Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae

Abstract
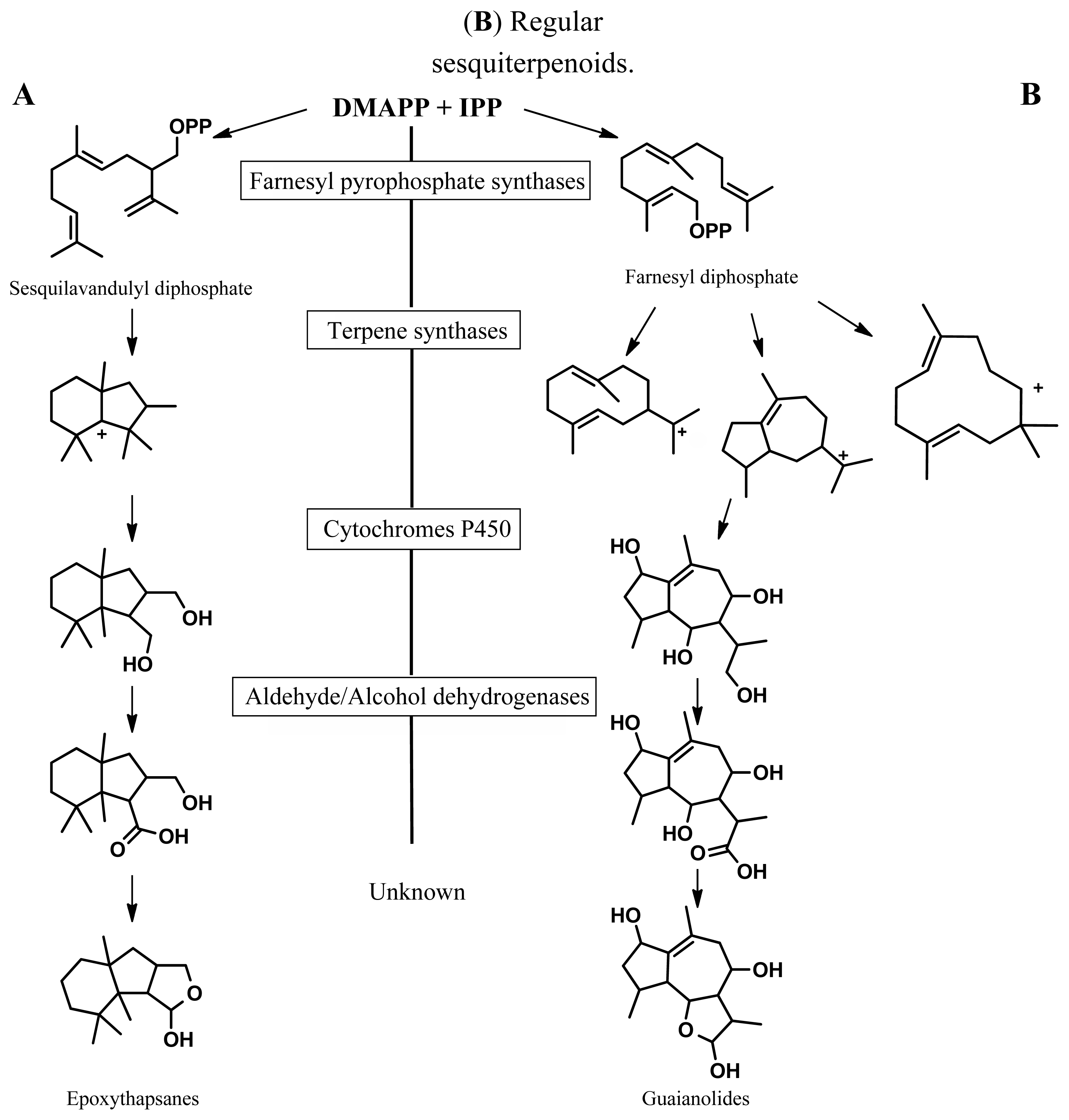
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Sequencing, de novo Assembly and General Metabolism
2.2. Candidate Genes for Farnesyl Diphosphate Synthase
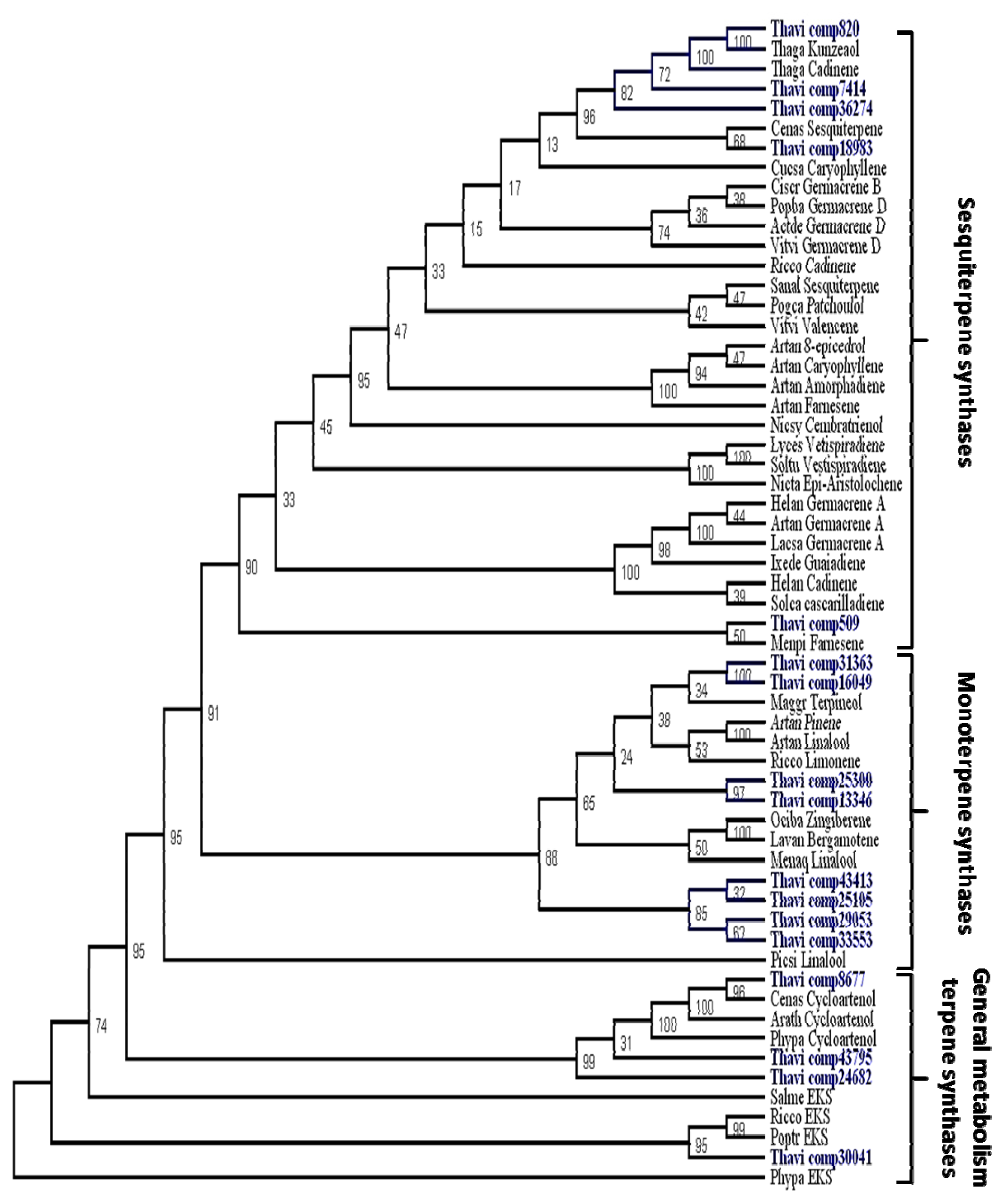
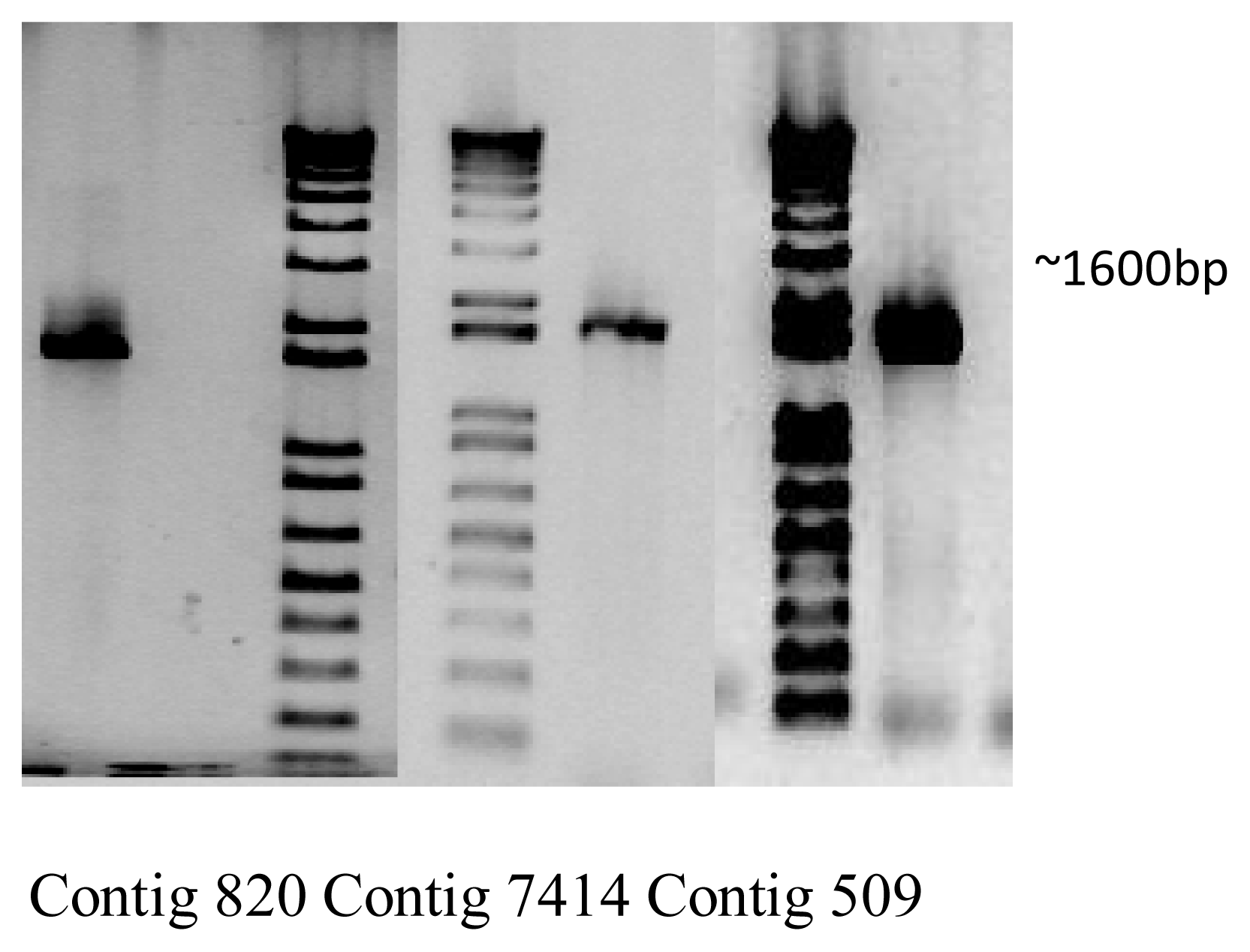
2.3. Candidate Genes for Terpene Synthases
2.4. Candidate Genes for Cytochromes P450 of the CYP71 Superfamily
2.5. Candidate Genes for Aldehyde Dehydrogenases Involved in Sesquiterpenoid Biosynthesis
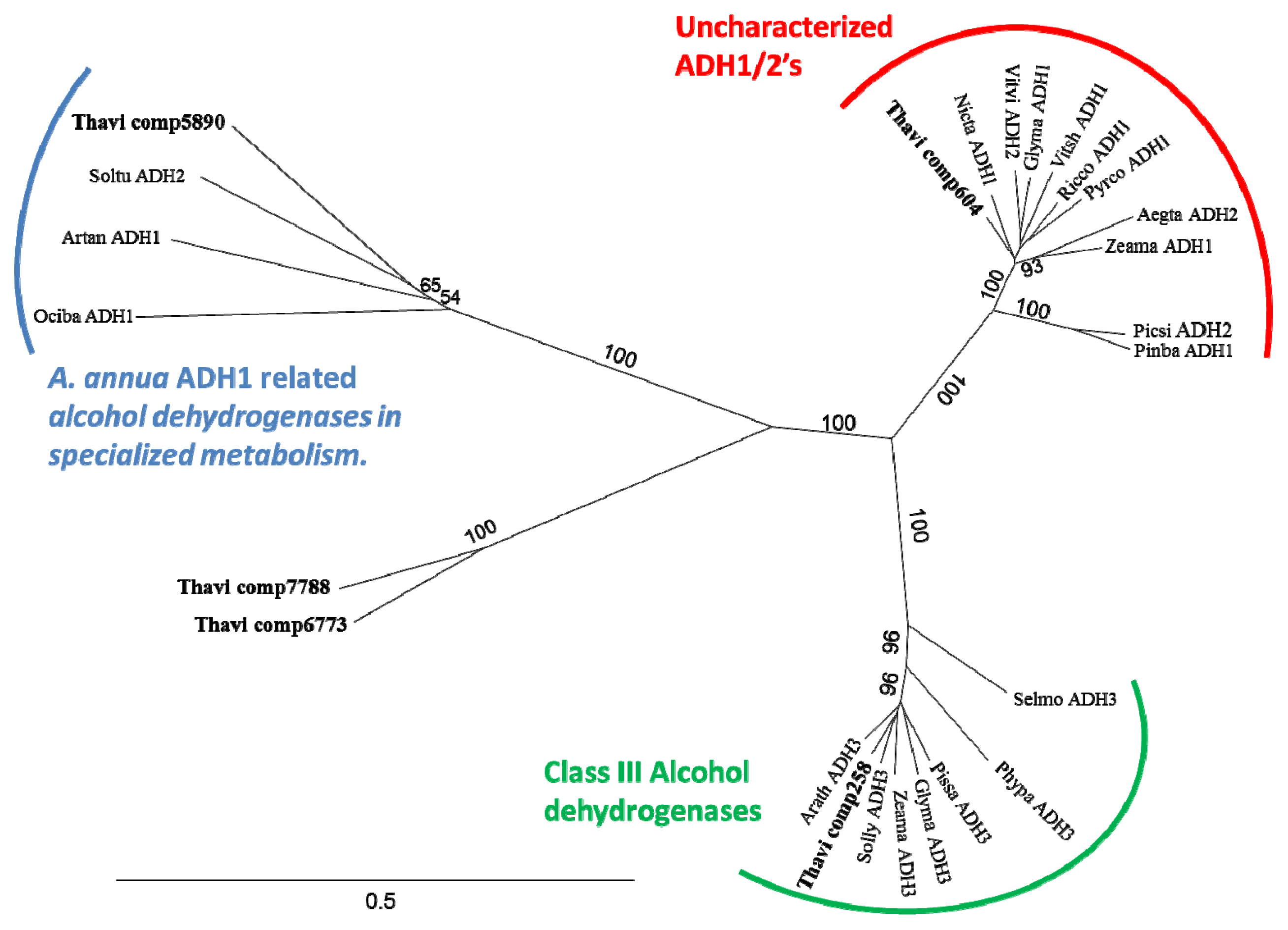
2.6. Candidate Genes for Alcohol Dehydrogenases Involved in Sesquiterpenoid Biosynthesis
3. Experimental Section
3.1. Plant Material and RNA Extraction
3.2. cDNA Library Construction and Sequencing
3.3. Sequence Analysis and Assembly
3.4. Mapping, Functional Annotation and Pathway Assignments
3.5. Alignment and Tree Building
3.6. Amplification and Sequencing of Representative Full-Length Terpene Synthases
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Simonsen, H.T.; Weitzel, C.; Christensen, S.B. Guaianolide Sesquiterpenoids—Their Pharmacology and Biosynthesis. In Handbook of Natural Products; Ramawat, K.G., Merillon, J.M., Eds.; Springer-Verlag: Berlin, Germany, 2013; Volume 134, in press. [Google Scholar]
