Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection

 , , , , ,
, , , , ,
Abstract
:1. Introduction
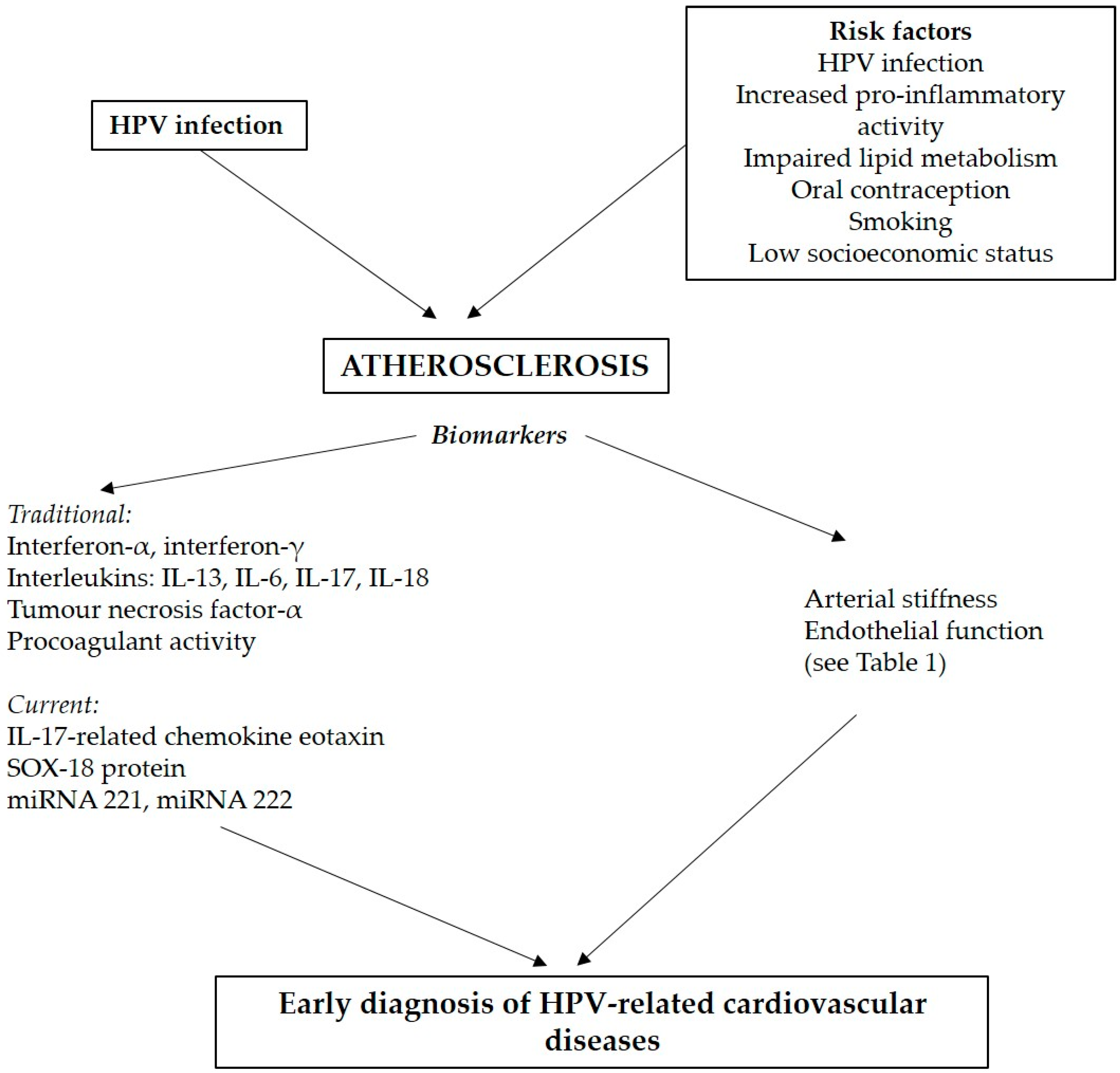
2. Cardiovascular Disease Associated with HPV Infection
2.1. Current Epidemiological Evidence
2.2. Pathophysiological Mechanisms
2.2.1. Infection-Induced Atherosclerosis
2.2.2. HPV as a Candidate Factor for Atypical Atherosclerosis
2.3. Other Risk Factors Associated with CVD and Carcinogenesis
2.4. Current Concepts of Atherosclerotic Mechanisms
3. Methods for Personalised Prediction of Cardiovascular Risk
3.1. Evaluation of Early Atherosclerotic Alterations
3.1.1. Endothelial Function
3.1.2. Arterial Stiffness
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HPV | human papillomavirus |
| CVD | cardiovascular diseases |
| HR-HPV | high-risk strains of human papillomavirus |
| DNA | deoxyribonucleic acid |
| BP | blood pressure |
| LDL | low-density lipoprotein |
| HDL | high-density lipoprotein |
| HIV | human immunodeficiency virus |
| EBV | Epstein-Barr virus |
| IMT | intima-media thickness |
| MetS | metabolic syndrome |
| SOX18 | SRY-related HMG–box 18 |
| miRNAs | micro RNAs |
| NO | nitric oxide |
| FMD | flow-mediated dilation in brachial artery |
| PAT | peripheral artery tonometry |
| PWV | pulse wave velocity |
| CAVI | cardio-ankle vascular index |
| CAVI0 | cardio-ankle vascular stiffness index 0 |
| CBP | central blood pressure |
| Ps | systolic blood pressure |
| Pd | diastolic blood pressure |
| a,b | CAVI constants |
| ρ | blood mass density |
| Pref | reference pressure |
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.X.; Lorincz, A.; Muñoz, N.; Meijer, C.J.L.M.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudela, E.; Holubekova, V.; Farkasova, A.; Danko, J. Determination of malignant potential of cervical intraepithelial neoplasia. Tumor. Biol. 2016, 37, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Baseman, J.G.; Koutsky, L.A. The epidemiology of human papillomavirus infections. J. Clin. Virol. 2005, 32, 16–24. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, H.; Clifford, G.; Li, N.; Franceschi, S. HPV infection in Europe. Eur. J. Cancer 2009, 45, 2632–2639. [Google Scholar] [CrossRef] [PubMed]
- Tachezy, R.; Smahelova, J.; Kaspirkova, J.; Salakova, M. Human Papillomavirus Type-Specific Prevalence in the Cervical Cancer Screening Population of Czech Women. PLoS ONE 2013, 8, e79156. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; de Sanjose, S. Chapter 1: Human Papillomavirus and Cervical Cancer--Burden and Assessment of Causality. JNCI Monogr. 2003, 2003, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondrusova, M.; Zubor, P.; Ondrus, D. Time trends in cervical cancer epidemiology in the Slovak Republic: Reflection on the non-implementation of screening with international comparisons. Neoplasma 2012, 59, 121–128. [Google Scholar] [CrossRef]
- Pfaendler, K.S.; Wenzel, L.; Mechanic, M.B.; Penner, K.R. Cervical cancer survivorship: Long-term quality of life and social support. Clin. Ther. 2015, 37, 39–48. [Google Scholar] [CrossRef]
- Kuo, H.K.; Fujise, K. Human papillomavirus and cardiovascular disease among U.S. women in the national health and nutrition examination survey, 2003 to 2006. J. Am. Coll. Cardiol. 2011, 58, 2001–2006. [Google Scholar] [CrossRef]
- Joo, E.J.; Kim, J.; Park, S.Y.; Cheong, H.S.; Chang, Y.; Seungho, R. High-risk Human Papillomavirus Infection and the Risk of Cardiovascular Disease: A Cohort Study. Presented at infectious diseases week, San Francisco, CA, USA, 3–7 October 2018. Abstract 2509. [Google Scholar]
