Azathioprine Has a Deleterious Effect on the Bone Health of Mice with DSS-Induced Inflammatory Bowel Disease

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
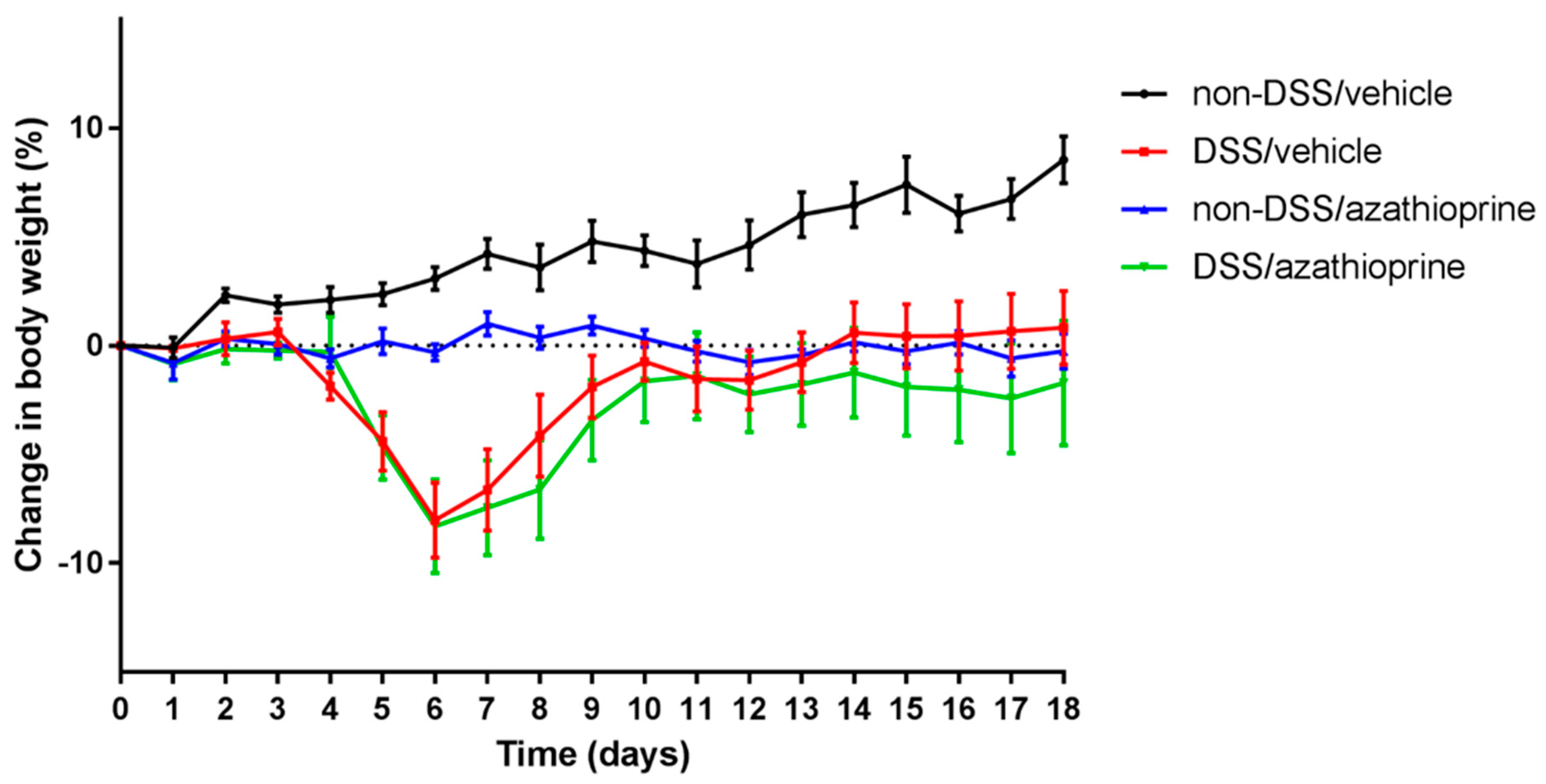
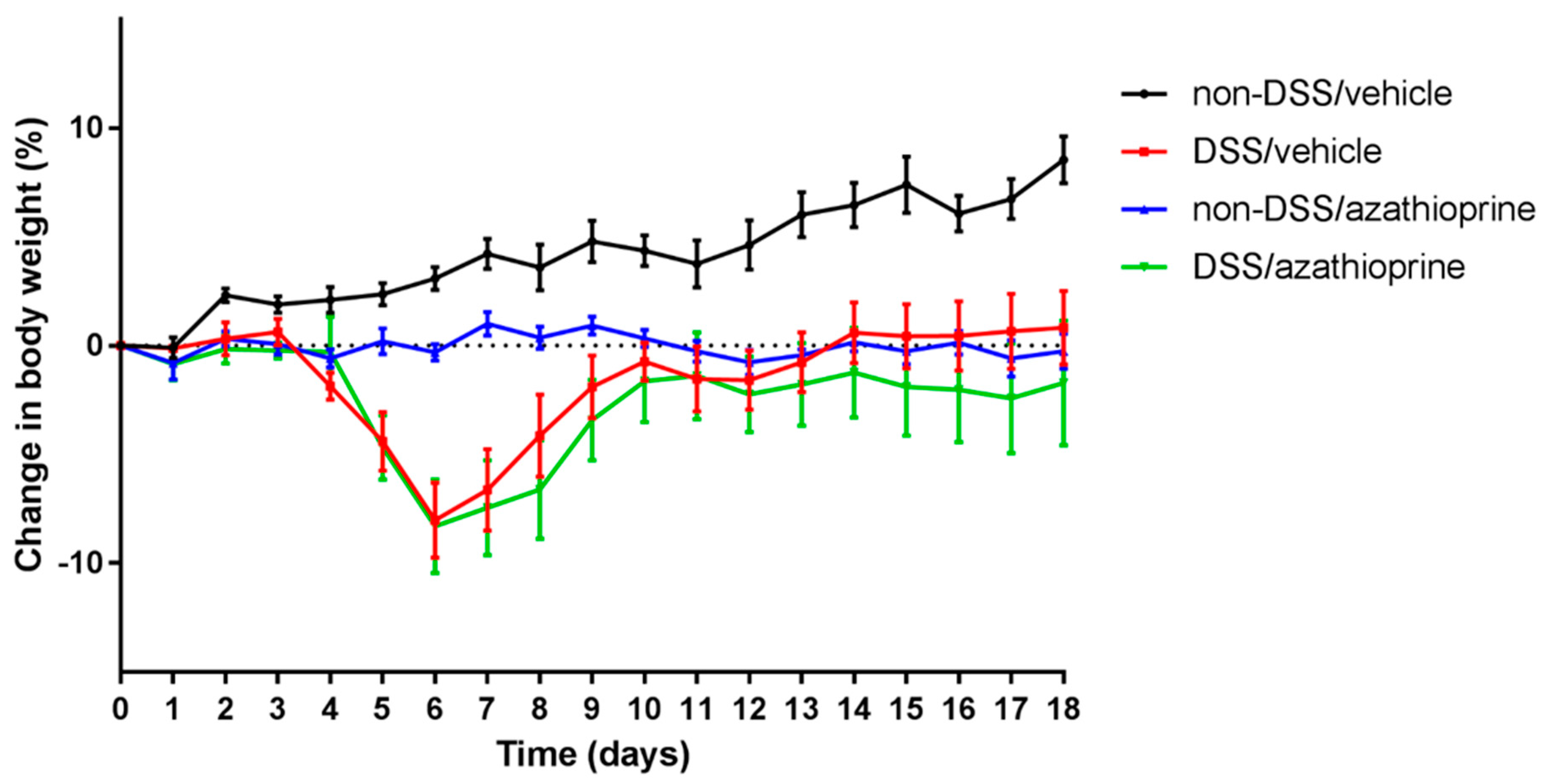
2.1. Effect of Azathioprine on Body Weight in DSS Treated Mice
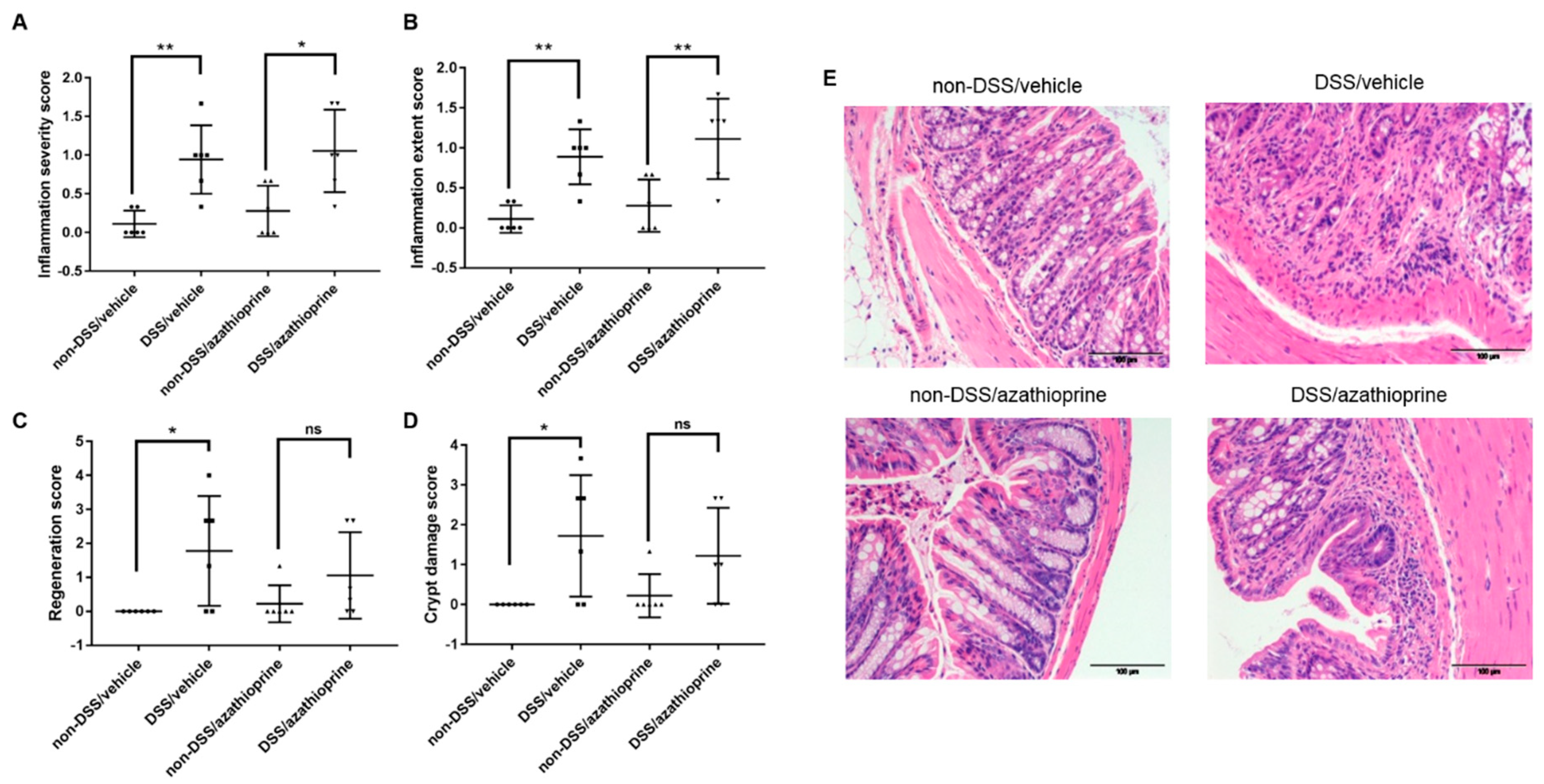
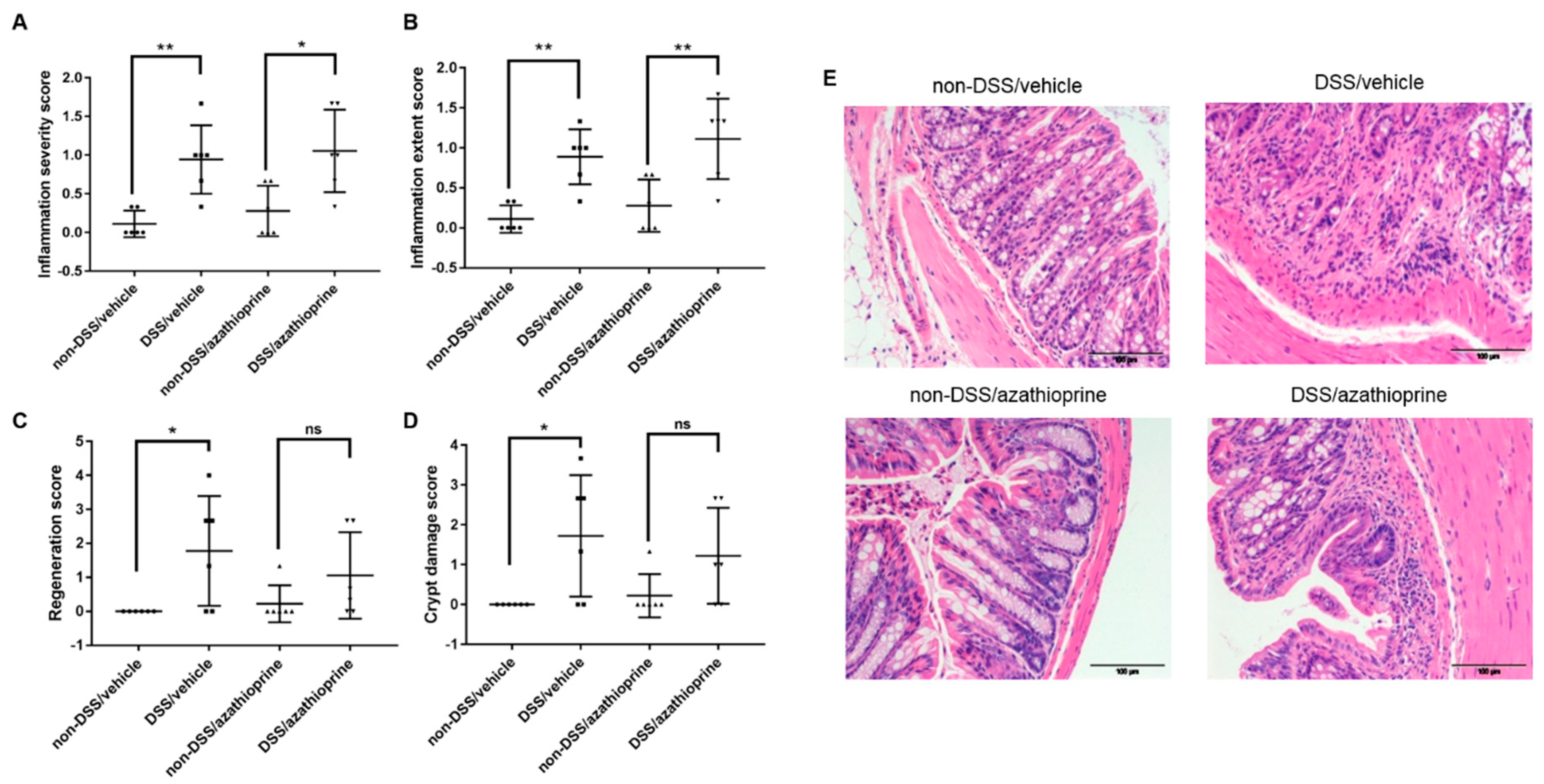
2.2. Effect of Azathioprine on Colon Pathology in DSS Treated Mice
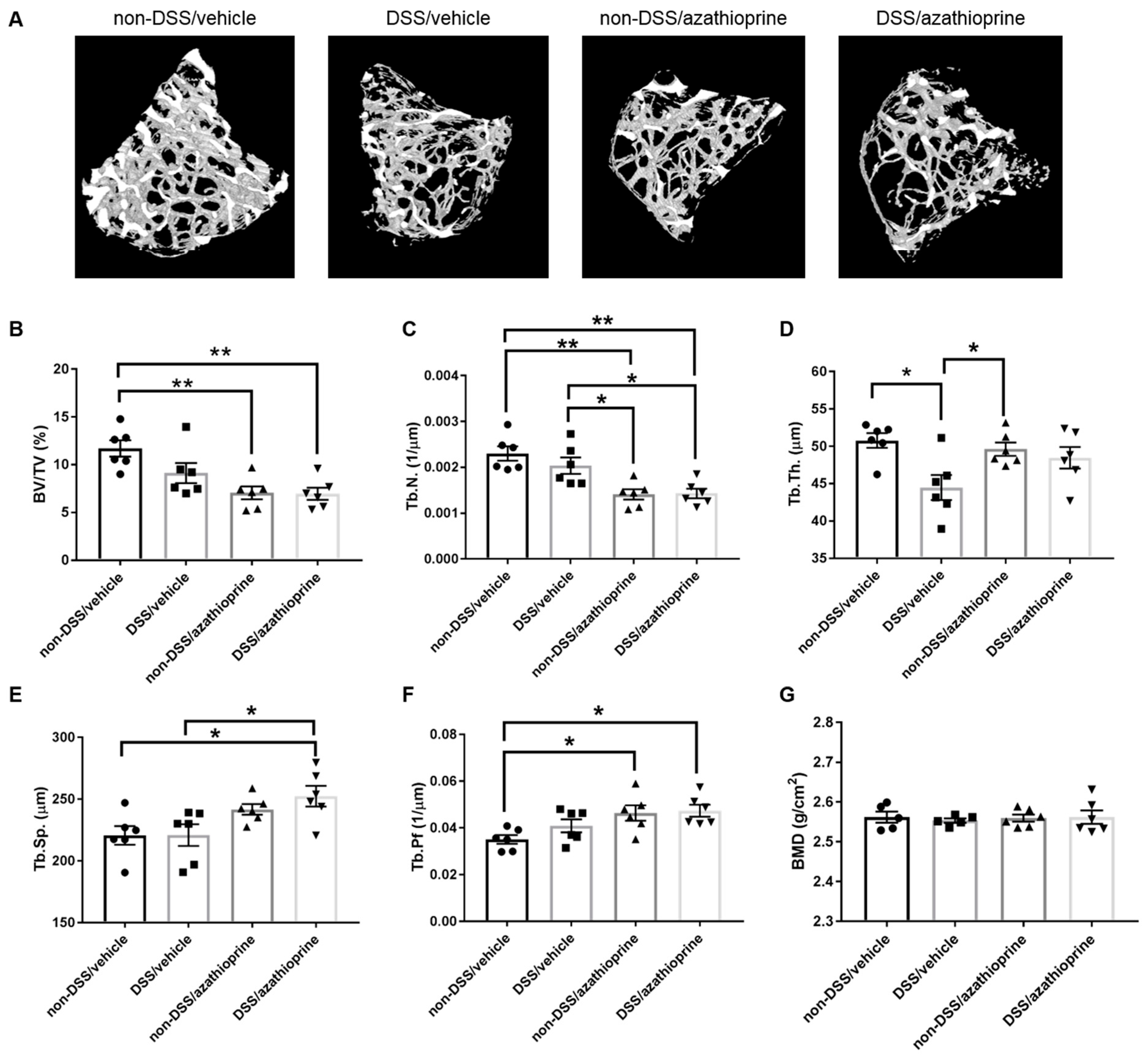
2.3. Effect of Azathioprine on Bone Phenotype in DSS Treated Mice
2.4. Induction of Autophagy by Azathioprine in DSS Treated Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Colon Pathology
4.3. Bone Histology and Immunohistochemistry
4.4. MicroCT
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- England, N. Nhs Standard Contract for Colorectal: Complex Inflammatory Bowel Disease (Adult). NHS Engl. 2013, 14, 1–6. [Google Scholar]
- Ali, T.; Lam, D.; Bronze, M.S.; Humphrey, M.B. Osteoporosis in Inflammatory Bowel Disease. Am. J. Med. 2009, 122, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.L. Inflammatory bowel diseases, celiac disease, and bone. Arch. Biochem. Biophys. 2010, 503, 54–65. [Google Scholar] [CrossRef]
