Introduction
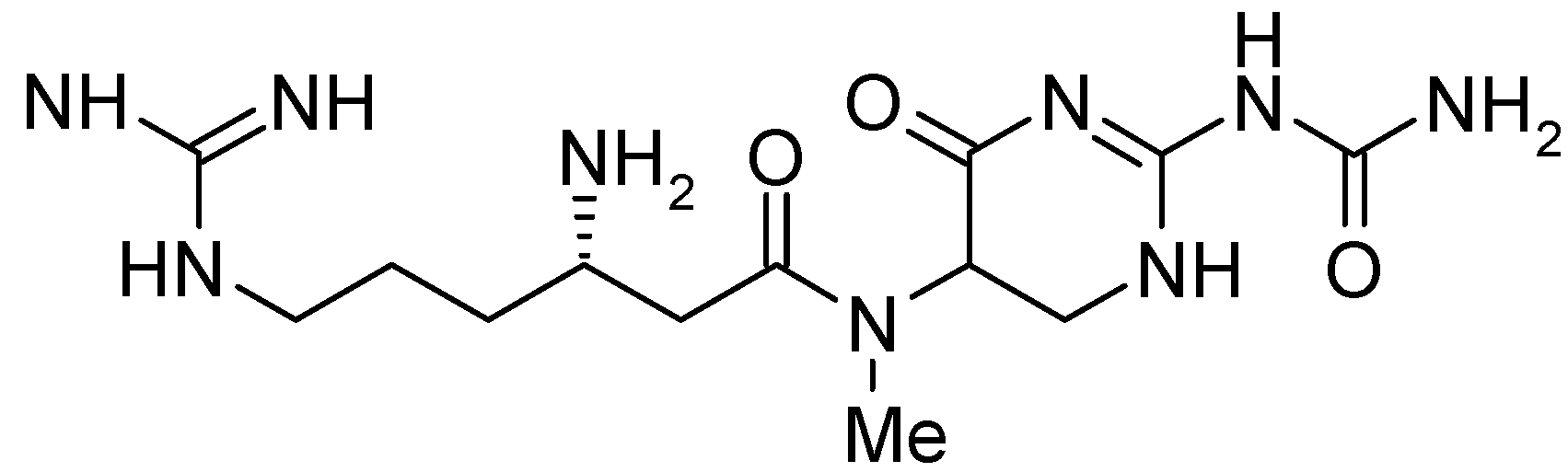
The natural dipeptide antibiotic TAN 1057 A,B (
1,
Figure 1) was first isolated and characterized by Takeda [
1]. To date, two independent total syntheses of this natural product have been disclosed [
2]. TAN 1057 A,B displays excellent
in vitro and
in vivo activity against staphylococci including
Methicillin Resistant Staphylococcus Aureus (MRSA) and represents a promising new antibiotic entity [
3]. However, TAN 1057 A,B suffers from strong toxic side effects (LD
50 = 50 mg/Kg) which are in our view prohibitive for therapeutic use in humans. Therefore, we launched a synthesis program with the goal of identifying analogs with significantly improved tolerance and concomitant retention of the promising antibiotic activity. In this communication we report a new strategy that allows for the parallel synthesis of novel derivatives of the natural product focusing on alteration of the β-arginine moiety.
Figure 1.
TAN 1057 A, B (1).
Figure 1.
TAN 1057 A, B (1).
Results and Discussion
The salient molecular architecture of TAN 1057 A,B defines its unique physical and chemical properties: the natural product is highly water soluble, displays almost no retention on RP-HPLC material under standard conditions [
4] and proves unstable towards nucleophiles and strong bases at elevated temperatures. This instability presumably results from the dihydropyrimidinone heterocycle which decomposes readily under the aforementioned conditions. However, early structure activity relationship (SAR) studies revealed that the heterocyclic core was essential for biological activity. Therefore, our program was primarily focused on alterations of the β-arginine side chain.