- Hegnauer, R. Chemotaxonomie der Pflanzen; Birkhiuser Verlag: Stuttgart, Germany, 1973; Volume 6. [Google Scholar]
- Holub, M.; Budesinsky, M. On Terpenes 292. Sesquiterpene lactones of the umbelliferae. Phytochemistry 1986, 25, 2015–2026. [Google Scholar]
- Drew, D.P.; Rasmussen, S.K.; Avato, P.; Simonsen, H.T. A Comparison of headspace solid-phase microextraction and classic hydrodistillation for the identification of volatile constituents from Thapsia spp. Provides Insights into guaianolide biosynthesis in Apiaceae. Phytochem. Anal 2012, 23, 44–51. [Google Scholar]
- Drew, D.P.; Krichau, N.; Reichwald, K.; Simonsen, H.T. Guaianolides in apiaceae: Perspectives on pharmacology and biosynthesis. Phytochem. Rev 2009, 8, 581–599. [Google Scholar]
- Christensen, S.B.; Andersen, A.; Smitt, U.W. Sesquiterpenoids from Thapsia species and medicinal chemistry of the thapsigargins. Fortschr. Chem. Org. Naturst 1997, 71, 129–167. [Google Scholar]
- Weitzel, C.; Rønsted, N.; Simonsen, H.T. Resurrecting deadly carrots. Towards a revision of ThapsiaL. (Apiaceae) based on phylogenetic analysis of nrITS sequences and chemical profiles. Bot. J. Linn. Soc. 2013. in print. [Google Scholar]
- Cool, L.G.; Vermillion, K.E.; Takeoka, G.R.; Wong, R.Y. Irregular sesquiterpenoids from Ligusticum grayi roots. Phytochemistry 2010, 71, 1545–1557. [Google Scholar]
