Mast Cells as Potential Accelerators of Human Atherosclerosis—From Early to Late Lesions


{kind=link}
{kind=link}
Abstract
1. Experimental Atherosclerosis: from Mouse to Mechanisms and Proofs of Concept—Still Far from Perfection
2. Atherogenesis—A Brief Outline of a Long Path of Events
3. Mast Cells and Their Presence in Human Atherosclerotic Lesions
3.1. General Definition of Mast Cells and Their Functions
3.2. The Abdominal Aorta
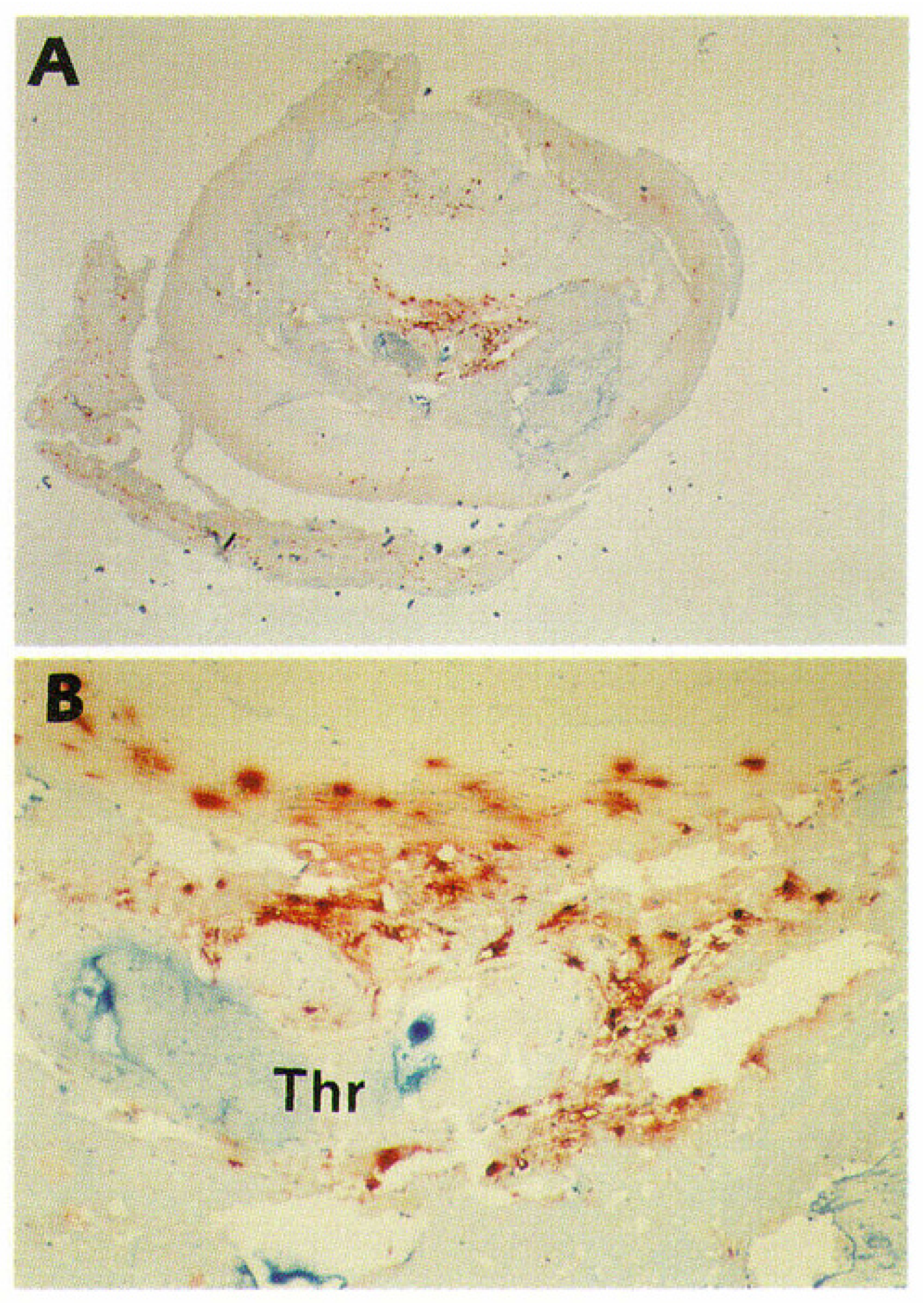
3.3. Epicardial Coronary Arteries
4. Grading Mast Cell Activation in Human Atherosclerotic Coronary Arteries
5. Potential Mast Cell Triggers in Human Atherosclerotic Coronary Arteries
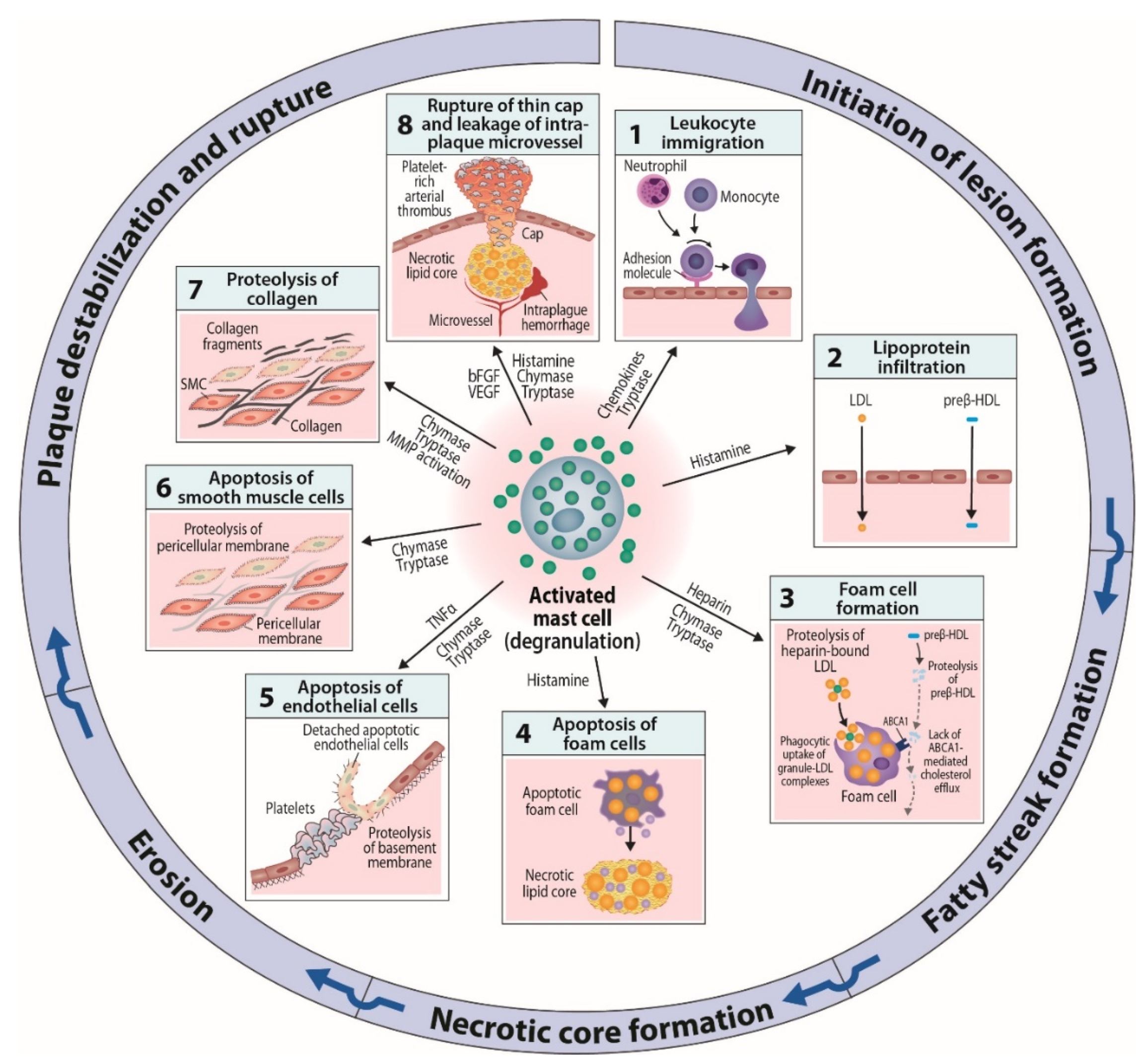
6. Mast Cells as Effector Cells in Atherogenesis
6.1. Actions Related to Early Atherogenesis
6.2. Actions Related to Advancing and Terminal Atherogenesis
7. Concluding Remarks: Allergic Atherosclerosis—Does One Exist?
Funding
Acknowledgments
Conflicts of Interest
References
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A., Jr.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar]
- Moghadasian, M.H. Experimental atherosclerosis: A historical overview. Life Sci. 2002, 70, 855–865. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setälä, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Piedrahita, J.A.; Zhang, S.H.; Hagaman, J.R.; Oliver, P.M.; Maeda, N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1992, 89, 4471–4475. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Wolters, P.J.; Yang, M.; Kitamoto, S.; Libby, P.; MacFarlane, L.A.; Mallen-St Clair, J.; Shi, G.P. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat. Med. 2007, 13, 719–724. [Google Scholar] [CrossRef]
- Bot, I.; de Jager, S.C.; Zernecke, A.; Lindstedt, K.A.; van Berkel, T.J.; Weber, C.; Biessen, E.A. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 2007, 115, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Öörni, K.; Pentikäinen, M.O.; Ala-Korpela, M.; Kovanen, P.T. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: Molecular mechanisms and effects on matrix interactions. J. Lipid Res. 2000, 41, 1703–1714. [Google Scholar] [PubMed]
- Boren, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Pentikäinen, M.O.; Öörni, K.; Ala-Korpela, M.; Kovanen, P.T. Modified LDL - trigger of atherosclerosis and inflammation in the arterial intima. J. Intern. Med. 2000, 247, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Sigari, F.; Segrado, T.; Hörkkö, S.; Hama, S.; Subbaiah, P.V.; Miwa, M.; Navab, M.; Witztum, J.L.; Reaven, P.D. All ApoB-containing lipoproteins induce monocyte chemotaxis and adhesion when minimally modified. Modulation of lipoprotein bioactivity by platelet-activating factor acetylhydrolase. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1437–1446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geng, Y.; Kodama, T.; Hansson, G.K. Differential expression of scavenger receptor isoforms during monocyte-macrophage differentiation and foam cell formation. Arterioscler Thromb. 1994, 14, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef]
- Buja, L.M.; Nikolai, N. Anitschkow and the lipid hypothesis of atherosclerosis. Cardiovasc. Pathol. 2014, 23, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J. et al.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Chroni, A.; Tietge, U.J.; Zanotti, I.; Escola-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, B.A.; Chapman, M.J.; Kontush, A.; Miller, N.E. HDL-targeted therapies: Progress, failures and future. Nat. Rev. Drug Discov. 2014, 13, 445–464. [Google Scholar] [CrossRef]
