Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity

 , and
, and
Abstract
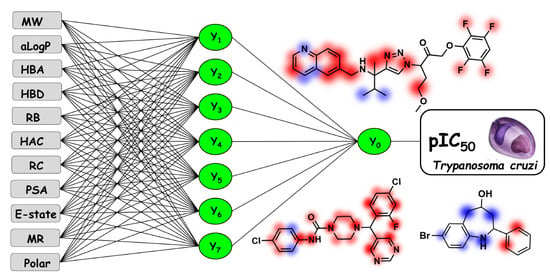
:
1. Introduction
2. Results
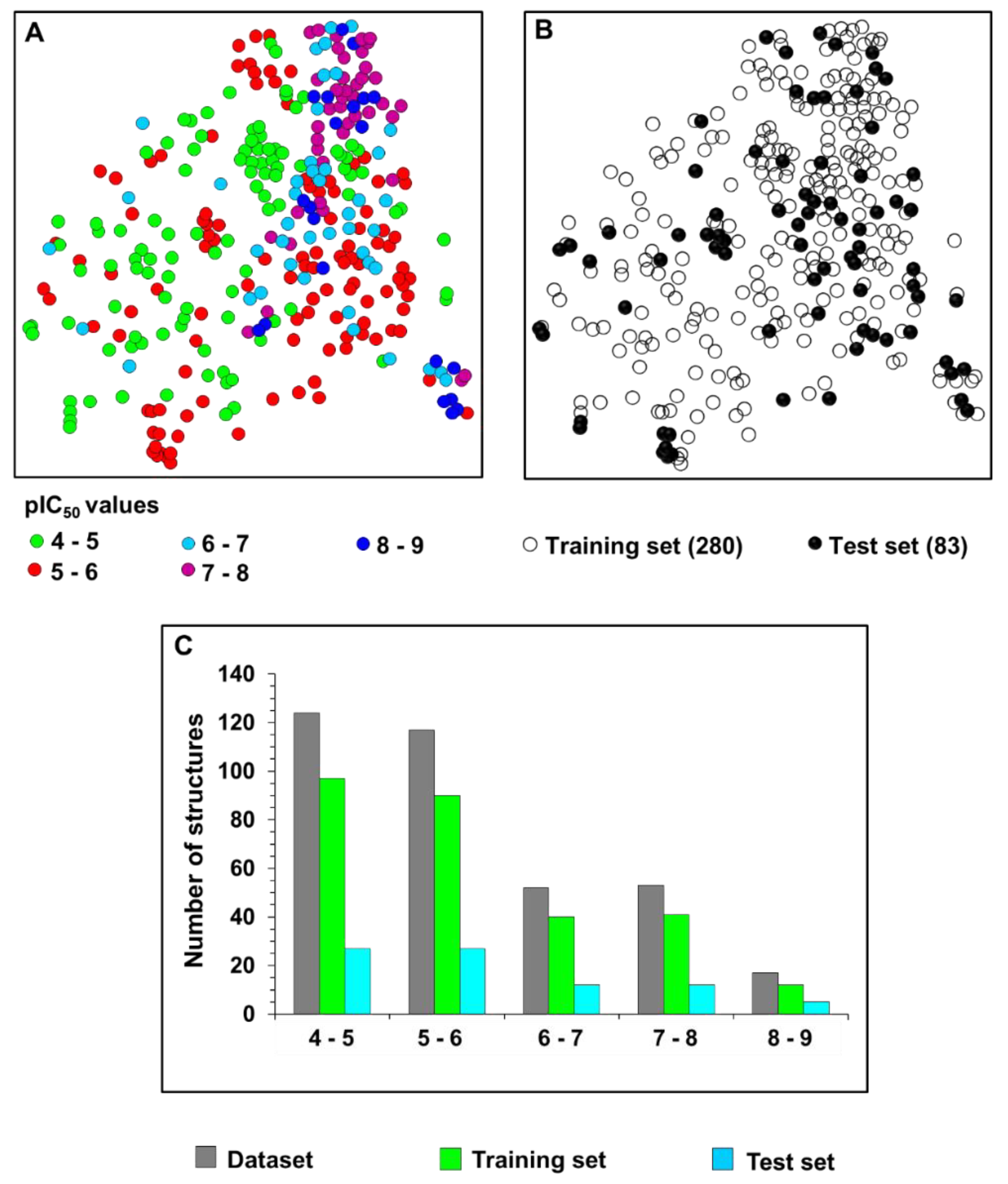
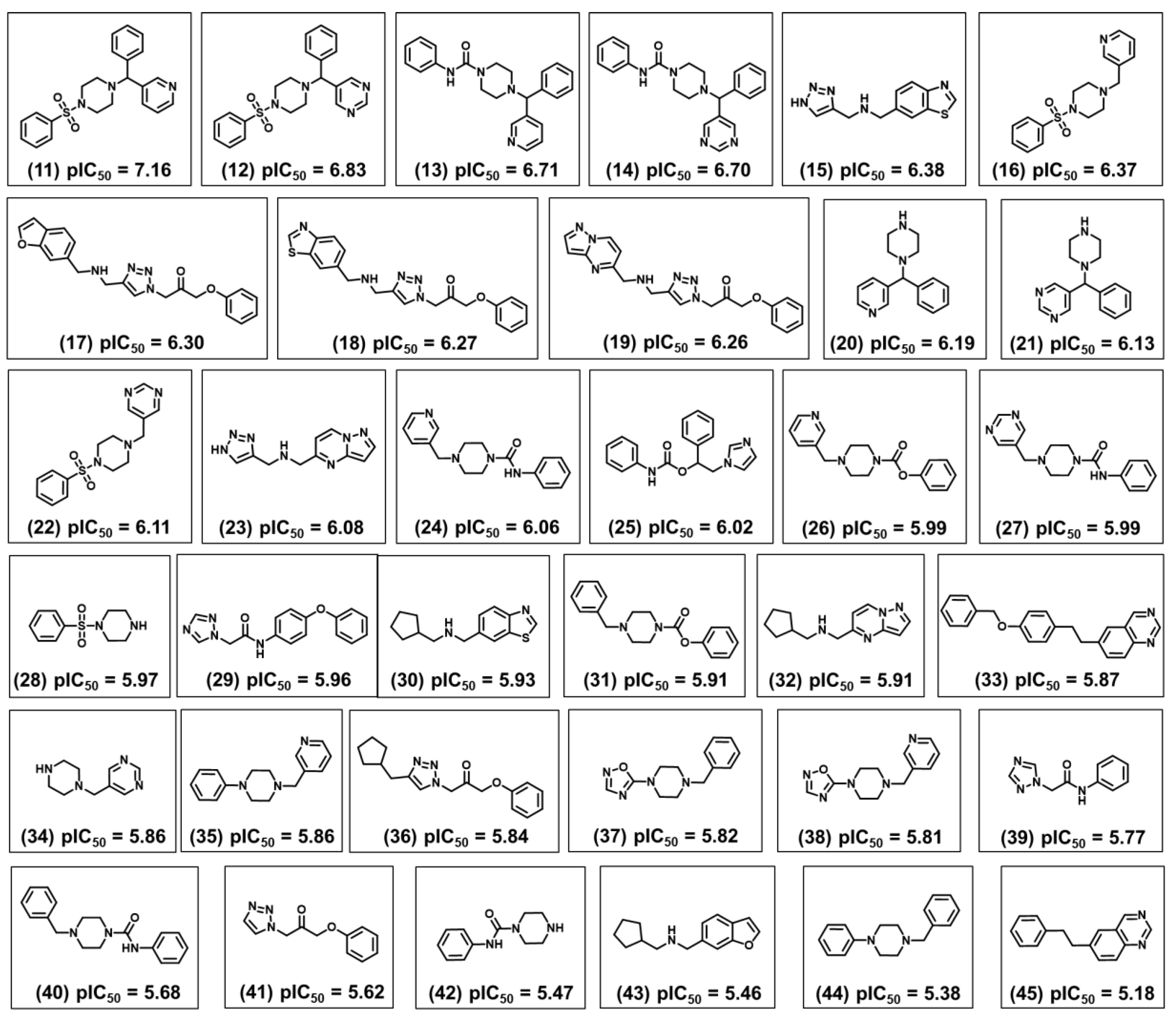
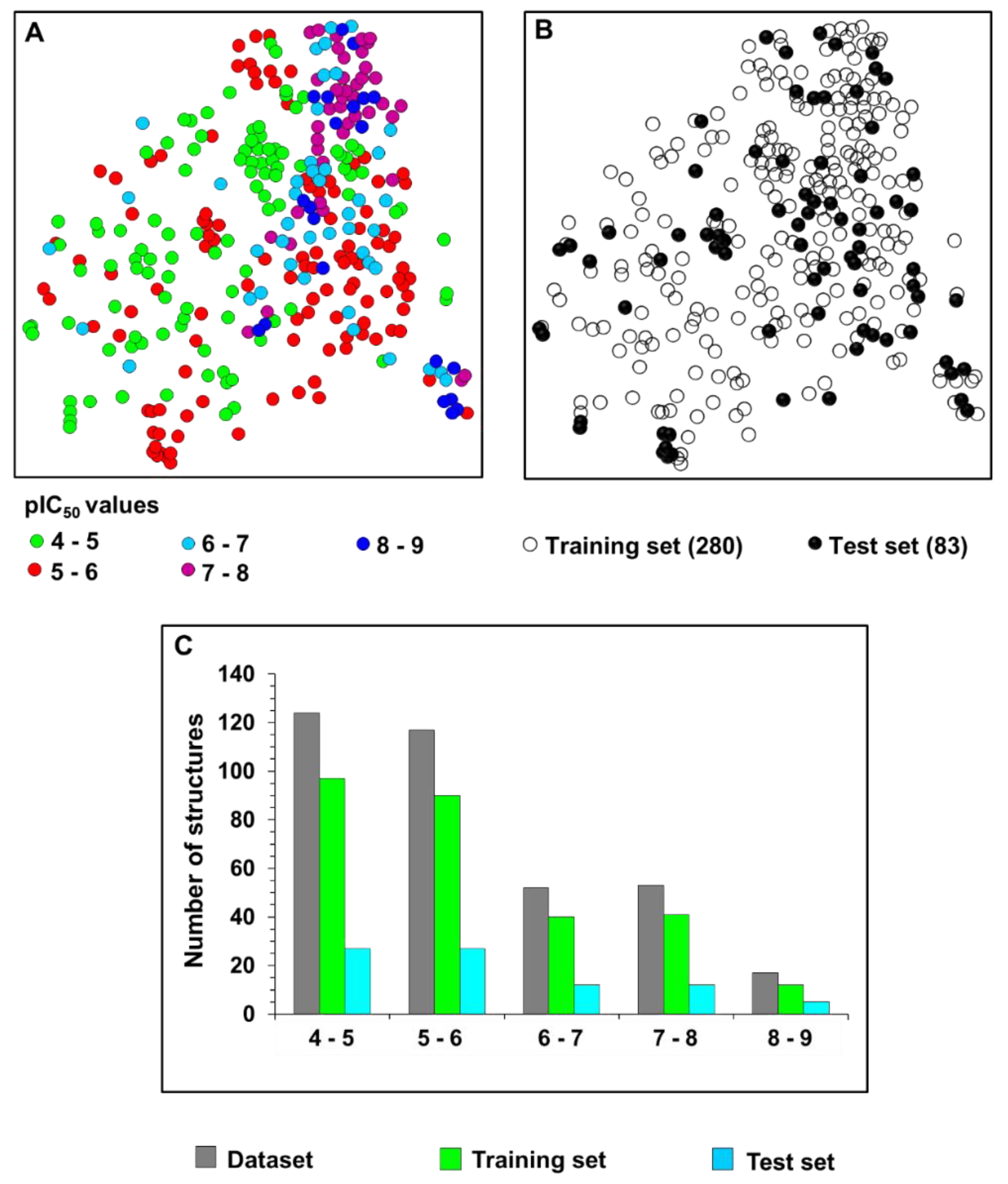
2.1. Chemical and Biological Landscape
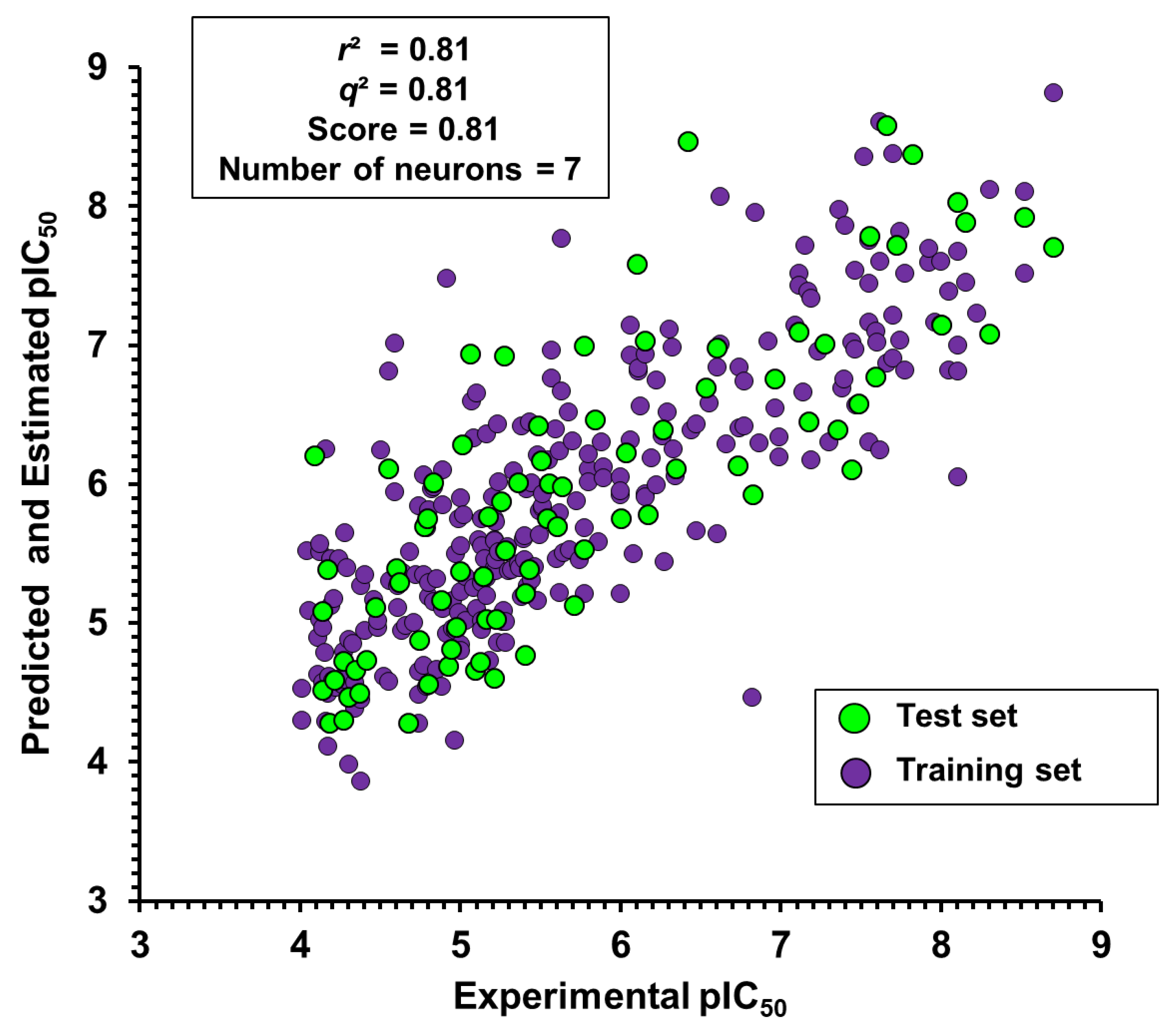
2.2. Artificial Neural Networks
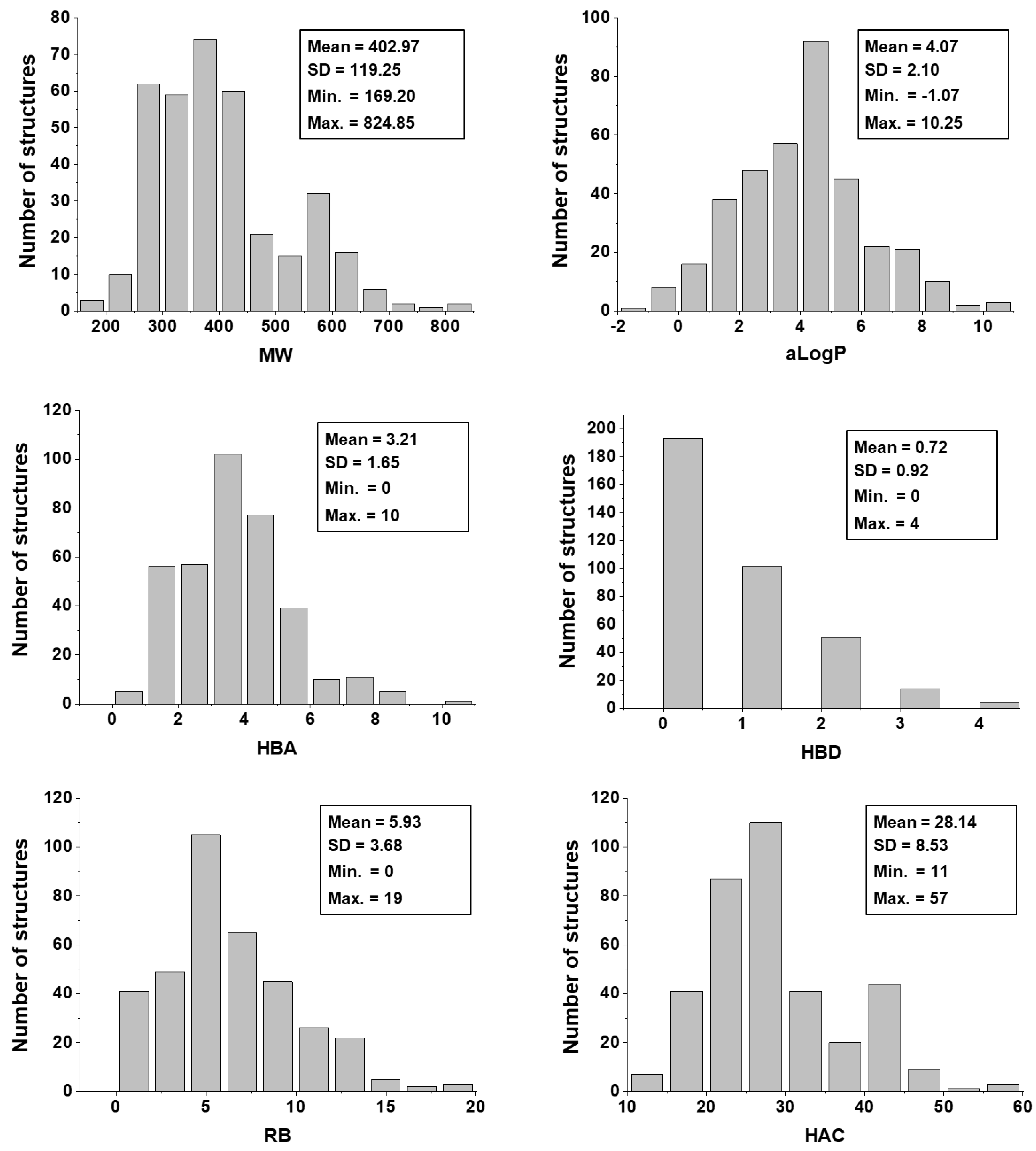