- Lawson, J.S.; Glenn, W.K.; Tran, D.D.; Ngan, C.C.; Duflou, J.A.; Whitaker, N.J. Identification of Human Papilloma Viruses in Atheromatous Coronary Artery Disease. Front. Cardiovasc. Med. 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.K.; Fujise, K. Reply. J. Am. Coll. Cardiol. 2012, 60, 82–83. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.J.; Huang, Y.S.; Tung, C.H.; Lee, C.C.; Lee, M.S.; Chiou, W.Y.; Lin, H.Y.; Hsu, F.C.; Tsai, C.H.; Su, Y.C.; et al. Increased risk of ischemic stroke in cervical cancer patients: A nationwide population-based study. Radiat. Oncol. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, G.D.; Britt, R.H.; Goffinet, D.R. Radiation-induced carotid artery disease. Cancer 1978, 41, 130–137. [Google Scholar] [CrossRef]
- Addison, D.; Seidelmann, S.B.; Janjua, S.A.; Emami, H.; Staziaki, P.V.; Hallett, T.R.; Szilveszter, B.; Lu, M.T.; Cambria, R.P.; Hoffmann, U.; et al. Human Papillomavirus Status and the Risk of Cerebrovascular Events Following Radiation Therapy for Head and Neck Cancer. J. Am. Heart Assoc. 2017, 6, e006453. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Gupta, N.; Singhal, S.; Nair, N. Carcinoma Cervix Presenting as Ischaemic Stroke in Young Female: A Case Report and Review of Literature. J. Clin. Diagn. Res. 2017, 11, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Khot, U.N.; Khot, M.B.; Bajzer, C.T.; Sapp, S.K.; Ohman, E.M.; Brener, S.J.; Ellis, S.G.; Lincoff, A.M.; Topol, E.J. Prevalence of Conventional Risk Factors in Patients with Coronary Heart Disease. JAMA 2003, 290, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Muhlestein, J.B. Chronic infection and coronary atherosclerosis: Will the hypothesis ever really pan out? J. Am. Coll. Cardiol. 2011, 58, 2007–2009. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, atherosclerosis, and coronary heart disease. Eur. Heart J. 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Lawson, J.S. Multiple Infectious Agents and the Origins of Atherosclerotic Coronary Artery Disease. Front. Cardiovasc. Med. 2016, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, R.; Pietro, M.D.; Filardo, S.; Turriziani, O. Infectious burden and atherosclerosis: A clinical issue. World J. Clin. Cases 2014, 2, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Fabricant, C.G.; Fabricant, J. Atherosclerosis induced by infection with Marek’s disease herpesvirus in chickens. Am. Heart J. 1999, 138, S465–S468. [Google Scholar] [CrossRef]
- Madan, M.; Bishayi, B.; Hoge, M.; Messas, E.; Amar, S. Doxycycline affects diet- and bacteria-associated atherosclerosis in an ApoE heterozygote murine model: Cytokine profiling implications. Atherosclerosis 2007, 190, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Prétet, J.L.; Mercier, M.; Riethmuller, D.; Aubin, F.; Vuitton, D.; Mougin, C. Human papillomavirus and cardiovascular disease. J. Am. Coll. Cardiol. 2012, 60, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Viereck, J.; Hua, N.; Phinikaridou, A.; Madrigal, A.G.; Gibson, F.C.; Hamilton, J.A.; Genco, C.A. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 2011, 215, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Stover, C.M.; Dupont, A.P. gingivalis in Periodontal Disease and Atherosclerosis—Scenes of Action for Antimicrobial Peptides and Complement. Front. Immunol. 2015, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, W.; Delvenne, P.; Arrese, J.E.; Nikkels, A.F.; Piérard, G.E.; Boniver, J. Inverse modulation of intraepithelial Langerhans’ cells and stromal macrophage/dendrocyte populations in human papillomavirus-associated squamous intraepithelial lesions of the cervix. Virchows Arch. 1995, 427, 41–48. [Google Scholar] [CrossRef]
- Tay, S.K.; Jenkins, D.; Maddox, P.; Hogg, N.; Singer, A. Tissue macrophage response in human papillomavirus infection and cervical intraepithelial neoplasia. Br. J. Obstet. Gynaecol. 1987, 94, 1094–1097. [Google Scholar] [CrossRef]
- Bonnez, W.; Reichman, R.C.; Strussenberg, J.; Roberts, N.J. In vitro interactions between bovine papillomavirus and human monocytes and macrophages. Intervirology 1991, 32, 246–252. [Google Scholar] [CrossRef]