- Brown, M.S.; Ho, Y.K.; Goldstein, J.L. The cholesteryl ester cycle in macrophage foam cells. Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J. Biol. Chem. 1980, 255, 9344–9352. [Google Scholar] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Fuster, V.; Moreno, P.R.; Fayad, Z.A.; Corti, R.; Badimon, J.J. Atherothrombosis and high-risk plaque: Part I: Evolving concepts. J. Am. Coll. Cardiol. 2005, 46, 937–954. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Lichtman, A.H. Monocyte-macrophages and T Cells in atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Marshall, J.S. Mast-cell responses to pathogens. Nat. Rev. Immunol. 2004, 4, 787–799. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef]
- Eklund, K.K. Mast cells in the pathogenesis of rheumatic diseases and as potential targets for anti-rheumatic therapy. Immunol. Rev. 2007, 217, 38–52. [Google Scholar] [CrossRef]
- Galinsky, D.S.; Nechushtan, H. Mast cells and cancer--no longer just basic science. Crit. Rev. Oncol. Hematol. 2008, 68, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.P.; Bot, I.; Kovanen, P.T. Mast cells in human and experimental cardiometabolic diseases. Nat. Rev. Cardiol. 2015, 12, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Rossi, F.W.; Galdiero, M.R.; Granata, F.; Criscuolo, G.; Spadaro, G.; de Paulis, A.; Marone, G. Physiological roles of mast cells: Collegium Internationale Allergologicum Update 2019. Int Arch. Allergy Immunol. 2019, 179, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Galli, S.J.; Kitamura, Y. Probing the roles of mast cells and basophils in natural and acquired immunity, physiology and disease. Trends Immunol. 2002, 23, 425–427. [Google Scholar] [CrossRef]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Irani, A.A.; Schechter, N.M.; Craig, S.S.; DeBlois, G.; Schwartz, L.B. Two types of human mast cells that have distinct neutral protease compositions. Proc. Natl. Acad. Sci. USA 1986, 83, 4464–4468. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Raap, U.; Rivellese, F.; Marone, G.; Gibbs, B.F. Human mast cells and basophils-How are they similar how are they different? Immunol. Rev. 2018, 282, 8–34. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef]
- Fulton, G.P.; Maynard, F.L.; Riley, J.F.; West, G.B. Humoral aspects of tissue mast cells. Physiol, Rev. 1957, 37, 221–232. [Google Scholar] [CrossRef]
- Bydlowski, S.P. Mast cell: Its mediators and effects on arterial wall metabolism. Gen. Pharmacol. 1986, 17, 625–631. [Google Scholar] [CrossRef]
- Atkinson, J.B.; Harlan, C.W.; Harlan, G.C.; Virmani, R. The association of mast cells and atherosclerosis: A morphologic study of early atherosclerotic lesions in young people. Hum. Pathol. 1994, 25, 154–159. [Google Scholar] [CrossRef]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Mast cells of two types differing in neutral protease composition in the human aortic intima. Demonstration of tryptase- and tryptase/chymase-containing mast cells in normal intimas, fatty streaks, and the shoulder region of atheromas. Arterioscler. Thromb. 1994, 14, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; Demetriou, D.; Grines, C.L.; Pica, M.; Shoukfeh, M.; O’Neill, W.W. Multiple complex coronary plaques in patients with acute myocardial infarction. N. Engl. J. Med. 2000, 343, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation 1994, 90, 1669–1678. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Kaartinen, M.; Paavonen, T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation 1995, 92, 1084–1088. [Google Scholar] [CrossRef]
