IgA: Structure, Function, and Developability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
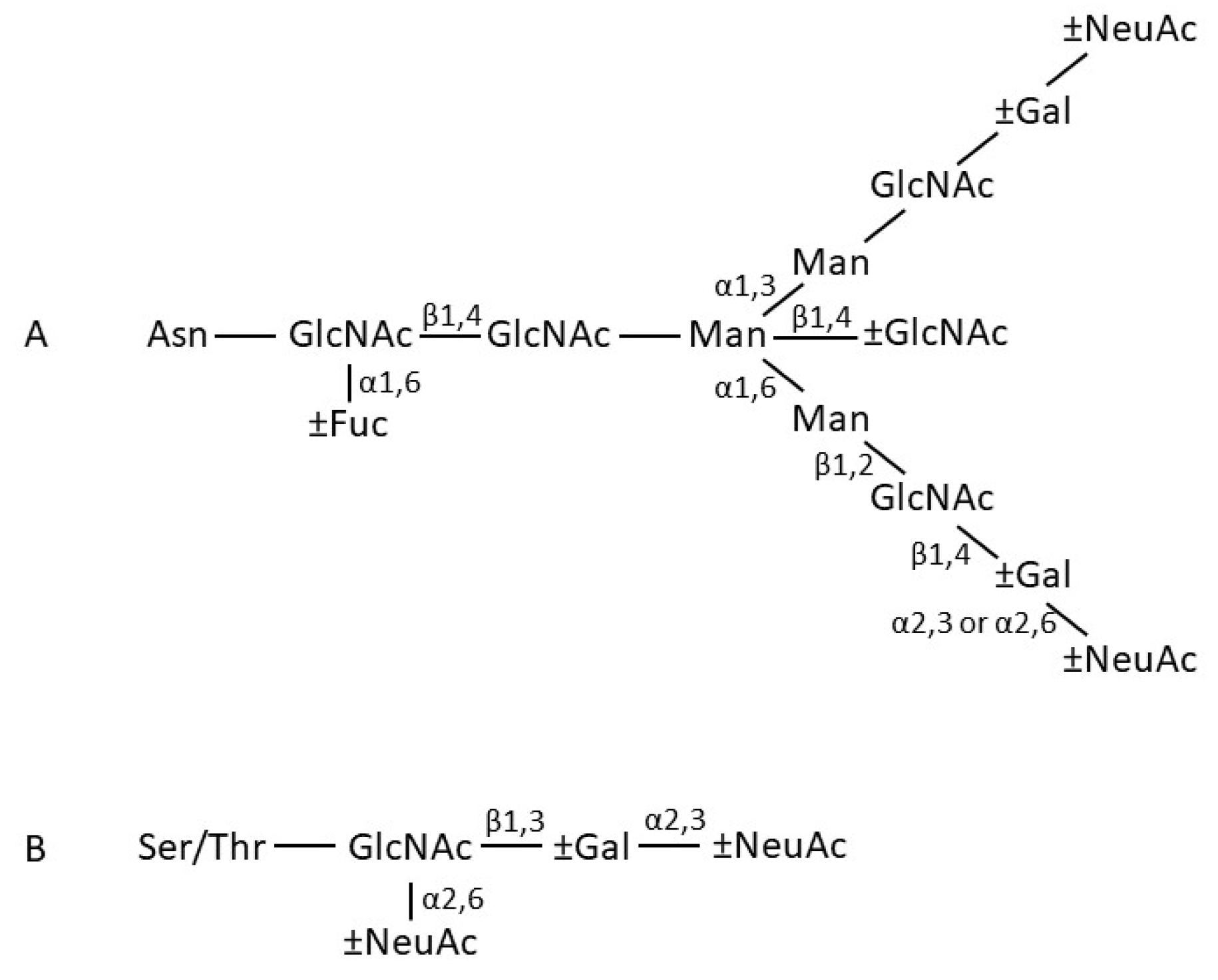