- Lima, C.A. Risk factors for osteoporosis in inflammatory bowel disease patients. World J. Gastrointest. Pathophysiol. 2015, 6, 210. [Google Scholar] [CrossRef]
- Blanck, S.; Aberra, F. Vitamin D deficiency is associated with ulcerative colitis disease activity. Dig. Dis. Sci. 2013, 58, 1698–1702. [Google Scholar] [CrossRef]
- Jørgensen, S.P.; Hvas, C.L.; Agnholt, J.; Christensen, L.A.; Heickendorff, L.; Dahlerup, J.F. Active Crohn’s disease is associated with low vitamin D levels. J. Crohns Colitis 2013, 7, e407–e413. [Google Scholar] [CrossRef] [Green Version]
- Silvennoinen, J.; Lamberg-Allardt, C.; Kärkkäinen, M.; Niemelä, S.; Lehtola, J. Dietary calcium intake and its relation to bone mineral density in patients with inflammatory bowel disease. J. Intern. Med. 1996, 240, 285–292. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Ludwiczek, O.; Gabriel, M.; Obrist, P.; Wolf, A.M.; Tilg, H. The RANKL/OPG system is activated in inflammatory bowel diseases and relates to the state or bone loss. Gut 2005, 54, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.N.; Sargent, M.; Leslie, W.D. Serum osteoprotegerin is increased in Crohn’s disease: A population-based case control study. Inflamm. Bowel Dis. 2005, 11, 325–330. [Google Scholar] [CrossRef]