- Kaartinen, M.; van der Wal, A.C.; van der Loos, C.M.; Piek, J.J.; Koch, K.T.; Becker, A.E.; Kovanen, P.T. Mast cell infiltration in acute coronary syndromes: Implications for plaque rupture. J. Am. Coll. Cardiol. 1998, 32, 606–612. [Google Scholar] [CrossRef]
- Lindstedt, K.A.; Kokkonen, J.O.; Kovanen, P.T. Soluble heparin proteoglycans released from stimulated mast cells induce uptake of low density lipoproteins by macrophages via scavenger receptor-mediated phagocytosis. J. Lipid Res. 1992, 33, 65–75. [Google Scholar]
- Lindstedt, K.A.; Kokkonen, J.O.; Kovanen, P.T. Regulation of the activity of secreted human lung mast cell tryptase by mast cell proteoglycans. Biochim. Biophys. Acta. 1998, 1425, 617–627. [Google Scholar] [CrossRef]
- Laine, P.; Kaartinen, M.; Penttilä, A.; Panula, P.; Paavonen, T.; Kovanen, P.T. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation 1999, 99, 361–369. [Google Scholar] [CrossRef]
- Kortelainen, M.L.; Porvari, K. Adventitial macrophage and lymphocyte accumulation accompanying early stages of human coronary atherogenesis. Cardiovasc. Pathol. 2014, 23, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef]
- Ohyama, K.; Matsumoto, Y.; Takanami, K.; Ota, H.; Nishimiya, K.; Sugisawa, J.; Tsuchiya, S.; Amamizu, H.; Uzuka, H.; Suda, A. Coronary adventitial and perivascular adipose tissue inflammation in patients with vasospastic angina. J. Am. Coll. Cardiol. 2018, 71, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Zelechowska, P.; Agier, J.; Kozlowska, E.; Brzezinska-Blaszczyk, E. Mast cells participate in chronic low-grade inflammation within adipose tissue. Obes. Rev. 2018, 19, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Sagi-Eisenberg, R. Anaphylactic degranulation of mast cells: Focus on compound exocytosis. J. Immunol. Res. 2019, 2019, 9542656. [Google Scholar] [CrossRef]
- Kounis, N.G.; Zavras, G.M. Histamine-induced coronary artery spasm: The concept of allergic angina. Br. J. Clin. Pract. 1991, 45, 121–128. [Google Scholar] [PubMed]
- Kounis, N.G. Kounis syndrome: An update on epidemiology, pathogenesis, diagnosis and therapeutic management. Clin. Chem. Lab. Med. 2016, 54, 1545–1559. [Google Scholar] [CrossRef]
- Niccoli, G.; Montone, R.A.; Sabato, V.; Crea, F. Role of allergic inflammatory cells in coronary artery disease. Circulation 2018, 138, 1736–1748. [Google Scholar] [CrossRef]
- Varricchi, G.; Loffredo, S.; Borriello, F.; Pecoraro, A.; Rivellese, F.; Genovese, A.; Spadaro, G.; Marone, G. Superantigenic activation of human cardiac mast cells. Int. J. Mol. Sci. 2019, 20, 1828. [Google Scholar] [CrossRef]
- Befus, A.D.; Mowat, C.; Gilchrist, M.; Hu, J.; Solomon, S.; Bateman, A. Neutrophil defensins induce histamine secretion from mast cells: Mechanisms of action. J. Immunol. 1999, 163, 947–953. [Google Scholar] [PubMed]
- Yu, Y.; Blokhuis, B.R.; Garssen, J.; Redegeld, F.A. Non-IgE mediated mast cell activation. Eur. J. Pharmacol. 2016, 778, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A. Mast cells and inflammation. Biochim. Biophys. Acta. 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T.; Bot, I. Mast cells in atherosclerotic cardiovascular disease - Activators and actions. Eur. J. Pharmacol. 2017, 816, 37–46. [Google Scholar] [CrossRef]