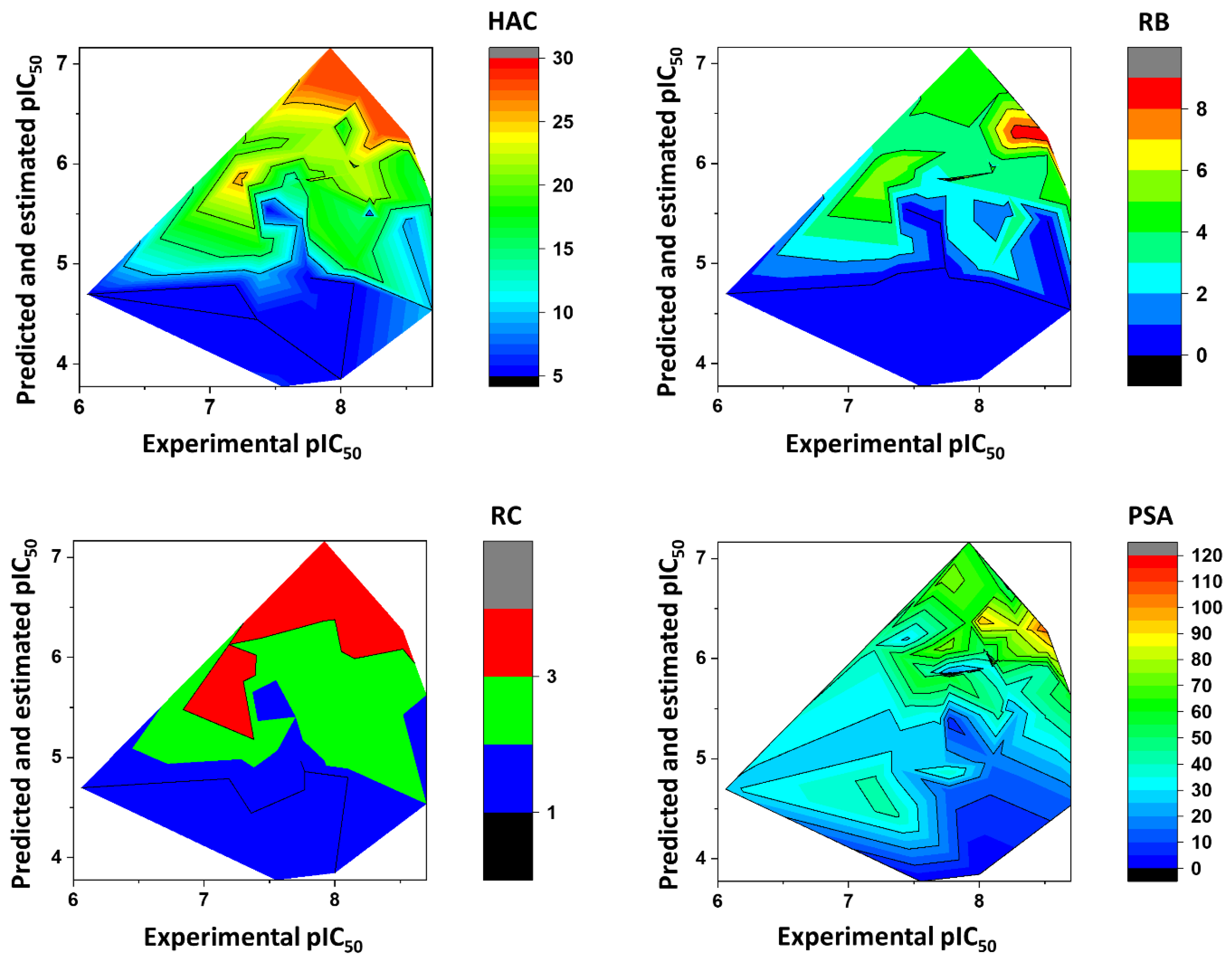
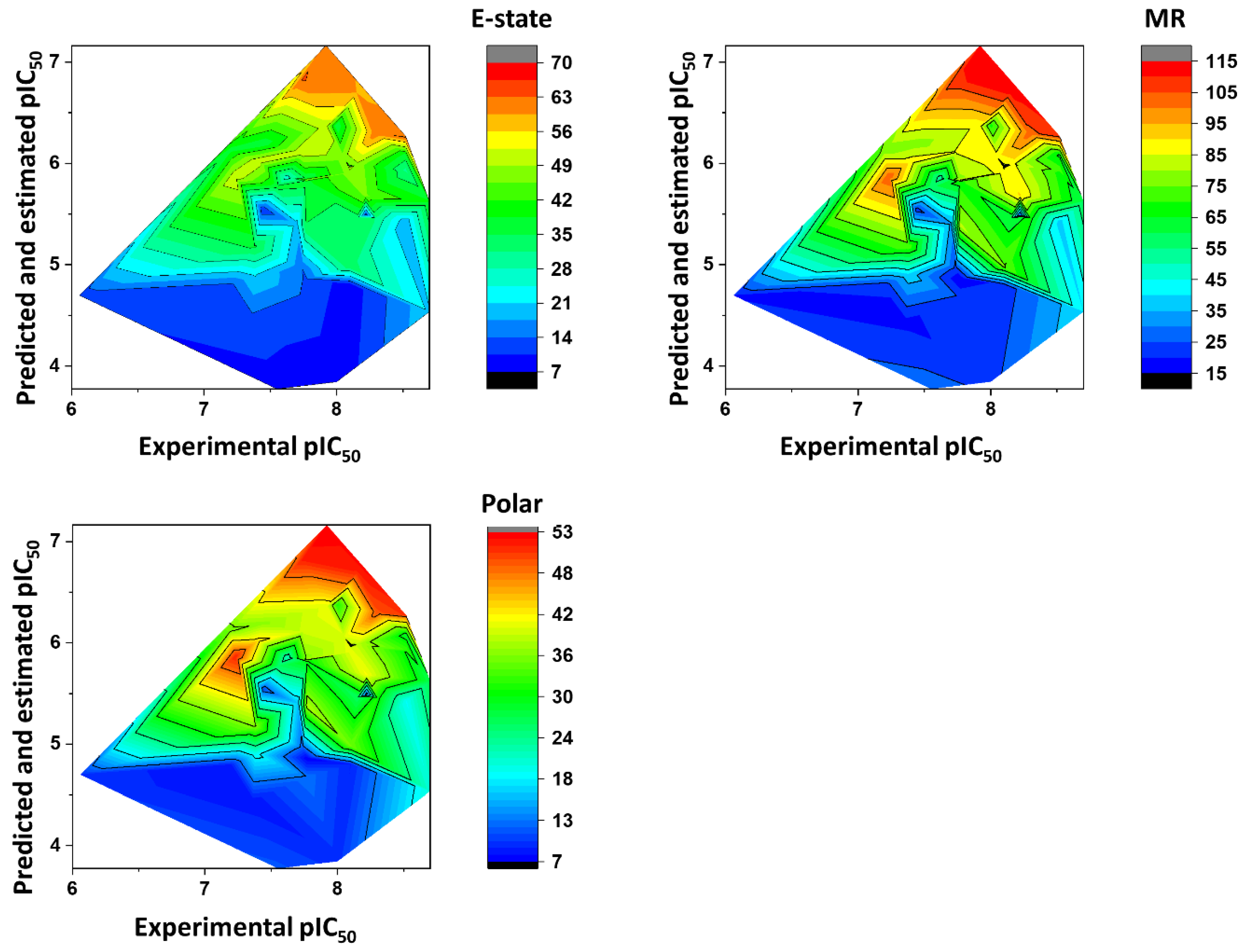
Impact of the Physicochemical Properties on the Trypanocidal Activity
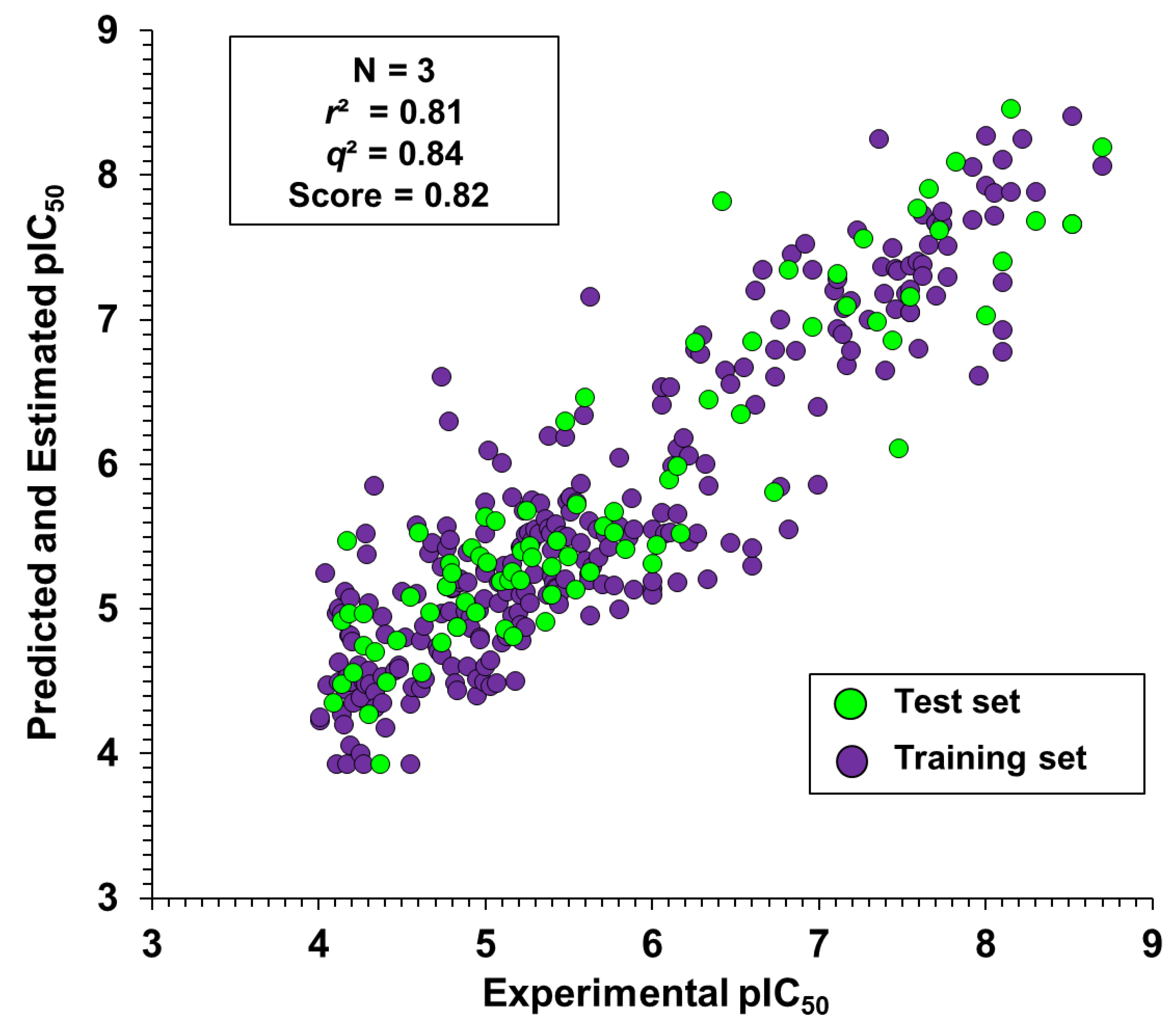
2.3. Kernel-Based Partial Least Squares
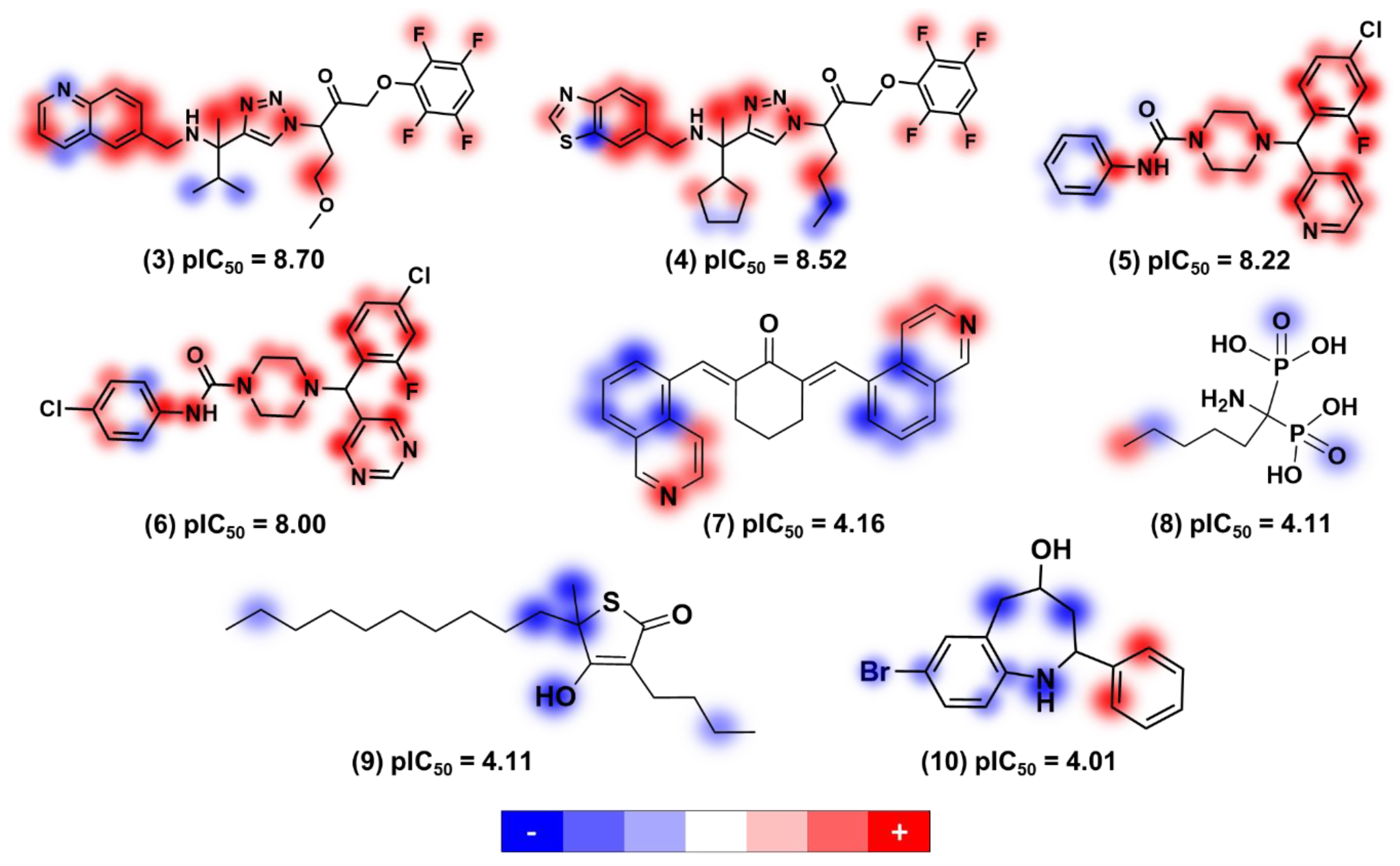
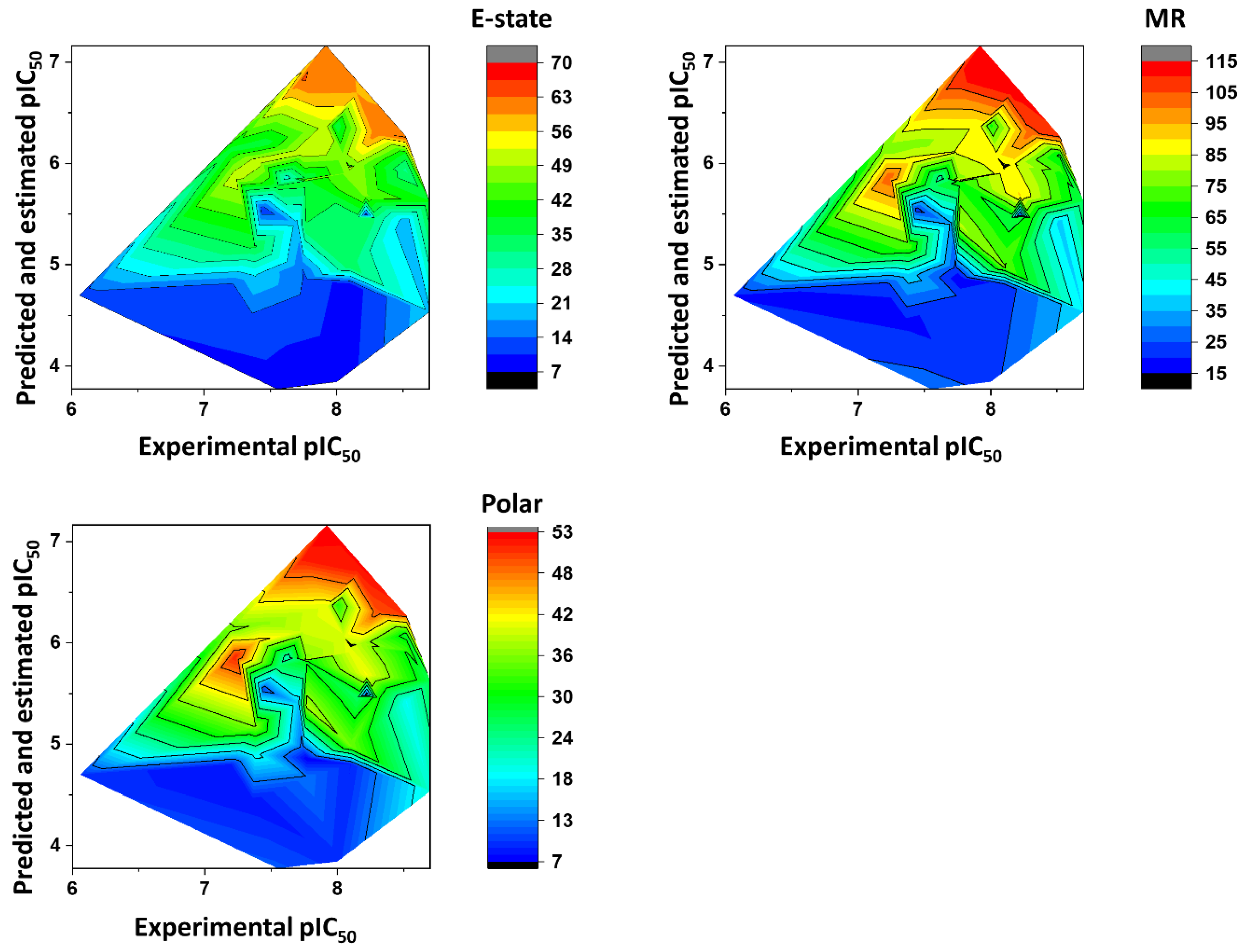
Contribution Maps
3. Physicochemical Profile of Favorable Fragments
4. Discussion
5. Materials and Methods