- Bonin, L.R.; Madden, K.; Shera, K.; Ihle, J.; Matthews, C.; Aziz, S.; Perez-Reyes, N.; McDougall, J.K.; Conroy, S.C. Generation and characterization of human smooth muscle cell lines derived from atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 575–587. [Google Scholar] [CrossRef]
- Mohammadi, A.; Pfeifer, J.D.; Lewis, J.S. Association between human papillomavirus DNA and temporal arteritis. BMC. Musculoskelet. Disord. 2012, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Füle, T.; Máthé, M.; Suba, Z.; Csapó, Z.; Szarvas, T.; Tátrai, P.; Paku, S.; Kovalszky, I. The presence of human papillomavirus 16 in neural structures and vascular endothelial cells. Virology 2006, 348, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.; Bennett, M. The role of p53 in atherosclerosis. Cell Cycle 2006, 5, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.; Gulati, N.; Shetty, D.; Jain, A. To analyze the concomitant expression of human papillomavirus-16 in the pathogenetic model of p53-dependant pathway in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2016, 20, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Merino, J.; Massimi, P.; Lizano, M.; Banks, L. The human papillomavirus (HPV) E6 oncoproteins promotes nuclear localization of active caspase 8. Virology 2014, 450, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Louw, L.; Seedat, R.; Claassen, A. HPV-induced recurrent laryngeal papillomatosis: Fatty acid role-players. Asia. Pac. J. Clin. Nutr. 2008, 17 (Suppl. 1), 208–211. [Google Scholar] [PubMed]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; Feingold, K.R., Ed.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Tania, M.; Khan, M.A.; Song, Y. Association of lipid metabolism with ovarian cancer. Curr. Oncol. 2010, 17, 6–11. [Google Scholar] [CrossRef]
- Gebrie, A.; Gnanasekaran, N.; Menon, M.; Sisay, M.; Zegeye, A. Evaluation of lipid profiles and hematological parameters in hypertensive patients: Laboratory-based cross-sectional study. SAGE Open. Med. 2018, 6, 1–11. [Google Scholar] [CrossRef]
- Zicha, J.; Kunes, J.; Devynck, M.A. Abnormalities of membrane function and lipid metabolism in hypertension: A review. Am. J. Hypertens. 1999, 12, 315–331. [Google Scholar] [CrossRef]
- Brito, L.M.O.; Brito, H.O.; Corrêa, R.G.C.F.; Neto, C.P.O.; Costa, J.P.L.; Monteiro, S.C.M.; Vidal, F.C.B.; Nascimento, M.D.S.B.; Neto, J.A.F.; Costa, R.M.G.; et al. Human Papillomavirus and Coronary Artery Disease in Climacteric Women: Is There an Association? Sci. World. J. 2019, 2019, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björnson, E.; Borén, J.; Mardinoglu, A. Personalized Cardiovascular Disease Prediction and Treatment-A Review of Existing Strategies and Novel Systems Medicine Tools. Front. Physiol. 2016, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Reckelhoff, J.F. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassuk, S.S.; Manson, J.E. Oral contraceptives and menopausal hormone therapy: Relative and attributable risks of cardiovascular disease, cancer, and other health outcomes. Ann. Epidemiol. 2015, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Castellsagué, X.; Chaturvedi, A.; Goodman, M.T.; Snijders, P.; Tommasino, M.; Arbyn, M.; Franceschi, S. Eurogin Roadmap: Comparative epidemiology of HPV infection and associated cancers of the head and neck and cervix. Int. J. Cancer 2014, 134, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Puolakka, E.; Pahkala, K.; Laitinen, T.T.; Magnussen, C.G.; Hutri-Kähönen, N.; Kähönen, M.; Lehtimäki, T.; Tossavainen, P.; Jokinen, E.; Sabin, M.A.; et al. Childhood Socioeconomic Status and Arterial Stiffness in Adulthood: The Cardiovascular Risk in Young Finns Study. Hypertension 2017, 70, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Constantini, S.; Finelli, C.; Capone, F.; Guerriero, E.; La Sala, N.; Gioia, S.; Castello, G. Carotid intima-media thickness is predicted by combined eotaxin levels and severity of hepatic steatosis at ultrasonography in obese patients with nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105610. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–556. [Google Scholar] [CrossRef]