- Baram, D.; Vaday, GG.; Salamon, P.; Drucker, I.; Hershkoviz, R.; Mekori, YA. Human mast cells release metalloproteinase-9 on contact with activated T cells: Juxtacrine regulation by TNF-α. J. Immunol. 2001, 167, 4008–4016. [Google Scholar] [CrossRef]
- Shefler, I.; Salamon, P.; Reshef, T.; Mor, A.; Mekori, YA. T cell-induced mast cell activation: A role for microparticles released from activated T cells. J. Immunol. 2010, 185, 4206–4212. [Google Scholar] [CrossRef]
- Yu, T.; He, Z.; Yang, M.; Song, J.; Ma, C.; Ma, S.; Feng, J.; Liu, B.; Wang, X.; Wei, Z. The development of methods for primary mast cells in vitro and ex vivo: An historical review. Exp. Cell Res. 2018, 369, 179–186. [Google Scholar] [CrossRef]
- Lappalainen, J.; Lindstedt, K.A.; Kovanen, P.T. A protocol for generating high numbers of mature and functional human mast cells from peripheral blood. Clin. Exp. Allergy. 2007, 37, 1404–1414. [Google Scholar] [CrossRef]
- Lappalainen, J.; Lindstedt, K.A.; Oksjoki, R.; Kovanen, P.T. OxLDL-IgG immune complexes induce expression and secretion of proatherogenic cytokines by cultured human mast cells. Atherosclerosis 2011, 214, 357–363. [Google Scholar] [CrossRef]
- Laine, P.; Pentikäinen, M.O.; Würzner, R.; Penttilä, A.; Paavonen, T.; Meri, S.; Kovanen, P.T. Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am. J. Cardiol. 2002, 90, 404–408. [Google Scholar] [CrossRef]
- Oksjoki, R.; Laine, P.; Helske, S.; Vehmaan-Kreula, P.; Mäyränpää, M.I.; Gasque, P.; Kovanen, P.T.; Pentikäinen, M.O. Receptors for the anaphylatoxins C3a and C5a are expressed in human atherosclerotic coronary plaques. Atherosclerosis 2007, 195, 90–99. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.R.; Wezel, A.; Schepers, A.; van Santbrink, P.J.; Woodruff, T.M.; Niessen, H.W.M.; Hamming, J.P.; Kuiper, J.; Bot, I.; Quax, P.H.A. Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc. Res. 2012, 97, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Laine, P.; Naukkarinen, A.; Heikkilä, L.; Penttilä, A.; Kovanen, P.T. Adventitial mast cells connect with sensory nerve fibers in atherosclerotic coronary arteries. Circulation 2000, 101, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.B.; Oates, J.A.; Robertson, D.; Robertson, R.M.; Roberts, L.J., 2nd; Virmani, R. Increased adventitial mast cells in a patient with coronary spasm. N. Engl. J. Med. 1985, 313, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Dichlberger, A.; Schlager, S.; Kovanen, P.T.; Schneider, W.J. Lipid droplets in activated mast cells - a significant source of triglyceride-derived arachidonic acid for eicosanoid production. Eur. J. Pharmacol. 2016, 785, 59–69. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Kempuraj, D.; Tagen, M.; Conti, P.; Kalogeromitros, D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol. Rev. 2007, 217, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Alevizos, M.; Karagkouni, A.; Panagiotidou, S.; Vasiadi, M.; Theoharides, T.C. Stress triggers coronary mast cells leading to cardiac events. Ann. Allergy Asthma Immunol. 2014, 112, 309–316. [Google Scholar] [CrossRef]
- Vicario, M.; Guilarte, M.; Alonso, C.; Yang, P.; Martinez, C.; Ramos, L.; Lobo, B.; Gonzalez, A.; Guila, M.; Pigrau, M. Chronological assessment of mast cell-mediated gut dysfunction and mucosal inflammation in a rat model of chronic psychosocial stress. Brain Behav. Immun. 2010, 24, 1166–1175. [Google Scholar] [CrossRef]