5.1. Selection and Construction of the Dataset
5.2. Characterization of the Chemical and Biological Space
5.3. Physicochemical Descriptors
5.4. Backpropagation Artificial Neural Networks
5.5. Molecular Fingerprints and 2D Contribution Maps
5.6. Heat Maps
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANN | Artificial Neural Network |
| KPLS | Kernel-based Partial Least Squares |
| IC50 | Concentration of compound that inhibits 50% of the growth of T. cruzi in phenotypic assays |
| pIC50 | −log IC50 |
| QSAR | Quantitative Structure-Activity Relationships |
| WHO | World Health Organization |
| MW | Molecular Weight |
| aLogP | Octanol-Water Partition Coefficient |
| HBD | Hydrogen Bond Donors |
| HBA | Hydrogen Bond Acceptors |
| RB | Rotatable Bonds |
| PSA | Polar Surface Area |
| E-state | Electrotopological State |
| MR | Molar Refractivity |
| Polar | Molecular Polarizability |
| T. cruzi | Trypanosoma cruzi |
| RC | Ring Count |
| HAC | Heavy Atom Count |
References
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2017, 6736, 82–94. [Google Scholar] [CrossRef]
- World Health Organization. American Trypanosomiasis (Chagas disease). Available online: http://www.who.int/chagas/en/ (accessed on 10 February 2019).