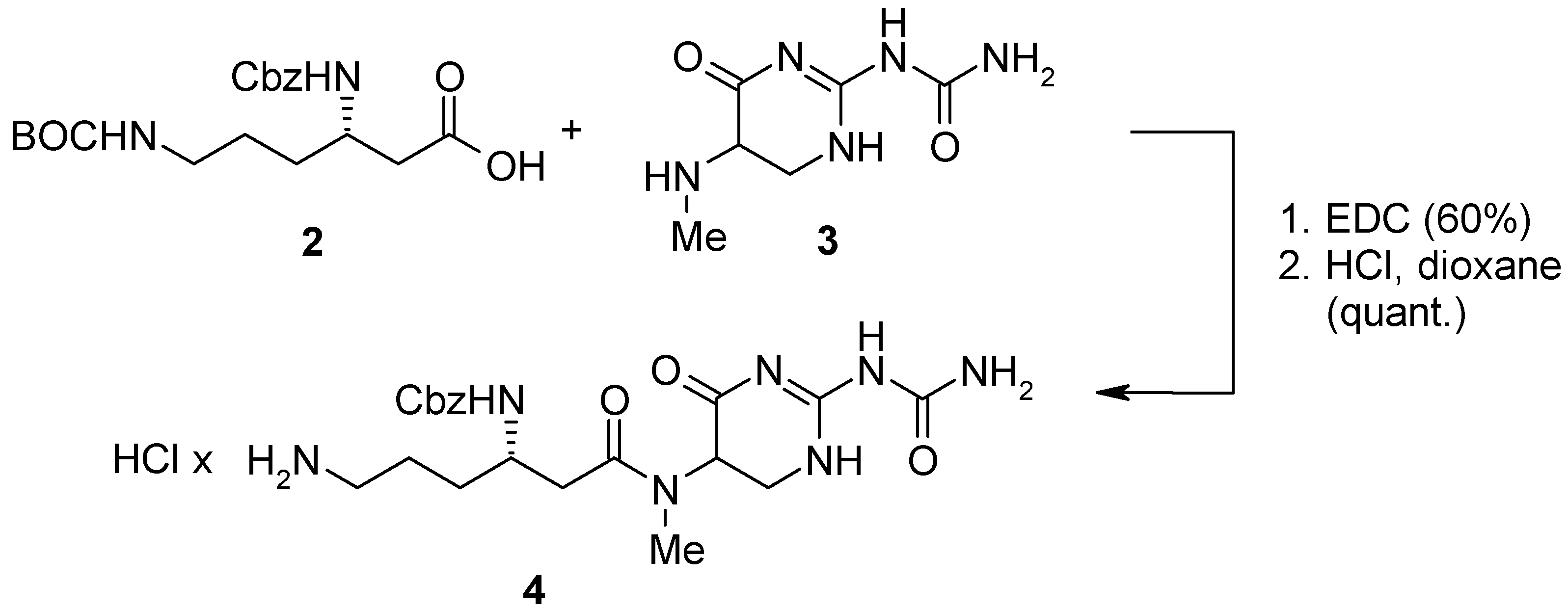
For the parallel synthesis of TAN 1057 analogs we envisioned a convergent strategy combining solid and liquid phase chemistry. Our synthesis commenced with the construction of amine
4 as outlined in
Scheme 1. Accordingly, orthogonally protected β-lysine
2 was coupled with the dihydropyrimidinone core
3 [2b] employing standard EDC-peptide coupling conditions followed by HCl mediated deprotection of the terminal BOC-group.
Scheme 1.
Synthesis of amine 4.
Scheme 1.
Synthesis of amine 4.
For the requisite installation of the guanidine moiety we employed the method published by
Dodd and
Wallace [
5] who described the use of immobilized isothioureas
6 as convenient guanylation agents (cf.
Scheme 2). Isothioureas
6 are readily available by a Mitsunobu reaction starting from commercially available resin
5.
For method evaluation we selected six structurally diverse primary alcohols R1OH (see
Table 1) and submitted them to the Mitsunobu protocol as outlined in
Scheme 2. Subsequently we used these polymeric intermediates
6 without further characterization to install the guanidine moiety at the terminus of
4. To our dismay, the conditions originally described to effect guanylation (excess amine, 50°C, DMF) failed in our hands to deliver protected intermediates
7. This was presumably due to the inherent instability of the heterocyclic moiety towards the given reaction conditions.
Scheme 2.