- Iwamoto, S.; Asada, Y.; Ebihara, N.; Hori, K.; Okayama, Y.; Kashiwakura, J.; Watanabe, Y.; Kawasaki, S.; Yokoi, N.; Inatomi, T. Interaction between conjunctival epithelial cells and mast cells induces CCL2 expression and piecemeal degranulation in mast cells. Invest. Ophthalmol. Vis. Sci. 2013, 54, 2465–2473. [Google Scholar] [CrossRef]
- McCurdy, J.D.; Olynych, T.J.; Maher, L.H.; Marshall, J.S. Cutting edge: Distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J. Immunol. 2003, 170, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Frossi, B.; D’Inca, F.; Crivellato, E.; Sibilano, R.; Gri, G.; Mongillo, M.; Danelli, L.; Maggi, L.; Pucillo, C.E. Single-cell dynamics of mast cell-CD4+ CD25+ regulatory T cell interactions. Eur. J. Immunol. 2011, 41, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, A.M. Basophils and mast cells: Piecemeal degranulation in situ and ex vivo: A possible mechanism for cytokine-induced function in disease. Immunol. Ser. 1992, 57, 169–271. [Google Scholar] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation 1996, 94, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- von der Thusen, J.H.; van Berkel, T.J.; Biessen, E.A. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation 2001, 103, 1164–1170. [Google Scholar] [CrossRef]
- Rozenberg, I.; Sluka, S.H.; Rohrer, L.; Hofmann, J.; Becher, B.; Akhmedov, A.; Soliz, J.; Mocharla, P.; Boren, J.; Johansen, P. Histamine H1 receptor promotes atherosclerotic lesion formation by increasing vascular permeability for low-density lipoproteins. Arterioscler Thromb. Vasc. Biol. 2010, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Sendo, T.; Oishi, R. Physiology and pathophysiology of proteinase-activated receptors (PARs): Role of tryptase/PAR-2 in vascular endothelial barrier function. J. Pharmacol. Sci. 2005, 97, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.D.; Maaninka, K.; Lappalainen, J.; Nurmi, K.; Metso, J.; Öörni, K.; Navab, M.; Fogelman, A.M.; Jauhiainen, M.; Lee-Rueckert, M.; et al. Carboxyl-terminal cleavage of apolipoprotein A-I by human mast cell chymase impairs its anti-inflammatory properties. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 274–284. [Google Scholar] [CrossRef]
- Maaninka, K.; Nguyen, S.D.; Mäyränpää, M.I.; Plihtari, R.; Rajamäki, K.; Lindsberg, P.J.; Kovanen, P.T.; Öörni, K. Human mast cell neutral proteases generate modified LDL particles with increased proteoglycan binding. Atherosclerosis 2018, 275, 390–399. [Google Scholar] [CrossRef]
- Kokkonen, J.O.; Kovanen, P.T. Stimulation of mast cells leads to cholesterol accumulation in macrophages in vitro by a mast cell granule-mediated uptake of low density lipoprotein. Proc. Natl. Acad. Sci. USA 1987, 84, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T. Mast cells in human fatty streaks and atheromas: Implications for intimal lipid accumulation. Curr. Opin. Lipidol. 1996, 7, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Extracellular mast cell granules carry apolipoprotein B-100-containing lipoproteins into phagocytes in human arterial intima. Functional coupling of exocytosis and phagocytosis in neighboring cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2047–2054. [Google Scholar] [CrossRef] [PubMed]