- Stein, J.H.; Korcarz, C.E.; Hurst, R.T.; Lonn, E.; Kendall, C.B.; Mohler, E.R.; Najjar, S.S.; Rembold, C.M.; Post, W.S. American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: A consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J. Am. Soc. Echocardiogr. 2008, 21, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Costantini, S.; Finelli, C.; Capone, F.; Guerriero, E.; La Sala, N.; Gioia, S.; Castello, G. Is serum Interleukin-17 associated with early atherosclerosis in obese patients? J. Transl. Med. 2014, 12, 214. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Q.; Guo, C.; Wang, X.; Cao, X.; Shi, Y.; Gao, F.; Ma, C.; Zhang, L. IL-17 induces apoptosis of vascular endothelial cells: A potential mechanism for human acute coronary syndrome. Clin. Immunol. 2011, 141, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Joo, E.J.; Chang, Y.; Kwon, M.J.; Cho, A.; Cheong, H.S.; Ryu, S. High-Risk Human Papillomavirus Infection and the Risk of Cardiovascular Disease in Korean Women. Circ. Res. 2019, 124, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, Q.; Yang, P.; Li, Y.; Yuan, H.; Wu, L.; Chen, Z. Metabolic syndrome and risk of cervical human papillomavirus incident and persistent infection. Medicine 2016, 95, e2905. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.; Nakagawa, M.; Moscicki, A.B. Cell-Mediated Immune Response to Human Papillomavirus Infection. Clin. Diagn. Lab. Immunol. 2001, 8, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Piñeres, A.J.; Hildesheim, A.; Herrero, R.; Trivett, M.; Williams, M.; Atmetlla, I.; Ramírez, M.; Villegas, M.; Schiffman, M.; Rodríguez, A.C.; et al. Persistent human papillomavirus infection is associated with a generalized decrease in immune responsiveness in older women. Cancer Res. 2006, 66, 11070–11076. [Google Scholar] [CrossRef] [PubMed]
- Kemp, T.J.; Hildesheim, A.; García-Piñeres, A.; Williams, M.C.; Shearer, G.M.; Rodriguez, A.C.; Schiffman, M.; Burk, R.; Freer, E.; Bonilla, J.; et al. Elevated systemic levels of inflammatory cytokines in older women with persistent cervical human papillomavirus infection. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1954–1959. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [Green Version]
- Olbromski, M.; Podhorska-Okołów, M.; Dzięgiel, P. Role of the SOX18 protein in neoplastic processes. Oncol. Lett. 2018, 16, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- García-Ramírez, M.; Martínez-González, J.; Juan-Babot, J.O.; Rodríguez, C.; Badimon, L. Transcription factor SOX18 is expressed in human coronary atherosclerotic lesions and regulates DNA synthesis and vascular cell growth. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2398–2403. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.G.; Isbir, S.; Kunt, A.T.; Isbir, T. Circulating microRNAs as novel biomarkers for atherosclerosis. In Vivo 2018, 32, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. Circulation 2014, 129, 49–73. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Coupland, C.; Vinogradova, Y.; Robson, J.; Minhas, R.; Sheikh, A.; Brindle, P. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ 2008, 336, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Conroy, R.M.; Pyörälä, K.; Fitzgerald, A.P.; Sans, S.; Menotti, A.; De Backer, G.; De Bacquer, D.; Ducimetière, P.; Jousilahti, P.; Keil, U.; et al. SCORE project group. Estimation of ten-year risk of fatal cardiovascular disease in Europe: The SCORE project. Eur. Heart J. 2003, 24, 987–1003. [Google Scholar] [CrossRef]