- Turk, N.; Cukovic-Cavka, S.; Korsic, M.; Turk, Z.; Vucelic, B. Proinflammatory cytokines and receptor activator of nuclear factor κB-ligand/osteoprotegerin associated with bone deterioration in patients with Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Radford-Smith, G.L.; Forwood, M.; Wong, J.; Taaffe, D.R. Body composition and muscle strength as predictors of bone mineral density in Crohn’s disease. J. Bone Miner. Metab. 2009, 27, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.P.; Prasad, P.; Malaty, H.M. Vitamin D deficiency and corticosteroid use are risk factors for low bone mineral density in inflammatory bowel disease patients. Dig. Dis. Sci. 2014, 59, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E. Glucocorticoid-Induced Osteoporosis: New Insights into the Pathophysiology and Treatments. Curr. Osteoporos. Rep. 2019, 17, 1–7. [Google Scholar] [CrossRef]
- Miheller, P.; Muzes, G.; Rácz, K.; Blázovits, A.; Lakatos, P.; Herszényi, L.; Tulassay, Z. Changes of OPG and RANKL concentrations in Crohn’s disease after infliximab therapy. Inflamm. Bowel Dis. 2007, 13, 1379–1384. [Google Scholar] [CrossRef]
- Franchimont, N.; Putzeys, V.; Collette, J.; Vermeire, S.; Rutgeerts, P.; De Vos, M.; Van Gossum, A.; Franchimont, D.; Fiasse, R.; Pelckmansj, P.; et al. Rapid improvement of bone metabolism after infliximab treatment in Crohn’s disease. Aliment. Pharmacol. Ther. 2004, 20, 607–614. [Google Scholar] [CrossRef]