- Kareinen, I.; Cedo, L.; Silvennoinen, R.; Laurila, P.P.; Jauhiainen, M.; Julve, J.; Blanco-Vaca, F.; Escola-Gil, J.C.; Kovanen, P.T.; Lee-Rueckert, M. Enhanced vascular permeability facilitates entry of plasma HDL and promotes macrophage-reverse cholesterol transport from skin in mice. J. Lipid Res. 2015, 56, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rueckert, M.; Kovanen, P.T. Extracellular modifications of HDL in vivo and the emerging concept of proteolytic inactivation of prebeta-HDL. Curr. Opin. Lipidol. 2011, 22, 394–402. [Google Scholar] [CrossRef]
- Mäyränpää, M.I.; Heikkilä, H.M.; Lindstedt, K.A.; Walls, A.F.; Kovanen, P.T. Desquamation of human coronary artery endothelium by human mast cell proteases: Implications for plaque erosion. Coron. Artery. Dis. 2006, 17, 611–621. [Google Scholar] [CrossRef]
- Lätti, S.; Leskinen, M.; Shiota, N.; Wang, Y.; Kovanen, P.T.; Lindstedt, K.A. Mast cell-mediated apoptosis of endothelial cells in vitro: A paracrine mechanism involving TNF-alpha-mediated down-regulation of bcl-2 expression. J. Cell Physiol. 2003, 195, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the mechanisms of acute coronary syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Wezel, A.; Lagraauw, H.M.; van der Velden, D.; de Jager, S.C.; Quax, P.H.; Kuiper, J.; Bot, I. Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis 2015, 241, 289–296. [Google Scholar] [CrossRef]
- Uryga, A.K.; Bennett, M.R. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Leskinen, M.J.; Kovanen, P.T.; Lindstedt, K.A. Regulation of smooth muscle cell growth, function and death in vitro by activated mast cells--a potential mechanism for the weakening and rupture of atherosclerotic plaques. Biochem. Pharmacol. 2003, 66, 1493–1498. [Google Scholar] [CrossRef]
- Wang, Y.; Shiota, N.; Leskinen, M.J.; Lindstedt, K.A.; Kovanen, P.T. Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: Transforming growth factor-beta1-dependent and -independent effects. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Saarinen, J.; Kalkkinen, N.; Welgus, H.G.; Kovanen, P.T. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J. Biol. Chem. 1994, 269, 18134–18140. [Google Scholar] [PubMed]
- Gruber, B.L.; Marchese, M.J.; Suzuki, K.; Schwartz, L.B.; Okada, Y.; Nagase, H.; Ramamurthy, N.S. Synovial procollagenase activation by human mast cell tryptase: Dependence upon matrix metalloproteinase 3 activation. J. Clin. Investig. 1989, 84, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Jackson, C.L.; Angelini, G.D.; George, S.J. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Marsch, E.; Sluimer, J.C.; Daemen, M.J. Hypoxia in atherosclerosis and inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Hulten, L.M.; Levin, M. The role of hypoxia in atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 409–414. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–18. [Google Scholar] [CrossRef]
- Ribatti, D.; Levi-Schaffer, F.; Kovanen, P.T. Inflammatory angiogenesis in atherogenesis—A double-edged sword. Ann. Med. 2008, 40, 606–621. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. Mast cells, angiogenesis, and tumour growth. Biochim. Biophys. Acta 2012, 1822, 2–8. [Google Scholar] [CrossRef]
- Gulliksson, M.; Carvalho, R.F.; Ullerås, E.; Nilsson, G. Mast cell survival and mediator secretion in response to hypoxia. PLoS ONE 2010, 5, e12360. [Google Scholar] [CrossRef] [PubMed]
- de Souza Junior, D.A.; Borges, A.C.; Santana, A.C.; Oliver, C.; Jamur, M.C. Mast cell proteases 6 and 7 stimulate angiogenesis by inducing endothelial cells to release angiogenic factors. PLoS ONE 2015, 10, e0144081. [Google Scholar] [CrossRef] [PubMed]