- D’Agostino, R.B.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General Cardiovascular Risk Profile for Use in Primary Care. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.; Muduli, S.K.; Kapoor, A.; Tewari, S.; Kumar, S.; Khanna, R.; Goel, P.K. Comparison of different cardiovascular risk score calculators for cardiovascular risk prediction and guideline recommended statin uses. Indian Heart J. 2017, 69, 458–463. [Google Scholar] [CrossRef]
- Rosolova, H.; Nussbaumerova, B. Cardio-metabolic risk prediction should be superior to cardiovascular risk assessment in primary prevention of cardiovascular diseases. EPMA J. 2011, 2, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Iso, H.A. Japanese health success story: Trends in cardiovascular diseases, their risk factors, and the contribution of public health and personalized approaches. EPMA J. 2011, 2, 49–57. [Google Scholar] [CrossRef]
- Izzo, J.L.; Shykoff, B.E. Arterial stiffness: clinical relevance, measurement and treatment. Rev. Cardiovasc. Med. 2001, 2, 29–40. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, M.F.; Hashimoto, J. Mechanical Factors in Arterial Aging: A Clinical Perspective. J. Am. Coll. Cardiol. 2007, 50, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avolio, A. Arterial Stiffness. Pulse 2013, 1, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Fleenor, B.S.; Berrones, A.J. Arterial Stiffness, 1st ed.; Springer International Publishing AG: Cham, Switzerland, 2015; p. 61. [Google Scholar]
- Payne, R.A.; Wilkinson, I.B.; Webb, D.J. Arterial stiffness and hypertension: Emerging concepts. Hypertension 2010, 55, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, J.; Nägele, M.P.; Ludovici, V.; Ruschitzka, F.; Sudano, I.; Flammer, A.J. Endothelial dysfunction in cardiovascular disease and Flammer syndrome—similarities and differences. EPMA J. 2017, 8, 99–109. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Triggle, C.R.; Samuel, S.M.; Ravishankar, S.; Marei, I. The endothelium: Influencing vascular smooth muscle in many ways. Can. J. Physiol. Pharmacol. 2012, 738, 713–738. [Google Scholar] [CrossRef]
- Brunner, H.; Cockcroft, J.R.; Deanfield, J.; Donald, A.; Ferrannini, E.; Halcox, J.; Kiowski, W.; Lüscher, T.F.; Mancia, G.; Natali, A.; et al. Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension Endothelial function and dysfunction. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J. Hypertens. 2005, 23, 233–246. [Google Scholar]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Anderson, T.J.; Phillips, S. Assessment and prognosis of peripheral artery measures of vascular function. Prog. Cardiovasc. Dis. 2014, 57, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.A.; Tschakovsky, M.E.; Augeri, A.L.; Polk, D.M.; Thompson, P.D.; Kiernan, F.J. Heterogenous vasodilator pathways underlie flow-mediated dilation in men and women. Am. J. Physiol. Circ. Physiol. 2011, 301, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Jurko, A.; Jurko, T.; Minarik, M.; Mestanik, M.; Mestanikova, A.; Micieta, V.; Visnovcova, Z.; Tonhajzerova, I. Endothelial function in children with white-coat hypertension. Heart Vessel. 2018, 33, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Townsend, R.R.; Wilkinson, I.B.; Schiffrin, E.L.; Avolio, A.P.; Chirinos, J.A.; Cockcroft, J.R.; Heffernan, K.S.; Lakatta, E.G.; McEniery, C.M.; Mitchell, G.F.; et al. American Heart Association Council on Hypertension. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement from the American Heart Association. Hypertension 2015, 66, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, S.E. Ageing of the conduit arteries. J. Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Shirai, K.; Song, M.; Suzuki, J.; Kurosu, T.; Oyama, T.; Nagayama, D.; Miyashita, Y.; Yamamura, S.; Takahashi, M. Contradictory effects of β1- and α1- aderenergic receptor blockers on cardio-ankle vascular stiffness index (CAVI)--CAVI independent of blood pressure. J. Atheroscler. Thromb. 2011, 18, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Erami, C.; Zhang, H.; Ho, J.G.; French, D.M.; Faber, J.E. α1-Adrenoceptor stimulation directly induces growth of vascular wall in vivo. Am. J. Physiol. Circ. Physiol. 2002, 283, 1577–1587. [Google Scholar] [CrossRef]