- Abreu, M.T.; Geller, J.L.; Vasiliauskas, E.A.; Kam, L.Y.; Vora, P.; Martyak, L.A.; Yang, H.; Hu, B.; Lin, Y.C.; Keenan, G.; et al. Treatment with infliximab is associated with increased markers of bone formation in patients with Crohn’s disease. J. Clin. Gastroenterol. 2006, 40, 55–63. [Google Scholar] [CrossRef]
- Krajcovicova, A.; Hlavaty, T.; Killinger, Z.; Miznerova, E.; Toth, J.; Letkovsky, J.; Nevidanska, M.; Cierny, D.; Koller, T.; Zelinkova, Z.; et al. Combination therapy with an immunomodulator and anti-TNFα agent improves bone mineral density in IBD patients. J. Crohns Colitis 2014, 8, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Denson, L.A. The role of the innate and adaptive immune system in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory bowel disease drugs: A focus on autophagy. J. Crohns Colitis 2017, 11, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perše, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Mukaisyo, K.I.; Sugihara, H.; Fujiyama, Y.; Hattori, T. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol. Rep. 2010, 24, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, G.; Gabet, Y.; Rachmilewitz, D.; Karmeli, F.; Bab, I.; Dresner-Pollak, R. Dextran sodium sulfate-induced colitis causes rapid bone loss in mice. Bone 2008, 43, 945–950. [Google Scholar] [CrossRef]