- Blair, R.J.; Meng, H.; Marchese, M.J.; Ren, S.; Schwartz, L.B.; Tonnesen, M.G.; Gruber, B.L. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J. Clin. Investig. 1997, 99, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Qian, N.; Li, X.; Wang, X.; Wu, C.; Yin, L.; Zhi, X. Tryptase promotes breast cancer angiogenesis through PAR-2 mediated endothelial progenitor cell activation. Oncol. Lett. 2018, 16, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, H.; Laine, P.; Pentikäinen, M.O.; Sajantila, A.; Kovanen, P.T. Mast cells in neovascularized human coronary plaques store and secrete basic fibroblast growth factor, a potent angiogenic mediator. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen P, T. Mast cells accompany microvessels in human coronary atheromas: Implications for intimal neovascularization and hemorrhage. Atherosclerosis 1996, 123, 123–131. [Google Scholar] [CrossRef]
- Kovanen, P.T. Mast cells and degradation of pericellular and extracellular matrices: Potential contributions to erosion, rupture and intraplaque haemorrhage of atherosclerotic plaques. Biochem. Soc. Trans. 2007, 35, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.C.; Kolodgie, F.D.; Bijnens, A.P.; Maxfield, K.; Pacheco, E.; Kutys, B.; Duimel, H.; Frederik, P.M.; van Hinsbergh, V.W.; Virmani, R. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions: Relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 2009, 53, 1517–1527. [Google Scholar] [CrossRef]
- Jeziorska, M.; McCollum, C.; Woolley, D.E. Mast cell distribution, activation, and phenotype in atherosclerotic lesions of human carotid arteries. J. Pathol. 1997, 182, 115–122. [Google Scholar] [CrossRef]
- Jeziorska, M.; Woolley, D.E. Local neovascularization and cellular composition within vulnerable regions of atherosclerotic plaques of human carotid arteries. J. Pathol. 1999, 188, 189–196. [Google Scholar] [CrossRef]
- Lehtonen-Smeds, E.M.; Mäyränpää, M.; Lindsberg, P.J.; Soinne, L.; Saimanen, E.; Järvinen, A.A.; Salonen, O.; Carpén, O.; Lassila, R.; Sarna, S.; et al. Carotid plaque mast cells associate with atherogenic serum lipids, high grade carotid stenosis and symptomatic carotid artery disease. Results from the Helsinki carotid endarterectomy study. Cerebrovasc. Dis. 2005, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Willems, S.; Vink, A.; Bot, I.; Quax, P.H.; de Borst, G.J.; de Vries, J.P.; van de Weg, S.M.; Moll, F.L.; Kuiper, J.; Kovanen, P.T. Mast cells in human carotid atherosclerotic plaques are associated with intraplaque microvessel density and the occurrence of future cardiovascular events. Eur. Heart. J. 2013, 34, 3699–3706. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.; Kovanen, P.T.; Bianconi, V.; Pirro, M.; Cicero, A.F.G.; Sahebkar, A. Mast cell tryptase - Marker and maker of cardiovascular diseases. Pharmacol. Ther. 2019, 199, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Morici, N.; Farioli, L.; Losappio, L.M.; Colombo, G.; Nichelatti, M.; Preziosi, D.; Micarelli, G.; Oliva, F.; Giannattasio, C.; Klugmann, S. Mast cells and acute coronary syndromes: Relationship between serum tryptase, clinical outcome and severity of coronary artery disease. Open Heart. 2016, 3, e000472. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T.; Mänttäri, M.; Palosuo, T.; Manninen, V.; Aho, K. Prediction of myocardial infarction in dyslipidemic men by elevated levels of immunoglobulin classes A, E, and G, but not M. Arch. Intern. Med. 1998, 158, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Patella, V.; Marino, I.; Lamparter, B.; Arbustini, E.; Adt, M.; Marone, G. Human heart mast cells. Isolation, purification, ultrastructure, and immunologic characterization. J. Immunol. 1995, 154, 2855–2865. [Google Scholar] [PubMed]
