Mitochondrial Network is Altered in Citrate-Resistant PC3 Cells
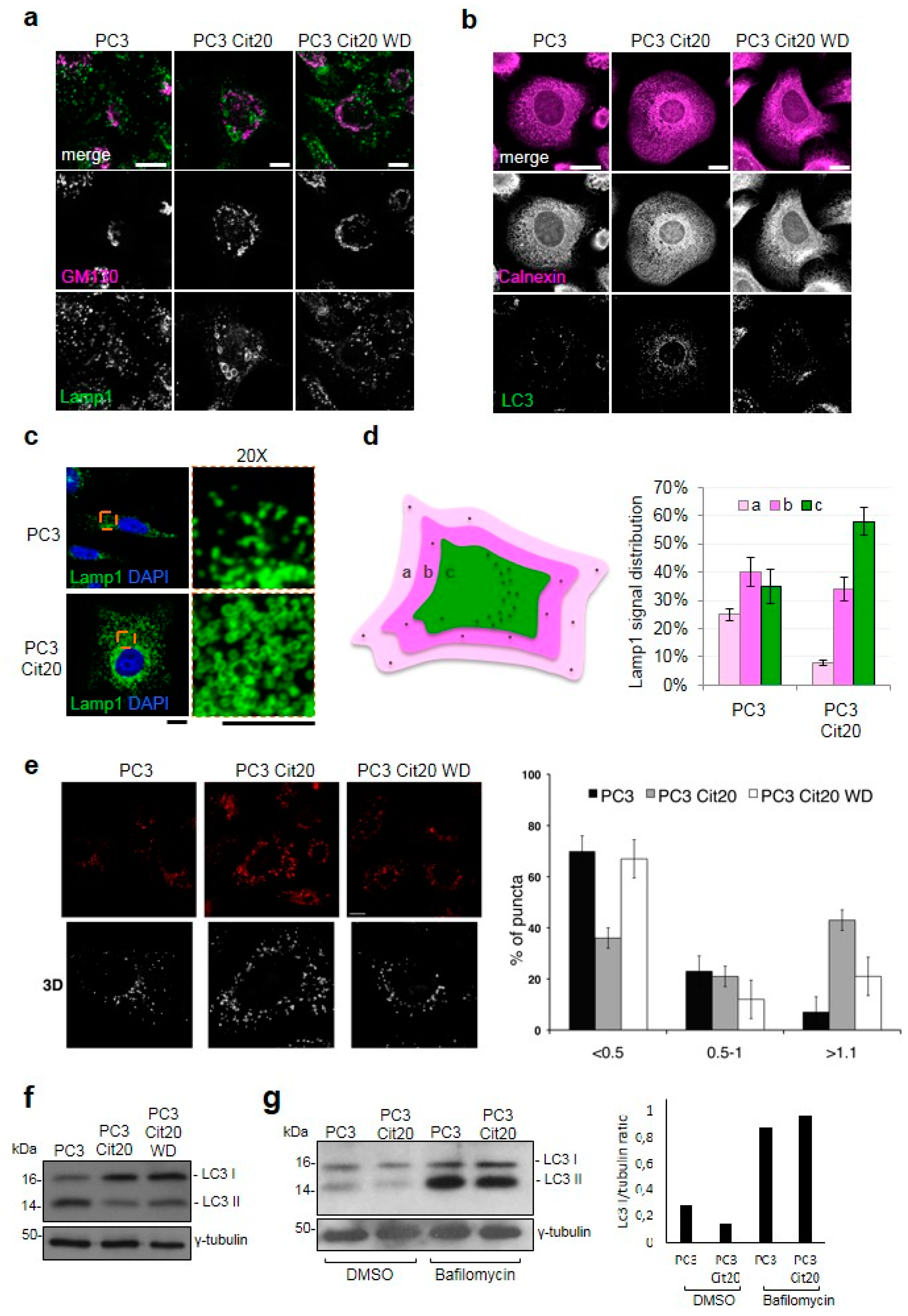
2.5. Exocytic and Endocytic Organelles Are Altered in Citrate-Resistant PC3 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antibodies
4.3. Plasmids and Cell Transfections
4.4. Cell Culture and Growth Curve
4.5. Determination of Total ROS and Mitochondrial ROS Production
4.6. Measurements of ECAR
4.7. Annexin V/PI Apoptotic Assay
4.8. Immunoblotting
4.9. Bafilomycin Treatment
4.10. Fluorescence Microscopy
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACO2 | m-aconitase |
| Cit | citrate |
| F2,6BP | fructose 2,6-bisphosphate |
| PCa | prostate cancer |
| PFK1 | phosphofructokinase 1 |
References
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Smerc, A.; Sodja, E.; Legisa, M. Posttranslational modification of 6-phosphofructo-1-kinase as an important feature of cancer metabolism. PLoS ONE 2011, 6, e19645. [Google Scholar] [CrossRef]
- Sola-Penna, M.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Zancan, P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 2010, 62, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Decreased zinc in the development and progression of malignancy: An important common relationship and potential for prevention and treatment of carcinomas. Expert. Opin. Ther. Targets 2017, 21, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzola, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef]
- Marberger, H.; Marberger, E.; Mann, T.; Lutwak-Mann, C. Citric acid in human prostatic secretion and metastasizing cancer of prostate gland. Br. Med. J. 1962, 1, 835–836. [Google Scholar] [CrossRef]
- Cooper, J.F.; Farid, I. The Role of Citric Acid in the Physiology of the Prostate. 3. Lactate/Citrate Ratios in Benign and Malignant Prostatic Homogenates as an Index of Prostatic Malignancy. J. Urol. 1964, 92, 533–536. [Google Scholar] [CrossRef]
- Giskeodegard, G.F.; Bertilsson, H.; Selnaes, K.M.; Wright, A.J.; Bathen, T.F.; Viset, T.; Halgunset, J.; Angelsen, A.; Gribbestad, I.S.; Tessem, M.B. Spermine and citrate as metabolic biomarkers for assessing prostate cancer aggressiveness. PLoS ONE 2013, 8, e62375. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B.; Narayan, P. Citrate in the diagnosis of prostate cancer. Prostate 1999, 38, 237–245. [Google Scholar] [CrossRef]
- Sarkar, B.K.; Chakraborty, C.; Sharma, A.R.; Bae, K.J.; Sharma, G.; Doss, G.P.; Dutta, D.; Ding, S.; Ganbold, B.; Nam, J.S.; et al. Novel biomarker for prostate cancer diagnosis by MRS. Front. Biosci. 2014, 19, 1186–1201. [Google Scholar] [CrossRef][Green Version]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target. Drug Resist. Updat. 2016, 29, 47–53. [Google Scholar] [PubMed]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef]
- Lincet, H.; Kafara, P.; Giffard, F.; Abeilard-Lemoisson, E.; Duval, M.; Louis, M.H.; Poulain, L.; Icard, P. Inhibition of Mcl-1 expression by citrate enhances the effect of Bcl-xL inhibitors on human ovarian carcinoma cells. J. Ovarian Res. 2013, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Su, M.; Wei, L.; Ji, F.; Wang, N.; Zhong, C.; Chen, T. BAY11-7082 inhibits proliferation and promotes apoptosis in breast carcinoma MCF-7 cells by inhibiting phosphorylation of ATP citrate lyase. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 1458–1462. [Google Scholar] [PubMed]