Parallel synthesis of TAN 1057 A,B analogs.
Scheme 2.
Parallel synthesis of TAN 1057 A,B analogs.
Thus, we surveyed a range of additional bases such as ethyl diisopropylamine, N-methylmorpholine, DBU, Schwesinger type bases or proton sponges to promote the desired transformation under milder conditions. Finally, the optimal conditions for the guanylation included the use of a slight excess of resin 6 with respect to the hydrochloride 4, DMF as solvent of choice, and DBU as base at room temperature for 3 days. The addition of transition metal salts like silver triflate or mercury chloride did not enhance the reaction rate significantly.
Subsequently, with an optimized coupling protocol in hand, all six polymeric isothioureas 6 were each reacted with 4 in a parallel setup. LC/MS analyses of the crude reaction mixtures revealed that protected intermediates 7 were formed as main products in all cases, however, the crude mixtures also contained byproducts arising from degradation of the dihydropyrimidinone. Purification of the crude products was achieved by automated preparative HPLC to deliver a range of modified natural product precursors 7 in acceptable yields around 40% (over 2 steps starting from 5) and sufficient HPLC-purities (>90% in all cases). Finally, the target molecules 8 were obtained from 7 upon deprotection of the amino groups using HBr in acetic acid followed by precipitation of the products from acetone-methanol mixtures.
The results for all TAN 1057 A,B analogs are depicted in
Table 1. In general, the overall yields (21 - 32%, 3 steps from
5) were acceptable taking into account that the dihydropyrimidinone core proved susceptible towards decomposition in the guanylation event. Moreover, HPLC analyses revealed that the majority of products were isolated in essentially pure form.
Table 1.
Analogs of TAN 1057 A,B by parallel synthesis.
Experimental
General procedure for the synthesis of 8 (90 μmol scale) as outlined in Scheme 2: To a suspension of 1.5 eq of functionalized resin
5 (purchased from
Novabiochem) in dry THF (3 mL), triphenylphosphine (9.4 eq) and the corresponding alcohol (9.4 eq) were added. Subsequently DIAD (9.0 eq.) was added dropwise and the resulting orange suspension was shaken at room temperature overnight. The solvent was filtered off and the resin was thoroughly washed with DMF (4 x 5mL), THF (3 x 5mL), MeOH (3 x 5mL) and dichloromethane (3x 5 mL).
The functionalized resin 6 thus obtained was suspended in DMF (2.5 mL) and the hydrochloride 4 (1.0 eq) was added in one portion. Finally, DBU (5 eq) was also added dropwise and the suspension was shaken at room temperature for 3 days. After filtration the resin was rinsed with DMF and the solvent was removed in vacuo. The crude products were purified using a Gilson automated preparative HPLC (conditions: Kromasil 100 C18, 5 μm, 50 x 20 mm; gradient: acetonitrile / water 15:85 to 90:10, room temperature, flow 25 mL / min, detection @ 220 nm) to yield 7 (35-45% yield, >90% HPLC purity in all cases).
Finally, at 0°C HBr (30% solution in acetic acid, 1 mL) was added to a solution of 7 in dichloromethane (2 mL) and the mixture was stirred at 0°C for 1 hour. The solvent was removed in vacuo. In the final step the products were redissolved in methanol (0.5 mL) and acetone was added until the hydrobromide salts precipitated from the solution. After filtration compounds 8 were obtained as white powders. The products were analyzed by HPLC (conditions: Kromasil 100 C18, 25 x 4 mm; gradient: A = water containing PIC B7 low UV, B = acetonitrile, 0-1 min 90% A, 1-9 min 90% A to 90% B, 9-13 min 90% B; room temperature, flow 2 mL / min, detection @ 210 nm), 1H-NMR (D4-methanol) and MS.





{kind=link}
{kind=link}
{kind=link}