- Dobie, R.; MacRae, V.E.; Pass, C.; Milne, E.M.; Ahmed, S.F.; Farquharson, C. Suppressor of cytokine signaling 2 (Socs2) deletion protects bone health of mice with DSS-induced inflammatory bowel disease. Dis. Model. Mech. 2018, 11, dmm028456. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Methotrexate, azathioprine, cyclosporine, and risk of fracture. Calcif. Tissue Int. 2006, 79, 69–75. [Google Scholar] [CrossRef]
- Schulte, C.M.S.; Dignass, A.U.; Goebell, H.; Roher, H.D.; Schulte, K.M. Genetic factors determine extent of bone loss in inflammatory bowel disease. Gastroenterology 2000, 119, 909–920. [Google Scholar] [CrossRef]
- Paganelli, M.; Albanese, C.; Borrelli, O.; Civitelli, F.; Canitano, N.; Viola, F.; Passariello, R.; Cucchiara, S. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 416–423. [Google Scholar] [CrossRef]
- Fernandez-Vojvodich, P.; Zaman, F.; Sävendahl, L. Interleukin-6 acts locally on the growth plate to impair bone growth. Ann. Rheum. Dis. 2013, 72, e24. [Google Scholar] [CrossRef]
- Wong, S.; Smyth, A.; McNeill, E.; Galloway, P.; Hassan, K.; McGrogan, P.; Ahmed, S. The growth hormone insulin‐like growth factor 1 axis in children and adolescents with inflammatory bowel disease and growth retardation. Clin. Endocrinol. 2010, 73, 220–228. [Google Scholar]