- Dias, J.C.; Ramos, A.N., Jr.; Gontijo, E.D.; Luquetti, A.; Shikanai-Yasuda, M.A.; Coura, J.R.; Torres, R.M.; Melo, J.R.; Almeida, E.A.; Oliveira, W., Jr.; et al. II Consenso Brasileiro em Doença de Chagas 2015. Epidemiol. Serv. Saude 2016, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Bournissen, F.G.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.L.G.; Andricopulo, A.D. Drugs and vaccines in the 21st century for neglected diseases. Lancet Infect. Dis. 2019, 19, 125–127. [Google Scholar] [CrossRef]
- Tyler, K.M.; Engman, D.M. The life cycle of Trypanosoma cruzi. Int. J. Parasitol. 2001, 31, 472–481. [Google Scholar] [CrossRef]
- Mandal, S. Epidemiological Aspects of Chagas Disease—A Review. J. Anc. Dis. Prev. Remedies 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Salto, M.L.; Bertello, L.E.; Vieira, M.; Docampo, R.; Moreno, S.N.J.; Lederkremer, R.M. De Formation and Remodeling of Inositolphosphoceramide during Differentiation of Trypanosoma cruzi from Trypomastigote to Amastigote. Eucaryotic Cell 2003, 2, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Lentini, G.; Pacheco, N.; dos, S.; Burleigh, B.A. Targeting host mitochondria: A role for the Trypanosoma cruzi amastigote flagellum. Cell. Microbiol. 2018, 20, 1–8. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Oliva, G.; Andricopulo, A.D. From Medicinal Chemistry to Human Health: Current Approaches to Drug Discovery for Cancer and Neglected Tropical Diseases. An. Acad. Bras. Cienc. 2018, 90, 645–661. [Google Scholar] [CrossRef]
- De Rycker, M.; Baragaña, B.; Duce, S.L.; Gilbert, I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature 2018, 559, 498–506. [Google Scholar] [CrossRef]
- Polinsky, A. Lead-Likeness and Drug-Likeness. Pract. Med. Chem. 2008, 244–254. [Google Scholar] [CrossRef]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Maltarollo, V.G.; Gertrudes, J.C.; Oliveira, P.R.; Honorio, K.M. Applying machine learning techniques for ADME-Tox prediction: A review. Expert Opin. Drug Metab. Toxicol. 2015, 11, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Preclinical Pharmacokinetics: An Approach Towards Safer and Efficacious Drugs. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M.; Handzlik, J.; Sanmartín, C.; Domínguez-Álvarez, E.; Spengler, G. Prediction of ADME properties for selenocompounds with anticancer and efflux pump inhibitory activity using preliminary computational methods. Acta Pharm. Hung. 2018, 88, 67–74. [Google Scholar]
- Honorio, K.M.; Garratt, R.C.; Polikatpov, I.; Andricopulo, A.D. 3D QSAR comparative molecular field analysis on nonsteroidal farnesoid X receptor activators. J. Mol. Graph. Modell. 2007, 25, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Schmidhuber, J. Deep Learning in neural networks: An overview. Neural Networks 2015, 61, 85–117. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, F.; Geladi, P.; Wold, S. Kernel-based PLS regression; Cross-validation and applications to spectral data. J. Chemom. 1994, 8, 377–389. [Google Scholar] [CrossRef]
- Lecun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating erros. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Gómez-Ayala, S.; Castrillón, J.A.; Palma, A.; Leal, S.M.; Escobar, P.; Bahsas, A. Synthesis, structural elucidation and in vitro antiparasitic activity against Trypanosoma cruzi and Leishmania chagasi parasites of novel tetrahydro-1-benzazepine derivatives. Bioorganic Med. Chem. 2010, 18, 4721–4739. [Google Scholar] [CrossRef]
- King-Keller, S.; Li, M.; Smith, A.; Zheng, S.; Kaur, G.; Yang, X.; Wang, B.; Docampo, R. Chemical validation of phosphodiesterase C as a chemotherapeutic target in Trypanosoma cruzi, the etiological agent of Chagas’ disease. Antimicrob. Agents Chemother. 2010, 54, 3738–3745. [Google Scholar] [CrossRef]
- Magaraci, F.; Jimenez Jimenez, C.; Rodrigues, C.; Rodrigues, J.C.F.; Vianna Braga, M.; Yardley, V.; De Luca-Fradley, K.; Croft, S.L.; De Souza, W.; Ruiz-Perez, L.M.; et al. Azasterols as Inhibitors of Sterol 24-Methyltransferase in Leishmania Species and Trypanosoma cruzi. J. Med. Chem. 2003, 46, 4714–4727. [Google Scholar] [CrossRef]
- Jones, S.M.; Urch, J.E.; Kaiser, M.; Brun, R.; Harwood, J.L.; Berry, C.; Gilbert, I.H. Analogues of thiolactomycin as potential antimalarial and anti-trypanosomal agents. J. Med. Chem. 2005, 48, 5932–5941. [Google Scholar] [CrossRef]
- Zuccotto, F.; Brun, R.; Gonzalez, P.D.; Ruiz, P.L.M.; Gilbert, I.H. The structure-based design and synthesis of selective inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorganic Med. Chem. Lett. 1999, 9, 1463–1468. [Google Scholar] [CrossRef]
- Jonckers, T.H.M.; Van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M.C.; Van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E.L.; et al. Synthesis, cytotoxicity, and antiplasmodial and antitrypanosomal activity of new neocryptolepine derivatives. J. Med. Chem. 2002, 45, 3497–3508. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J.B. Bisphosphonates derived from fatty acids are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. Bioorganic Med. Chem. Lett. 2003, 13, 3231–3235. [Google Scholar] [CrossRef]
- Da Rosa, R.; de Moraes, M.H.; Zimmermann, L.A.; Schenkel, E.P.; Steindel, M.; Bernardes, L.S.C. Design and synthesis of a new series of 3,5-disubstituted isoxazoles active against Trypanosoma cruzi and Leishmania amazonensis. Eur. J. Med. Chem. 2017, 128, 25–35. [Google Scholar] [CrossRef]
- De Azeredo, C.M.O.; Ávila, E.P.; Pinheiro, D.L.J.; Amarante, G.W.; Soares, M.J. Biological activity of the azlactone derivative EPA-35 against Trypanosoma cruzi. FEMS Microbiol. Lett. 2017, 364, 1–7. [Google Scholar] [CrossRef]
- Guerra, A.; Gonzalez-Naranjo, P.; Campillo, N.E.; Varela, J.; Lavaggi, M.L.; Merlino, A.; Cerecetto, H.; González, M.; Gomez-Barrio, A.; Escario, J.A.; et al. Novel Imidazo[4,5-c][1,2,6]thiadiazine 2,2-dioxides as antiproliferative Trypanosoma cruzi drugs: Computational screening from neural network, synthesis and in vivo biological properties. Eur. J. Med. Chem. 2017, 136, 223–234. [Google Scholar] [CrossRef]
- Sykes, M.L.; Avery, V.M. Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 215–228. [Google Scholar] [CrossRef]
- De Menezes, D.; da, R.; Calvet, C.M.; Rodrigues, G.C.; de Souza Pereira, M.C.; Almeida, I.R.; de Aguiar, A.P.; Supuran, C.T.; Vermelho, A.B. Hydroxamic acid derivatives: A promising scaffold for rational compound optimization in Chagas disease. J. Enzyme Inhib. Med. Chem. 2016, 31, 964–973. [Google Scholar] [CrossRef]
- Olmo, F.; Urbanová, K.; Rosales, M.J.; Martín-Escolano, R.; Sánchez-Moreno, M.; Marín, C. An in vitro iron superoxide dismutase inhibitor decreases the parasitemia levels of Trypanosoma cruzi in BALB/c mouse model during acute phase. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 110–116. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; O’Shea, I.P.; Wilkinson, S.R.; Kaiser, M. 3-Nitrotriazole-based piperazides as potent antitrypanosomal agents. Eur. J. Med. Chem. 2015, 103, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; O’Shea, I.P.; Wilkinson, S.R.; Kaiser, M.; Chatelain, E.; Ioset, J.R. Discovery of potent nitrotriazole-based antitrypanosomal agents: In vitro and in vivo evaluation. Bioorganic Med. Chem. 2015, 23, 6467–6476. [Google Scholar] [CrossRef] [Green Version]