- Bouvier, F.; Rahier, A.; Camara, B. Biogenesis, molecular regulation and function of plant isoprenoids. Prog. Lipid Res 2005, 44, 357–429. [Google Scholar]
- Thulasiram, H.V.; Erickson, H.K.; Poulter, C.D. A Common mechanism for branching, cyclopropanation, and cyclobutanation reactions in the isoprenoid biosynthetic pathway. J. Am. Chem. Soc 2008, 130, 1966–1971. [Google Scholar]
- Tang, L.; Li, J.; Khalil, R.; Yang, Y.; Fan, J.; Liu, M.; Li, Z. Cloning and functional analysis of CDS_CCI2: A Tanacetum cinerariaefolium chrysanthemyl diphosphate synthase gene. Plant Growth Regul 2012, 67, 161–169. [Google Scholar]
- Genspera Genspera, Inc. Available online: http://www.genspera.com (accessed on 1 January 2012).
- Rubal, J.J.; Moreno-Dorado, F.J.; Guerra, F.M.; Jorge, Z.D.; Saouf, A.; Akssira, M.; Mellouki, F.; Romero-Garrido, R.; Massanet, G.M. A pyran-2-one and four meroterpenoids from Thapsia transtagana and their implication in the biosynthesis of transtaganolides. Phytochemistry 2007, 68, 2480–2486. [Google Scholar]
- Pickel, B.; Drew, D.P.; Manczak, T.; Weitzel, C.; Simonsen, H.T.; Ro, D.-K. Molecular characterization of kunzeaol synthase from Thapsia garganica and its implication in Thapsigargin biosynthesis. Biochem. J 2012, 448, 261–271. [Google Scholar]