- Wang, T.A.; Zhang, X.D.; Guo, X.Y.; Xian, S.L.; Lu, Y.F. 3-bromopyruvate and sodium citrate target glycolysis, suppress survivin, and induce mitochondrial-mediated apoptosis in gastric cancer cells and inhibit gastric orthotopic transplantation tumor growth. Oncol. Rep. 2016, 35, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, X.; Zhang, H.; Lan, J.; Huang, G.; Varin, E.; Lincet, H.; Poulain, L.; Icard, P. Citrate induces apoptotic cell death: A promising way to treat gastric carcinoma? Anticancer Res. 2011, 31, 797–805. [Google Scholar] [PubMed]
- Xia, Y.; Zhang, X.; Bo, A.; Sun, J.; Li, M. Sodium citrate inhibits the proliferation of human gastric adenocarcinoma epithelia cells. Oncol. Lett. 2018, 15, 6622–6628. [Google Scholar] [CrossRef]
- Hanai, J.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J. Cell Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Spinelli, E.; Gandolfo, F.; Lemma, V.; Di Fruscio, G.; Padoan, R.; Morescalchi, F.; D’Agostino, M.; Savoldi, G.; Semeraro, F.; et al. Familial exudative vitreoretinopathy caused by a homozygous mutation in TSPAN12 in a cystic fibrosis infant. Ophthalmic. Genet. 2014, 35, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.I.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef]
- Zhang, X.; Varin, E.; Allouche, S.; Lu, Y.; Poulain, L.; Icard, P. Effect of citrate on malignant pleural mesothelioma cells: A synergistic effect with cisplatin. Anticancer Res. 2009, 29, 1249–1254. [Google Scholar] [PubMed]
- Shah, S.; Carriveau, W.J.; Li, J.; Campbell, S.L.; Kopinski, P.K.; Lim, H.W.; Daurio, N.; Trefely, S.; Won, K.J.; Wallace, D.C.; et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7, 43713–43730. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Nilchian, A.; Orrenius, S.; Zhivotovsky, B.; Gogvadze, V. Citrate kills tumor cells through activation of apical caspases. Cell Mol. Life Sci. 2012, 69, 4229–4237. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Piao, M.J.; Kang, K.A.; Shilnikova, K.; Hyun, Y.J.; Oh, S.K.; Jeong, Y.J.; Chae, S.; Hyun, J.W. A Benzylideneacetophenone Derivative Induces Apoptosis of Radiation-Resistant Human Breast Cancer Cells via Oxidative Stress. Biomol. Ther. 2017, 25, 404–410. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, T.L.; Tsai, W.H.; Tsai, H.J.; Hu, K.H.; Chang, H.C.; Yeh, C.W.; Chen, Y.C.; Liao, C.C.; Chang, W.T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep. 2012, 2, 785. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Liang, J.Y.; Li, T.L.; Guan, Z.X.; Zou, J.; Franklin, R.; Costello, L.C. Zinc induces mitochondria apoptogenesis in prostate cells. Mol. Urol. 2000, 4, 31–36. [Google Scholar]
- Halabe Bucay, A. Hypothesis proved...citric acid (citrate) does improve cancer: A case of a patient suffering from medullary thyroid cancer. Med. Hypotheses 2009, 73, 271. [Google Scholar] [CrossRef]
- Bucay, A.H. Clinical report: A patient with primary peritoneal mesothelioma that has improved after taking citric acid orally. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 241. [Google Scholar] [CrossRef] [PubMed]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest. Urol. 1979, 17, 16–23. [Google Scholar] [PubMed]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Dettmer, K.; Rummele, P.; Schmidt, K.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.; et al. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Anil Kumar, K.; Reddanna, P. The Paradox of Akt-mTOR Interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef]