- Neitz, R.J.; Bryant, C.; Chen, S.; Gut, J.; Hugo Caselli, E.; Ponce, S.; Chowdhury, S.; Xu, H.; Arkin, M.R.; Ellman, J.A.; et al. Tetrafluorophenoxymethyl ketone cruzain inhibitors with improved pharmacokinetic properties as therapeutic leads for Chagas’ disease. Bioorganic Med. Chem. Lett. 2015, 25, 4834–4837. [Google Scholar] [CrossRef]
- Sangenito, L.S.; d’Avila-Levy, C.M.; Branquinha, M.H.; Santos, A.L.S. Nelfinavir and lopinavir impair Trypanosoma cruzi trypomastigote infection in mammalian host cells and show anti-amastigote activity. Int. J. Antimicrob. Agents 2016, 48, 703–711. [Google Scholar] [CrossRef]
- Santos, G.B.; Krogh, R.; Magalhaes, L.G.; Andricopulo, A.D.; Pupo, M.T.; Emery, F.S. Semisynthesis of new aphidicolin derivatives with high activity against Trypanosoma cruzi. Bioorganic Med. Chem. Lett. 2016, 26, 1205–1208. [Google Scholar] [CrossRef]
- De Vita, D.; Moraca, F.; Zamperini, C.; Pandolfi, F.; Di Santo, R.; Matheeussen, A.; Maes, L.; Tortorella, S.; Scipione, L. In vitro screening of 2-(1H-imidazol-1-yl)-1-phenylethanol derivatives as antiprotozoal agents and docking studies on Trypanosoma cruzi CYP51. Eur. J. Med. Chem. 2016, 113, 28–33. [Google Scholar] [CrossRef]
- Eberle, C.; Burkhard, J.A.; Stump, B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Synthesis, inhibition potency, binding mode, and antiprotozoal activities of fluorescent inhibitors of trypanothione reductase based on mepacrine-conjugated diaryl sulfide scaffolds. ChemMedChem 2009, 4, 2034–2044. [Google Scholar] [CrossRef]
- Palma, A.; Yépes, A.F.; Leal, S.M.; Coronado, C.A.; Escobar, P. Synthesis and in vitro activity of new tetrahydronaphtho[1,2-b]azepine derivatives against Trypanosoma cruzi and Leishmania chagasi parasites. Bioorganic Med. Chem. Lett. 2009, 19, 2360–2363. [Google Scholar] [CrossRef]
- Herrera, C.; Vallejos, G.A.; Loaiza, R.; Zeledón, R.; Urbina, A.; Sepúlveda-Boza, S. In vitro activity of thienyl-2-nitropropene compounds against Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2009, 104, 980–985. [Google Scholar] [CrossRef]
- Rosso, V.S.; Szajnman, S.H.; Malayil, L.; Galizzi, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of new 2-alkylaminoethyl-1,1- bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorganic Med. Chem. 2011, 19, 2211–2217. [Google Scholar] [CrossRef]
- Eberle, C.; Lauber, B.S.; Fankhauser, D.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Improved Inhibitors of Trypanothione Reductase by Combination of Motifs: Synthesis, Inhibitory Potency, Binding Mode, and Antiprotozoal Activities. ChemMedChem 2011, 6, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Sealey-Cardona, M.; Cammerer, S.; Jones, S.; Ruiz-Pérez, L.M.; Brun, R.; Gilbert, I.H.; Urbina, J.A.; González-Pacanowska, D. Kinetic characterization of squalene synthase from Trypanosoma cruzi: Selective inhibition by quinuclidine derivatives. Antimicrob. Agents Chemother. 2007, 51, 2123–2129. [Google Scholar] [CrossRef]
- Franck, X.; Fournet, A.; Prina, E.; Mahieux, R.; Hocquemiller, R.; Figadère, B. Biological evaluation of substituted quinolines. Bioorganic Med. Chem. Lett. 2004, 14, 3635–3638. [Google Scholar] [CrossRef]
- Khabnadideh, S.; Pez, D.; Musso, A.; Brun, R.; Ruiz Pérez, L.M.; González-Pacanowska, D.; Gilbert, I.H. Design, synthesis and evaluation of 2,4-diaminoquinazolines as inhibitors of trypanosomal and leishmanial dihydrofolate reductase. Bioorganic Med. Chem. 2005, 13, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorganic Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Szajnman, S.H.; García Liñares, G.E.; Li, Z.H.; Jiang, C.; Galizzi, M.; Bontempi, E.J.; Ferella, M.; Moreno, S.N.J.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 2-alkylaminoethyl-1,1-bisphosphonic acids against Trypanosoma cruzi and Toxoplasma gondii targeting farnesyl diphosphate synthase. Bioorganic Med. Chem. 2008, 16, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Brun, R.; Kaiser, M.; Neumann, S. Synthesis and antiprotozoal activities of simplified analogs of naphthylisoquinoline alkaloids. Eur. J. Med. Chem. 2008, 43, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.C.; Escobar, P.; Leal, S.M.; Bahsas, A.; Cobo, J.; Nogueras, M.; Palma, A. Synthesis of novel polysubstituted (2SR,4RS)-2-heteroaryltetrahydro-1,4- epoxy-1-benzazepines and cis-2-heteroaryl-4-hydroxytetrahydro-1H-1-benzazepines as antiparasitic agents. Eur. J. Med. Chem. 2014, 86, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Olmo, F.; Clares, M.P.; Marín, C.; González, J.; Inclán, M.; Soriano, C.; Urbanová, K.; Tejero, R.; Rosales, M.J.; Krauth-Siegel, R.L.; et al. Synthetic single and double aza-scorpiand macrocycles act as inhibitors of the antioxidant enzymes iron superoxide dismutase and trypanothione reductase in Trypanosoma cruzi with promising results in a murine model. RSC Adv. 2014, 4, 65108–65120. [Google Scholar] [CrossRef]
- Braga, S.F.P.; Alves, É.V.P.; Ferreira, R.S.; Fradico, J.R.B.; Lage, P.S.; Duarte, M.C.; Ribeiro, T.G.; Júnior, P.A.S.; Romanha, A.J.; Tonini, M.L.; et al. Synthesis and evaluation of the antiparasitic activity of bis-(arylmethylidene) cycloalkanones. Eur. J. Med. Chem. 2014, 71, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Lepesheva, G.I.; Rosenzweig, H.S.; Kaiser, M.; Aguilera-Venegas, B.; Wilkinson, S.R.; Chatelain, E.; Ioset, J.R. Novel 3-nitrotriazole-based amides and carbinols as bifunctional antichagasic agents. J. Med. Chem. 2015, 58, 1307–1319. [Google Scholar] [CrossRef]
- Keenan, M.; Alexander, P.W.; Diao, H.; Best, W.M.; Khong, A.; Kerfoot, M.; Thompson, R.C.A.; White, K.L.; Shackleford, D.M.; Ryan, E.; et al. Design, structure-activity relationship and in vivo efficacy of piperazine analogues of fenarimol as inhibitors of Trypanosoma cruzi. Bioorganic Med. Chem. 2013, 21, 1756–1763. [Google Scholar] [CrossRef]
- Carvalho, S.A.; Feitosa, L.O.; Soares, M.; Costa, T.E.M.M.; Henriques, M.G.; Salomão, K.; De Castro, S.L.; Kaiser, M.; Brun, R.; Wardell, J.L.; et al. Design and synthesis of new (E)-cinnamic N-acylhydrazones as potent antitrypanosomal agents. Eur. J. Med. Chem. 2012, 54, 512–521. [Google Scholar] [CrossRef]
- Galiana-Roselló, C.; Bilbao-Ramos, P.; Dea-Ayuela, M.A.; Rolón, M.; Vega, C.; Bolás-Fernández, F.; García-España, E.; Alfonso, J.; Coronel, C.; González-Rosende, M.E. In vitro and in vivo antileishmanial and trypanocidal studies of new N-benzene- and N-naphthalenesulfonamide derivatives. J. Med. Chem. 2013, 56, 8984–8998. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Dixit, S.S.; Földesi, A.; Chattopadhyaya, J. New antiprotozoal agents: Their synthesis and biological evaluations. Bioorganic Med. Chem. Lett. 2013, 23, 2750–2758. [Google Scholar] [CrossRef] [PubMed]
- Silva-Júnior, E.F.; Silva, E.P.S.; França, P.H.B.; Silva, J.P.N.; Barreto, E.O.; Silva, E.B.; Ferreira, R.S.; Gatto, C.C.; Moreira, D.R.M.; Siqueira-Neto, J.L.; et al. Design, synthesis, molecular docking and biological evaluation of thiophen-2-iminothiazolidine derivatives for use against Trypanosoma cruzi. Bioorganic Med. Chem. 2016, 24, 4228–4240. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Canvas Schrödinger Release 2016-3. Available online: https://www.schrodinger.com/canvas (accessed on 9 January 2019).