- Chirinos, J.A. Arterial Stiffness: Basic Concepts and Measurement Techniques. J. Cardiovasc. Transl. Res. 2012, 5, 243–255. [Google Scholar] [CrossRef]
- Wohlfahrt, P.; Krajčoviechová, A.; Seidlerová, J.; Mayer, O.; Bruthans, J.; Filipovský, J.; Laurent, S.; Cífková, R. Arterial stiffness parameters: How do they differ? Atherosclerosis 2013, 231, 359–364. [Google Scholar] [CrossRef]
- Lim, J.; Pearman, M.E.; Park, W.; Alkatan, M.; Machin, D.R.; Tanaka, H. Impact of blood pressure perturbations on arterial stiffness. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, 1540–1545. [Google Scholar] [CrossRef]
- Tanaka, H. Various Indices of Arterial Stiffness: Are They Closely Related or Distinctly Different? Pulse 2018, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S. How to assess vascular aging? J. Hypertens. Res. 2018, 4, 39–52. [Google Scholar]
- Tan, I.; Spronck, B.; Kiat, H.; Barin, E.; Reesink, K.D.; Delhaas, T.; Avolio, A.P.; Butlin, M. Heart Rate Dependency of Large Artery Stiffness Novelty and Significance. Hypertension 2016, 68, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Spronck, B.; Heusinkveld, M.H.; Vanmolkot, F.H.; Roodt, J.O.; Hermeling, E.; Delhaas, T.; Kroon, A.A.; Reesink, K.D. Pressure-dependence of arterial stiffness: Potential clinical implications. J. Hypertens. 2015, 33, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Spronck, B.; Mestanik, M.; Tonhajzerova, I.; Jurko, A.; Jurko, T.; Avolio, A.P.; Butlin, M. Direct means of obtaining CAVI0—a corrected cardio-ankle vascular stiffness index (CAVI)—from conventional CAVI measurements or their underlying variables. Physiol. Meas. 2017, 38, N128–N137. [Google Scholar] [CrossRef] [PubMed]
- Spronck, B.; Delhaas, T.; Butlin, M.; Reesink, K.D.; Avolio, A.P. Options for dealing with pressure dependence of pulse wave velocity as a measure of arterial stiffness: An update of cardio-ankle vascular index (CAVI) and CAVI0. Pulse (Basel) 2017, 5, 106–114. [Google Scholar] [CrossRef]
- Jurko, A.J.r.; Minarik, M.; Jurko, T.; Tonhajzerova, I. White coat hypertension in pediatrics. Ital. J. Pediatr. 2016, 42, 1–5. [Google Scholar] [CrossRef]
- Shirai, K.; Utino, J.; Otsuka, K.; Takata, M. A novel blood pressure-independent arterial wall stiffness parameter; cardio-ankle vascular index (CAVI). J. Atheroscler. Thromb. 2006, 13, 101–107. [Google Scholar] [CrossRef]
- Spronck, B.; Avolio, A.P.; Tan, I.; Butlin, M.; Reesink, K.D.; Delhaas, T. Arterial stiffness index beta and cardio-ankle vascular index inherently depend on blood pressure but can be readily corrected. J. Hypertens. 2017, 35, 98–104. [Google Scholar] [CrossRef]
- Segers, P. A lesson in vigilance: Pressure dependency of a presumed pressure-independent index of arterial stiffness. J. Hypertens. 2017, 35, 33–35. [Google Scholar] [CrossRef]
- Mestanik, M.; Jurko, A.; Spronck, B.; Avolio, A.P.; Butlin, M.; Jurko, T.; Visnovcova, Z.; Mestanikova, A.; Langer, P.; Tonhajzerova, I. Improved assessment of arterial stiffness using corrected cardio-ankle vascular index (CAVI0) in overweight adolescents with white-coat and essential hypertension. Scand. J. Clin. Lab. Investig. 2017, 77, 665–672. [Google Scholar] [CrossRef]