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: Current Evidence, Gaps in Knowledge, and Future Directions. Endocr. Rev. 2016, 37, 62–110. [Google Scholar] [CrossRef] [PubMed]
- Cegiela, U.; Kaczmarczyk-Sedlak, I.; Pytlik, M.; Folwarczna, J.; Nowińska, B.; Fronczek-Sokół, J. Alendronate prevents development of the skeletal changes induced by azathioprine in rats. Acta Pol. Pharm. Drug Res. 2013, 70, 309–315. [Google Scholar]
- Chaabane, W.; Appell, M.L. Interconnections between apoptotic and autophagic pathways during thiopurine-induced toxicity in cancer cells: The role of reactive oxygen species. Oncotarget 2016, 7, 75616–75634. [Google Scholar] [CrossRef] [Green Version]
- Henderson, P.; Stevens, C. The Role of Autophagy in Crohn’s Disease. Cells 2012, 1, 492–519. [Google Scholar] [CrossRef] [Green Version]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1977. [Google Scholar] [CrossRef]
- Lee, K.-W.; Yook, J.-Y.; Son, M.-Y.; Kim, M.-J.; Koo, D.-B.; Han, Y.-M.; Cho, Y.S. Rapamycin promotes the osteoblastic differentiation of human embryonic stem cells by blocking the mTOR pathway and stimulating the BMP/Smad pathway. Stem Cells Dev. 2010, 19, 557–568. [Google Scholar] [CrossRef]