- Andrejc, D.; Legisa, M. Kallikrein-related peptidase 6 can cleave human-muscle-type 6-phosphofructo-1-kinase into highly active shorter fragments. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Marin-Hernandez, A.; Rodriguez-Enriquez, S.; Vital-Gonzalez, P.A.; Flores-Rodriguez, F.L.; Macias-Silva, M.; Sosa-Garrocho, M.; Moreno-Sanchez, R. Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J. 2006, 273, 1975–1988. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Royaux, I.; Swinnen, J.V.; Smans, K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol. Cancer Ther. 2012, 11, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Islam, M.S.; Tian, J.; Lui, V.W.; Xiao, D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014, 349, 15–25. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na(+)-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Mycielska, M.E.; Milenkovic, V.M.; Wetzel, C.H.; Rummele, P.; Geissler, E.K. Extracellular Citrate in Health and Disease. Curr. Mol. Med. 2015, 15, 884–891. [Google Scholar] [CrossRef]
- Kline, E.E.; Treat, E.G.; Averna, T.A.; Davis, M.S.; Smith, A.Y.; Sillerud, L.O. Citrate concentrations in human seminal fluid and expressed prostatic fluid determined via 1H nuclear magnetic resonance spectroscopy outperform prostate specific antigen in prostate cancer detection. J. Urol. 2006, 176, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.B.; Juang, H.H.; Zou, J.; Costello, L.C. Regulation of citrate metabolism by androgen in the LNCaP human prostate carcinoma cell line. Endocrine 1995, 3, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zeng, F.; Li, S.; Li, G.; Lai, X.; Wang, Q.J.; Deng, F. Crosstalk of protein kinase C epsilon with Smad2/3 promotes tumor cell proliferation in prostate cancer cells by enhancing aerobic glycolysis. Cell Mol. Life Sci. 2018, 75, 4583–4598. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ulmert, D.; O’Brien, M.F.; Bjartell, A.S.; Lilja, H. Prostate kallikrein markers in diagnosis, risk stratification and prognosis. Nat. Rev. Urol. 2009, 6, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Briollais, L.; Ozcelik, H.; Xu, J.; Kwiatkowski, M.; Lalonde, E.; Sendorek, D.H.; Fleshner, N.E.; Recker, F.; Kuk, C.; Olkhov-Mitsel, E.; et al. Germline Mutations in the Kallikrein 6 Region and Predisposition for Aggressive Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, 4. [Google Scholar] [CrossRef]
- Edelmann, A.; Kirchberger, J.; Naumann, M.; Kopperschlager, G. Generation of catalytically active 6-phosphofructokinase from Saccharomyces cerevisiae in a cell-free system. Eur. J. Biochem. 2000, 267, 4825–4830. [Google Scholar] [CrossRef]
- Paliouras, M.; Diamandis, E.P. Intracellular signaling pathways regulate hormone-dependent kallikrein gene expression. Tumor Biol. 2008, 29, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Paliouras, M.; Diamandis, E.P. An AKT activity threshold regulates androgen-dependent and androgen-independent PSA expression in prostate cancer cell lines. Biol. Chem. 2008, 389, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Cui, J.; Wang, L.; Wu, K.; Hong, X.; Zou, Y.; Zhao, S.; Ke, H. Excessive mitochondrial fragmentation triggered by erlotinib promotes pancreatic cancer PANC-1 cell apoptosis via activating the mROS-HtrA2/Omi pathways. Cancer Cell Int. 2018, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Song, Y.S. Mitochondrial dynamics altered by oxidative stress in cancer. Free Radic. Res. 2016, 50, 1065–1070. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, 6. [Google Scholar] [CrossRef]