- Yu, R.; Wen, W. Artemisinin biosynthesis and its regulatory enzymes: Progress and perspective. Pharmacogn. Rev 2011, 5, 189–194. [Google Scholar]
- Ikezawa, N.; Göpfert, J.C.; Nguyen, D.T.; Kim, S.-U.; O’Maille, P.E.; Spring, O.; Ro, D.-K. Lettuce costunolide synthase (CYP71BL2) and its homolog (CYP71BL1) from sunflower catalyze distinct regio- and stereoselective hydroxylations in sesquiterpene lactone metabolism. J. Biol. Chem 2011, 286, 21601–21611. [Google Scholar]
- Tutin, T.G.; Heywood, V.H.; Burges, N.A.; Moore, D.M.; Valentine, D.H.; Walters, S.M.; Webb, D.A. Flora Europaea; Cambridge University Press: Cambridge, UK, 1986; Volume 2. [Google Scholar]
- Smitt, U.W.; Jäger, A.K.; Adsersen, A.; Gudiksen, L. Comparative studies in phytochemistry and fruit anatomy of Thapsia garganica and T.transtagana, Apiaceae (Umbelliferae). Bot. J. Linn. Soc 1995, 117, 281–292. [Google Scholar]
- Rouy, G. Illustrationes Plantarum Europae Rariarum; Fascicule I: Paris, France, 1895. [Google Scholar]
- Avato, P.; Trabace, G.; Smitt, U.W. Essential oils from fruits of three types of Thapsia villosa. Phytochemistry 1996, 43, 609–612. [Google Scholar]