- Kawakami, T.; Kitaura, J. Mast cell survival and activation by IgE in the absence of antigen: A consideration of the biologic mechanisms and relevance. J. Immunol. 2005, 175, 4167–4173. [Google Scholar] [CrossRef]
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J. Flow cytometry-based characterization of mast cells in human atherosclerosis. Cells 2019, 8, 334. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Paju, S.; Koponen, J.; Viikari, J.S.A.; Taittonen, L.; Laitinen, T.; Burgner, D.P.; Kähönen, M.; Hutri-Kähönen, N.; Raitakari, O.T. Association of childhood oral infections with cardiovascular risk factors and subclinical atherosclerosis in adulthood. JAMA Netw. Open 2019, 2, e192523. [Google Scholar] [CrossRef]
- Tang, W.H.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef]
- Progulske-Fox, A.; Kozarov, E.; Dorn, B.; Dunn, W., Jr.; Burks, J.; Wu, Y. Porphyromonas gingivalis virulence factors and invasion of cells of the cardiovascular system. J. Periodontal. Res. 1999, 34, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kozarov, E.V.; Dorn, B.R.; Shelburne, C.E.; Dunn, W.A., Jr.; Progulske-Fox, A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, e17–e18. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4592–4598. [Google Scholar] [CrossRef]
- Grayston, J.T.; Kuo, C.C.; Coulson, A.S.; Campbell, L.A.; Lawrence, R.D.; Lee, M.J.; Strandness, E.D.; Wang, S.P. Chlamydia pneumoniae (TWAR) in atherosclerosis of the carotid artery. Circulation 1995, 92, 3397–3400. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Idahosa, C.; Roy, S.; Lee, D.; Subramanian, H.; Dhingra, A.; Boesze-Battaglia, K.; Korostoff, J.; Ali, H. Differential regulation of mas-related G Protein-coupled receptor X2-mediated mast cell degranulation by antimicrobial host defense peptides and Porphyromonas gingivalis lipopolysaccharide. Infect. Immun. 2017, 85, e00246–17. [Google Scholar] [CrossRef] [PubMed]
- Lassila, R.; Lindstedt, K.; Kovanen P, T. Native macromolecular heparin proteoglycans exocytosed from stimulated rat serosal mast cells strongly inhibit platelet-collagen interactions. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Lassila, R.; Jouppila, A. Mast cell–derived heparin proteoglycans as a model for a local antithrombotic. Semin. Thromb. Hemost. 2014, 40, 837–844. [Google Scholar] [PubMed]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovanen, P.T. Mast Cells as Potential Accelerators of Human Atherosclerosis—From Early to Late Lesions. Int. J. Mol. Sci. 2019, 20, 4479. https://doi.org/10.3390/ijms20184479
Kovanen PT. Mast Cells as Potential Accelerators of Human Atherosclerosis—From Early to Late Lesions. International Journal of Molecular Sciences. 2019; 20(18):4479. https://doi.org/10.3390/ijms20184479
Chicago/Turabian StyleKovanen, Petri T. 2019. "Mast Cells as Potential Accelerators of Human Atherosclerosis—From Early to Late Lesions" International Journal of Molecular Sciences 20, no. 18: 4479. https://doi.org/10.3390/ijms20184479
APA StyleKovanen, P. T. (2019). Mast Cells as Potential Accelerators of Human Atherosclerosis—From Early to Late Lesions. International Journal of Molecular Sciences, 20(18), 4479. https://doi.org/10.3390/ijms20184479