- Ciano, M.; Allocca, S.; Ciardulli, M.C.; Della Volpe, L.; Bonatti, S.; D’Agostino, M. Differential phosphorylation-based regulation of alphaB-crystallin chaperone activity for multipass transmembrane proteins. Biochem Biophys Res. Commun. 2016, 479, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Picascia, A.; Stanzione, R.; Chieffi, P.; Kisslinger, A.; Dikic, I.; Tramontano, D. Proline-rich tyrosine kinase 2 regulates proliferation and differentiation of prostate cells. Mol. Cell Endocrinol. 2002, 186, 81–87. [Google Scholar] [CrossRef]
- Conte, A.; Kisslinger, A.; Procaccini, C.; Paladino, S.; Oliviero, O.; de Amicis, F.; Faicchia, D.; Fasano, D.; Caputo, M.; Matarese, G.; et al. Convergent Effects of Resveratrol and PYK2 on Prostate Cells. Int. J. Mol. Sci. 2016, 17, 1542. [Google Scholar] [CrossRef]
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M.; et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity 2016, 44, 712. [Google Scholar] [CrossRef]
- D’Agostino, M.; Crespi, A.; Polishchuk, E.; Generoso, S.; Martire, G.; Colombo, S.F.; Bonatti, S. ER reorganization is remarkably induced in COS-7 cells accumulating transmembrane protein receptors not competent for export from the endoplasmic reticulum. J. Membr. Biol. 2014, 247, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Risselada, H.J.; Endter, L.J.; Comte-Miserez, V.; Mayer, A. SNARE-mediated membrane fusion arrests at pore expansion to regulate the volume of an organelle. EMBO J. 2018, 37, 19. [Google Scholar] [CrossRef]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladino, S.; Valentino, A.; Scialo, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonium improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor gamma coactivator 1alpha expression. J. Cell Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Paladino, S.; Bianco, G.; Fasano, D.; Gerlini, R.; Tornincasa, M.; Renna, M.; Fusco, A.; Tramontano, D.; Pierantoni, G.M. High mobility group A1 protein modulates autophagy in cancer cells. Cell Death Differ. 2017, 24, 1948–1962. [Google Scholar] [CrossRef] [PubMed]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Rinaldo, C.; Moncada, A.; Gradi, A.; Ciuffini, L.; D’Eliseo, D.; Siepi, F.; Prodosmo, A.; Giorgi, A.; Pierantoni, G.M.; Trapasso, F.; et al. HIPK2 controls cytokinesis and prevents tetraploidization by phosphorylating histone H2B at the midbody. Mol. Cell 2012, 47, 87–98. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caiazza, C.; D’Agostino, M.; Passaro, F.; Faicchia, D.; Mallardo, M.; Paladino, S.; Pierantoni, G.M.; Tramontano, D. Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line. Int. J. Mol. Sci. 2019, 20, 2613. https://doi.org/10.3390/ijms20112613
Caiazza C, D’Agostino M, Passaro F, Faicchia D, Mallardo M, Paladino S, Pierantoni GM, Tramontano D. Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line. International Journal of Molecular Sciences. 2019; 20(11):2613. https://doi.org/10.3390/ijms20112613
Chicago/Turabian StyleCaiazza, Carmen, Massimo D’Agostino, Fabiana Passaro, Deriggio Faicchia, Massimo Mallardo, Simona Paladino, Giovanna Maria Pierantoni, and Donatella Tramontano. 2019. "Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line" International Journal of Molecular Sciences 20, no. 11: 2613. https://doi.org/10.3390/ijms20112613
APA StyleCaiazza, C., D’Agostino, M., Passaro, F., Faicchia, D., Mallardo, M., Paladino, S., Pierantoni, G. M., & Tramontano, D. (2019). Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line. International Journal of Molecular Sciences, 20(11), 2613. https://doi.org/10.3390/ijms20112613