- Acquadro, A.; Magurno, F.; Portis, E.; Lanteri, S. dbEST-derived microsatellite markers in celery (Apium graveolens L. var. dulce). Mol. Ecol. Notes 2006, 6, 1080–1082. [Google Scholar]
- Ruhlman, T.; Lee, S.-B.; Jansen, R.; Hostetler, J.; Tallon, L.; Town, C.; Daniell, H. Complete plastid genome sequence of Daucus carota: Implications for biotechnology and phylogeny of angiosperms. BMC Genomics 2006, 7, 222. [Google Scholar]
- Cavagnaro, P.; Chung, S.-M.; Szklarczyk, M.; Grzebelus, D.; Senalik, D.; Atkins, A.; Simon, P. Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol. Genet. Genomics 2009, 281, 273–288. [Google Scholar]
- Iorizzo, M.; Senalik, D.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.; Matvienko, M.; Ashrafi, H.; Van Deynze, A.; Simon, P. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 2011, 12, 389. [Google Scholar]
- Sui, C.; Zhang, J.; Wei, J.; Chen, S.; Li, Y.; Xu, J.; Jin, Y.; Xie, C.; Gao, Z.; Chen, H.; et al. Transcriptome analysis of Bupleurum chinense focusing on genes involved in the biosynthesis of saikosaponins. BMC Genomics 2011, 12, 539. [Google Scholar]
- Nguyen, D.T.; Göpfert, J.C.; Ikezawa, N.; MacNevin, G.; Kathiresan, M.; Conrad, J.; Spring, O.; Ro, D.-K. Biochemical conservation and evolution of germacrene a oxidase in asteraceae. J. Biol. Chem 2010, 285, 16588–16598. [Google Scholar]