- SYBYL-X. Available online: https://www.certara.com/pressreleases/certara-enhances-sybyl-x-drug-design-and-discovery-software-suite/ (accessed on 9 January 2019).
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA Data Mining Software: An Update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Cybenko, G. Approximation by Superpositions of a Sigmoidal Function. Math. Control. Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- An, Y.; Sherman, W.; Dixon, S.L. Kernel-based partial least squares: Application to fingerprint-based QSAR with model visualization. J. Chem. Inf. Model. 2013, 53, 2312–2321. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set | Test Set | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LR | Score | r² | MAE | RMSE | RAE | RRSE | q² | MAE | RMSE | RAE | RRSE |
| 0.1 | 0.80 | 0.79 | 0.65 | 0.82 | 65 | 68 | 0.85 | 0.6 | 0.75 | 59 | 61 |
| 0.2 | 0.76 | 0.80 | 0.58 | 0.76 | 59 | 64 | 0.78 | 0.66 | 0.84 | 65 | 68 |
| 0.3 | 0.77 | 0.79 | 0.60 | 0.77 | 60 | 65 | 0.78 | 0.67 | 0.89 | 66 | 72 |
| 0.4 | 0.75 | 0.80 | 0.58 | 0.77 | 59 | 64 | 0.77 | 0.69 | 0.94 | 68 | 76 |
| Training Set | Test Set | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MP | Score | r² | MAE | RMSE | RAE | RRSE | q² | MAE | RMSE | RAE | RRSE |
| 0.1 | 0.79 | 0.78 | 0.64 | 0.81 | 64 | 68 | 0.84 | 0.58 | 0.73 | 57 | 59 |
| 0.2 | 0.80 | 0.79 | 0.65 | 0.82 | 65 | 68 | 0.85 | 0.6 | 0.75 | 59 | 61 |
| 0.3 | 0.80 | 0.79 | 0.66 | 0.83 | 66 | 70 | 0.85 | 0.62 | 0.77 | 61 | 63 |
| 0.4 | 0.80 | 0.79 | 0.67 | 0.84 | 67 | 71 | 0.85 | 0.62 | 0.78 | 62 | 63 |
| Training Set | Test Set | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NN | Score | r² | MAE | RMSE | RAE | RRSE | q² | MAE | RMSE | RAE | RRSE |
| 1 | - | 0.51 | 0.86 | 1.03 | 87 | 68 | - | - | - | - | - |
| 2 | - | 0.72 | 0.69 | 0.85 | 69 | 72 | - | - | - | - | - |
| 3 | - | 0.75 | 0.69 | 0.86 | 69 | 72 | - | - | - | - | - |
| 4 | - | 0.77 | 0.68 | 0.84 | 68 | 70 | - | - | - | - | - |
| 5 | 0.78 | 0.78 | 0.64 | 0.81 | 65 | 68 | 0.80 | 0.64 | 0.82 | 63 | 66 |
| 6# | 0.80 | 0.79 | 0.65 | 0.82 | 65 | 68 | 0.85 | 0.60 | 0.75 | 59 | 61 |
| 7 | 0.81 | 0.81 | 0.57 | 0.73 | 56 | 62 | 0.81 | 0.59 | 0.76 | 58 | 62 |
| 8 | 0.76 | 0.79 | 0.68 | 0.85 | 68 | 71 | 0.77 | 0.69 | 0.90 | 68 | 73 |
| 9 | 0.80 | 0.80 | 0.65 | 0.82 | 65 | 68 | 0.82 | 0.64 | 0.81 | 63 | 66 |
| 10 | 0.77 | 0.82 | 0.58 | 0.75 | 58 | 63 | 0.79 | 0.62 | 0.83 | 61 | 67 |
| Neuron | MW | aLogP | HBA | HBD | RB | HAC | RC | PSA | E-state | MR | Polar |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.51 | 1.94 | 0.90 | 3.34 | 0.73 | 0.46 | −0.27 | −0.28 | −0.34 | 0.99 | 0.59 |
| 2 | 2.77 | −3.96 | −2.61 | −0.11 | −0.23 | 2.98 | −2.21 | −2.82 | 1.04 | 2.39 | 3.68 |
| 3 | −1.42 | 1.79 | 0.49 | 2.12 | −0.33 | −0.20 | 2.44 | 1.55 | −0.28 | −1.07 | −0.60 |
| 4 | 0.59 | 0.06 | 0.07 | 1.05 | 0.14 | 0.29 | 0.44 | 0.31 | 0.25 | 0.06 | 0.76 |
| 5 | 1.39 | −4.91 | −6.71 | −0.28 | −0.79 | 3.33 | 4.58 | 1.08 | −0.20 | 4.85 | 5.49 |
| 6 | 0.55 | 2.75 | 0.94 | 3.22 | −2.00 | −0.55 | 0.66 | −5.24 | −3.30 | 1.49 | −0.76 |
| 7 | 1.60 | 3.78 | 5.40 | 2.38 | −2.72 | −2.20 | −2.52 | −0.14 | 0.92 | −1.10 | −2.80 |
| Fingerprint | Score | q2 | r2 | RMSE | SD | N |
|---|---|---|---|---|---|---|
| Dendritic | 0.76 | 0.82 | 0.89 | 0.40 | 0.53 | 3 |
| Linear | 0.78 | 0.83 | 0.89 | 0.41 | 0.51 | 3 |
| Radial | 0.80 | 0.81 | 0.80 | 0.54 | 0.54 | 2 |
| Molprint2D | 0.82 | 0.84 | 0.81 | 0.52 | 0.50 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, A.S.; Ferreira, L.L.G.; de Oliveira, A.S.; Andricopulo, A.D. Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity. Int. J. Mol. Sci. 2019, 20, 2801. https://doi.org/10.3390/ijms20112801
de Souza AS, Ferreira LLG, de Oliveira AS, Andricopulo AD. Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity. International Journal of Molecular Sciences. 2019; 20(11):2801. https://doi.org/10.3390/ijms20112801
Chicago/Turabian Stylede Souza, Anacleto S., Leonardo L. G. Ferreira, Aldo S. de Oliveira, and Adriano D. Andricopulo. 2019. "Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity" International Journal of Molecular Sciences 20, no. 11: 2801. https://doi.org/10.3390/ijms20112801
APA Stylede Souza, A. S., Ferreira, L. L. G., de Oliveira, A. S., & Andricopulo, A. D. (2019). Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity. International Journal of Molecular Sciences, 20(11), 2801. https://doi.org/10.3390/ijms20112801