- Li, X.; Chang, B.; Wang, B.; Bu, W.; Zhao, L.; Liu, J.; Meng, L.; Wang, L.; Xin, Y.; Wang, D.; et al. Rapamycin promotes osteogenesis under inflammatory conditions. Mol. Med. Rep. 2017, 16, 8923–8929. [Google Scholar] [CrossRef] [Green Version]
- Singha, U.K.; Jiang, Y.; Yu, S.; Luo, M.; Lu, Y.; Zhang, J.; Xiao, G. Rapamycin inhibits osteoblast proliferation and differentiation in MC3T3-E1 cells and primary mouse bone marrow stromal cells. J. Cell. Biochem. 2008, 103, 434–446. [Google Scholar] [CrossRef]
- Williams, K.L.; Fuller, C.R.; Dieleman, L.A.; DaCosta, C.M.; Haldeman, K.M.; Sartor, R.B.; Lund, P.K. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology 2001, 120, 925–937. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Palmen, M.J.H.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.M.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Zhou, T.; Shu, J.; Mao, J.-H. Quantifying Chromogen Intensity in Immunohistochemistry via Reciprocal Intensity. Available online: www.CancerInCytes.org (accessed on 19 November 2019).
- Staines, K.A.; Javaheri, B.; Hohenstein, P.; Fleming, R.; Ikpegbu, E.; Unger, E.; Hopkinson, M.; Buttle, D.J.; Pitsillides, A.A.; Farquharson, C. Hypomorphic conditional deletion of E11/Podoplanin reveals a role in osteocyte dendrite elongation. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, S.; Hooper, K.M.; Milne, E.M.; Farquharson, C.; Stevens, C.; Staines, K.A. Azathioprine Has a Deleterious Effect on the Bone Health of Mice with DSS-Induced Inflammatory Bowel Disease. Int. J. Mol. Sci. 2019, 20, 6085. https://doi.org/10.3390/ijms20236085
Morgan S, Hooper KM, Milne EM, Farquharson C, Stevens C, Staines KA. Azathioprine Has a Deleterious Effect on the Bone Health of Mice with DSS-Induced Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2019; 20(23):6085. https://doi.org/10.3390/ijms20236085
Chicago/Turabian StyleMorgan, Stephanie, Kirsty M. Hooper, Elspeth M. Milne, Colin Farquharson, Craig Stevens, and Katherine A. Staines. 2019. "Azathioprine Has a Deleterious Effect on the Bone Health of Mice with DSS-Induced Inflammatory Bowel Disease" International Journal of Molecular Sciences 20, no. 23: 6085. https://doi.org/10.3390/ijms20236085
APA StyleMorgan, S., Hooper, K. M., Milne, E. M., Farquharson, C., Stevens, C., & Staines, K. A. (2019). Azathioprine Has a Deleterious Effect on the Bone Health of Mice with DSS-Induced Inflammatory Bowel Disease. International Journal of Molecular Sciences, 20(23), 6085. https://doi.org/10.3390/ijms20236085