- Ro, D.K.; Paradise, E.M.; Quellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar]
- De Kraker, J.W.; Franssen, M.C.R.; Joerink, M.; de Groot, A.; Bouwmeester, H.J. Biosynthesis of costunolide, dihydrocostunolide, and leucodin. Demonstration of cytochrome P450-catalyzed formation of the lactone ring present in sesquiterpene lactones of chicory. Plant Physiol 2002, 129, 257–268. [Google Scholar]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar]
- Zerbino, D.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008, 18, 821–829. [Google Scholar]
- Gaasterland, T.; Sensen, C.W. Fully automated genome analysis that reflects user needs and preferences. A detailed introduction to the MAGPIE system architecture. Biochimie 1996, 78, 302–310. [Google Scholar]
- Hemmerlin, A.; Hoeffler, J.F.; Meyer, O.; Tritsch, D.; Kagan, I.A.; Grosdemange-Billiard, C.; Rohmer, M.; Bach, T.J. Cross-talk between the cytosolic mevalonate and the plastidial methylerythritol phosphate pathways in tobacco bright yellow-2 cells. J. Biol. Chem 2003, 278, 26666–26676. [Google Scholar]
- Tholl, D.; Lee, S. Terpene specialized metabolism in Arabidopsis thaliana. Arabidopsis Book 2011, 9, e0143. [Google Scholar]
- Hamberger, B. Personal Communication, University of Copenhagen: Frederiksberg 1871, Denmark, 2012.