- Jurko, T.; Mestanik, M.; Jurko, A., Jr.; Spronck, B.; Avolio, A.; Mestanikova, A.; Sekaninova, N.; Tonhajzerova, I. Pediatric Reference Values For Arterial Stiffness Parameters Cardio-Ankle Vascular Index (CAVI) And CAVI0. J. Am. Soc. Hypertens. 2018, 12, e35–e43. [Google Scholar] [CrossRef]
- Wohlfahrt, P.; Cífková, R.; Movsisyan, N.; Kunzová, Š.; Lešovský, J.; Homolka, M.; Soška, V.; Dobšák, P.; Lopez-Jimenez, F.; Sochor, O. Reference values of cardio-ankle vascular index in a random sample of a white population. J. Hypertens. 2017, 35, 2238–2244. [Google Scholar] [CrossRef]
- Namekata, T.; Suzuki, K.; Ishizuka, N.; Shirai, K. Establishing baseline criteria of cardio-ankle vascular index as a new indicator of arteriosclerosis: A cross-sectional study. BMC Cardiovasc. Disord. 2011, 11, 1–10. [Google Scholar] [CrossRef]
- Safar, M.E.; Blacher, J.; Jankowski, P. Arterial stiffness, pulse pressure, and cardiovascular disease-is it possible to break the vicious circle? Atherosclerosis 2011, 218, 263–271. [Google Scholar] [CrossRef]
- McEniery, C.M.; Cockcroft, J.R.; Roman, M.J.; Franklin, S.S.; Wilkinson, I.B. Central blood pressure: Current evidence and clinical importance. Eur. Heart J. 2014, 35, 1719–1725. [Google Scholar] [CrossRef]
- Vamsi, V.; Golub, A.; Pezić, M.; Fekete, P.; Findri, P.; Prkačin, I. Central blood pressure and pulse wave velocity in patients with resistant hypertension. Signa Vitae 2018, 14, 28–30. [Google Scholar] [CrossRef]
- Pini, R.; Cavallini, M.C.; Palmieri, V.; Marchionni, N.; Di Bari, M.; Devereux, R.B.; Masotti, G.; Roman, M.J. Central but not brachial blood pressure predicts cardiovascular events in an unselected geriatric population: The ICARe Dicomano Study. J. Am. Coll. Cardiol. 2008, 51, 2432–2439. [Google Scholar] [CrossRef]
- Niiranen, T.J.; Kalesan, B.; Mitchell, G.F.; Vasan, R.S. Relative Contributions of Pulse Pressure and Arterial Stiffness to Cardiovascular Disease. Hypertension 2019, 73, 712–717. [Google Scholar] [CrossRef]
- Yadav, D.; Kim, S.J.; Kim, J.R.; Cho, K.H. Correlation among lipid parameters, pulse wave velocity and central blood pressure in young Korean population. Clin. Exp. Hypertens. 2019, 41, 20–27. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Hwang, S.J.; Vasan, R.S.; Larson, M.G.; Pencina, M.J.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J. Arterial stiffness and cardiovascular events: The Framingham Heart Study. Circulation 2010, 121, 505–511. [Google Scholar] [CrossRef]
- Laurent, S.; Sharman, J.; Boutouyrie, P. Central versus peripheral blood pressure: Finding a solution. J. Hypertens. 2016, 34, 1497–1499. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P. The structural factor of hypertension: Large and small artery alterations. Circ. Res. 2015, 116, 1007–1021. [Google Scholar] [CrossRef]
- Gori, T.; Muxel, S.; Damaske, A.; Radmacher, M.C.; Fasola, F.; Schaefer, S.; Schulz, A.; Jabs, A.; Parker, J.D.; Münzel, T. Endothelial function assessment: Flow-mediated dilation and constriction provide different and complementary information on the presence of coronary artery disease. Eur. Heart J. 2012, 33, 363–371. [Google Scholar] [CrossRef]
- Hamburg, N.M.; Keyes, M.J.; Larson, M.G.; Vasan, R.S.; Schnabel, R.; Pryde, M.M.; Mitchell, G.F.; Sheffy, J.; Vita, J.A.; Benjamin, E.J. Cross-sectional relations of digital vascular function to cardiovascular risk factors in The Framingham Heart Study. Circulation 2008, 117, 2467–2474. [Google Scholar] [CrossRef]
- Aristizábal-Ocampo, D.; Espíndola-Fernández, D.; Gallo-Villegas, J. Pulse wave velocity reference values in 3,160 adults referred to a hypertension clinic for 24-hour ambulatory blood pressure monitoring. Clin. Exp. Hypertens. 2018, 2, 1–7. [Google Scholar] [CrossRef]
- Park, Y.C.; Kang, H.C.; Lee, D.C.; Kim, S.H.; Kim, J.K. Correlation between Abnormal Pap Smear Finding and Brachial-ankle Pulse Wave Velocity in Korean Women. J. Lifestyle Med. 2013, 3, 68–72. [Google Scholar]