- Bak, S.; Beisson, F.; Bishop, G.; Hamberger, B.; Höfer, R.; Paquette, S.; Werck-Reichhart, D. Cytochromes P450. Arabidopsis Book 2011, 9, e0144. [Google Scholar]
- Nelson, D.; Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J 2011, 66, 194–211. [Google Scholar]
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Covello, P.S. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany 2009, 87, 635–642. [Google Scholar]
- Kotchoni, S.O.; Jimenez-Lopez, J.C.; Kayodé, A.P.P.; Gachomo, E.W.; Baba-Moussa, L. The soybean aldehyde dehydrogenase (ALDH) protein superfamily. Gene 2012, 495, 128–133. [Google Scholar]
- Polichuk, D.R.; Zhang, Y.; Reed, D.W.; Schmidt, J.F.; Covello, P.S. A glandular trichome-specific monoterpene alcohol dehydrogenase from Artemisia annua. Phytochemistry 2010, 71, 1264–1269. [Google Scholar]
- Polichuk, D.; Teoh, K.H.; Zhang, Y.; Ellens, K.W.; Reed, D.W.; Covello, P.S. Nucleotide Sequence Encoding an Alcohol Dehydrogenase from Artemisia annuaand uses thereof. U.S. Patent 20110162097, 2010. [Google Scholar]
- Meisel, L.; Fonseca, B.; Gonzalez, S.; Baeza-Yates, R.; Cambiazo, V.; Campos, R.; Gonzalez, M.; Orellana, A.; Retamales, J.; Silva, H. A rapid and efficient method for purifying high quality total RNA from peaches (Prunus persica) for functional genomics analyses. Biol. Res 2005, 38, 83–88. [Google Scholar]
- Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 December 2011).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Bioinforma. Action 2011, 17, 10–12. [Google Scholar]
- Eddy, S.R. Multiple Alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol 1995, 3, 114–120. [Google Scholar]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 2007, 35, W182–W185. [Google Scholar]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999, 27, 29–34. [Google Scholar]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol 2003, 52, 696–704. [Google Scholar]
- Nørholm, M.H. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol 2010, 10, 21. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCA cycle enzyme | Arabidopsis query | Contig (identity) | Reads |
|---|---|---|---|
| Citrate synthase | NP_850415 | Comp7488 (84%) | 12696 |
| Aconitase | NP_567763 | Comp1618 (85%) | 46037 |
| Isocitrate dehydrogenase | NP_175836 | Comp214 (85%) | 194838 |
| 2-oxoglutarate dehydrogenase | NP_191101 | Comp359 (85%) | 162552 |
| Dihydrolipoyllysine succinyltransferase | NP_200318 | Comp2028 (71%) | 35422 |
| Dihydrolipoyl dehydrogenase | NP_567487 | Comp12031 (84%) | 7805 |
| Succinyl-CoA synthetase | NP_001119263 | Comp3388 (90%) | 28993 |
| Succinate dehydrogenase flavoprotein subunit | NP_201477 | Comp2812 (94%) | 29604 |
| Succinate dehydrogenase iron-sulfur subnunit | NP_001118718 | Comp3447 (76%) | 21231 |
| Fumarase | NP_001078075 | Comp8438 (87%) | 13149 |
| Malate dehydrogenase | NP_190336 | Comp4342 (77%) | 19283 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Drew, D.P.; Dueholm, B.; Weitzel, C.; Zhang, Y.; Sensen, C.W.; Simonsen, H.T. Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae. Int. J. Mol. Sci. 2013, 14, 9080-9098. https://doi.org/10.3390/ijms14059080
Drew DP, Dueholm B, Weitzel C, Zhang Y, Sensen CW, Simonsen HT. Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae. International Journal of Molecular Sciences. 2013; 14(5):9080-9098. https://doi.org/10.3390/ijms14059080
Chicago/Turabian StyleDrew, Damian Paul, Bjørn Dueholm, Corinna Weitzel, Ye Zhang, Christoph W. Sensen, and Henrik Toft Simonsen. 2013. "Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae" International Journal of Molecular Sciences 14, no. 5: 9080-9098. https://doi.org/10.3390/ijms14059080
APA StyleDrew, D. P., Dueholm, B., Weitzel, C., Zhang, Y., Sensen, C. W., & Simonsen, H. T. (2013). Transcriptome Analysis of Thapsia laciniata Rouy Provides Insights into Terpenoid Biosynthesis and Diversity in Apiaceae. International Journal of Molecular Sciences, 14(5), 9080-9098. https://doi.org/10.3390/ijms14059080