- Zuo, J.; Chang, G.; Tan, I.; Butlin, M.; Chu, S.L.; Avolio, A. Central aortic pressure improves prediction of cardiovascular events compared to peripheral blood pressure in short-term follow-up of a hypertensive cohort. Clin. Exp. Hypertens. 2018, 16, 1–8. [Google Scholar] [CrossRef]
- Spronck, B.; Mestanik, M.; Tonhajzerova, I.; Jurko, A.; Tan, I.; Butlin, M.; Avolio, A.P. Easy conversion of cardio-ankle vascular index into CAVI0 influence of scale coefficients. J. Hypertens. 2019, 37. [Google Scholar] [CrossRef]
- Philip, R.; Alpert, B.S.; Schwingshackl, A.; Huang, X.; Blakely, D.; Rovnaghi, C.R.; Tran, Q.T.; Velasquez, A.; Arevalo, A.; Anand, K.J. Inverse relationship between cardio-ankle vascular index and body mass index in healthy children. J. Pediatr. 2015, 167, 361–365. [Google Scholar] [CrossRef]


{kind=link}
| Biomarker | Selected Major Studies | Significance |
|---|---|---|
| Endothelial Function | ||
| Flow-mediated dilation (FMD) as an index of macrovascular endothelial function | Gori et al., 2012 (451 subjects) adults [117] | Predicting the presence of coronary artery disease in patients before coronary angiography |
| Peripheral arterial tonometry (PAT) as an index of reactive hyperaemia | Framingham study (Hamburg et al., 2008 (1957 subjects)) adults [118] | PAT is related to metabolic cardiovascular risk factors (smoking, cholesterol, body mass index) |
| Arterial Stiffness | ||
| Pulse wave velocity (PWV) the current “gold standard” arterial stiffness metric | Aristizábal-Ocampo et al., 2018 (3160 subjects) adults [119] | Increased PWV values related to age and blood pressure |
| Park et al., 2013 (1779 subjects) adult women [120] | Increased PWV values in women with atypical cervical cells | |
| Central blood pressure (CBP) | Zuo et al. 2018 (675 subjects) adults [121] | CBP improved prediction of CVD compared to peripheral pressure hypertensive patients |
| Cardio-ankle vascular index (CAVI) [122] Cardio-ankle vascular stiffness index (CAVI0) [99] [122] | Wohlfart et al., 2017 (2160 subjects) adults [106] | Increased CAVI in smokers and patients with dyslipidaemia |
| Namekata et al., 2011 (32,627 subjects) adults [107] | Group with increased cardiovascular disease (hypertension, hyperglycaemia, abnormal lipid metabolism, ischemic and sclerotic changes) showed increased CAVI | |
| Philip et al., 2015 (292 subjects) children [123] | Lower CAVI in overweight children suggesting vascular adaptation to obesity |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonhajzerova, I.; Olexova, L.B.; Jurko, A.; Spronck, B.; Jurko, T.; Sekaninova, N.; Visnovcova, Z.; Mestanikova, A.; Kudela, E.; Mestanik, M. Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection. Int. J. Mol. Sci. 2019, 20, 3720. https://doi.org/10.3390/ijms20153720
Tonhajzerova I, Olexova LB, Jurko A, Spronck B, Jurko T, Sekaninova N, Visnovcova Z, Mestanikova A, Kudela E, Mestanik M. Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection. International Journal of Molecular Sciences. 2019; 20(15):3720. https://doi.org/10.3390/ijms20153720
Chicago/Turabian StyleTonhajzerova, Ingrid, Lucia B. Olexova, Alexander Jurko, Bart Spronck, Tomas Jurko, Nikola Sekaninova, Zuzana Visnovcova, Andrea Mestanikova, Erik Kudela, and Michal Mestanik. 2019. "Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection" International Journal of Molecular Sciences 20, no. 15: 3720. https://doi.org/10.3390/ijms20153720
APA StyleTonhajzerova, I., Olexova, L. B., Jurko, A., Spronck, B., Jurko, T., Sekaninova, N., Visnovcova, Z., Mestanikova, A., Kudela, E., & Mestanik, M. (2019). Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection. International Journal of Molecular Sciences, 20(15), 3720. https://doi.org/10.3390/ijms20153720