2. IgA structure
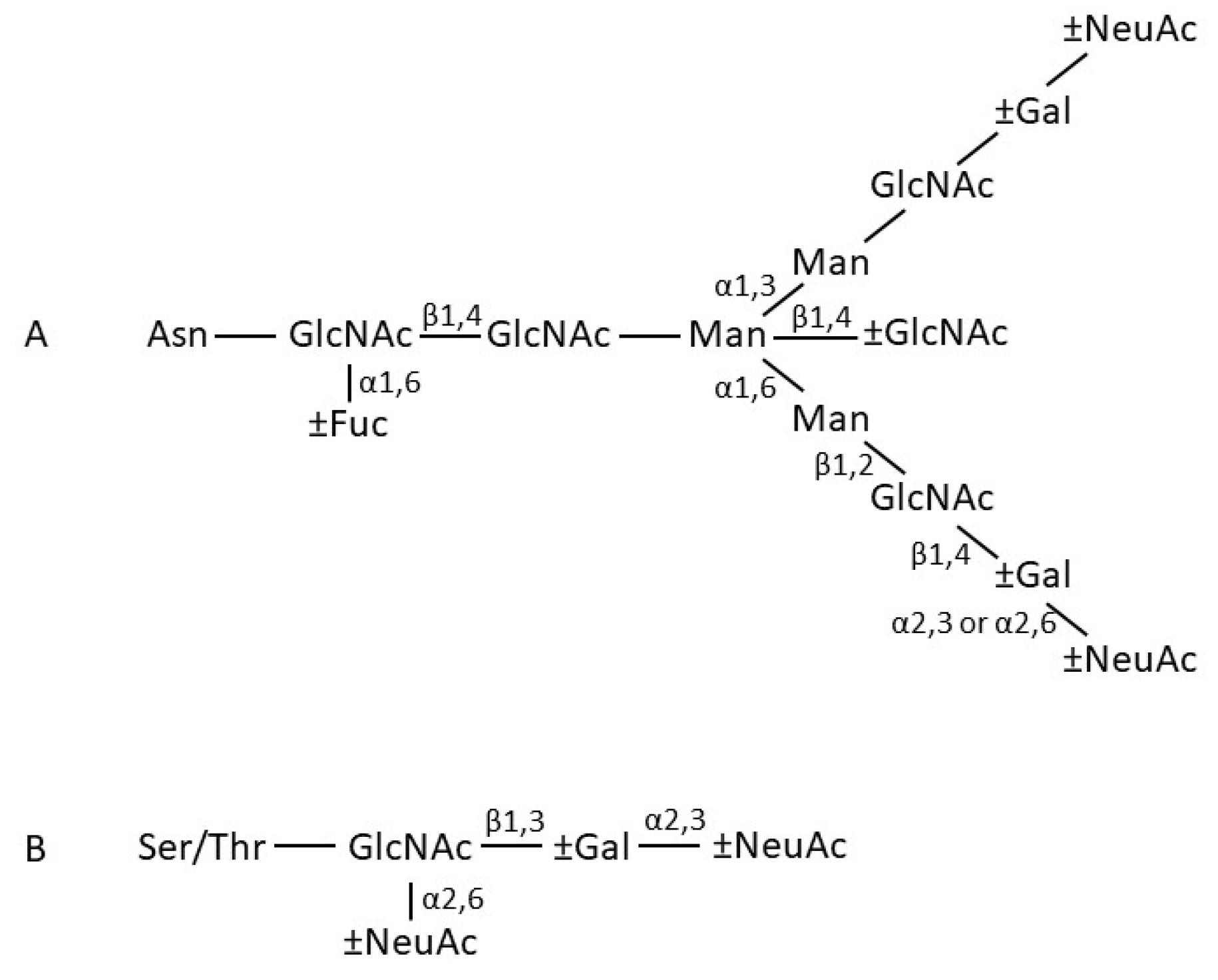
2.1. General Features
2.2. IgA Fab Region
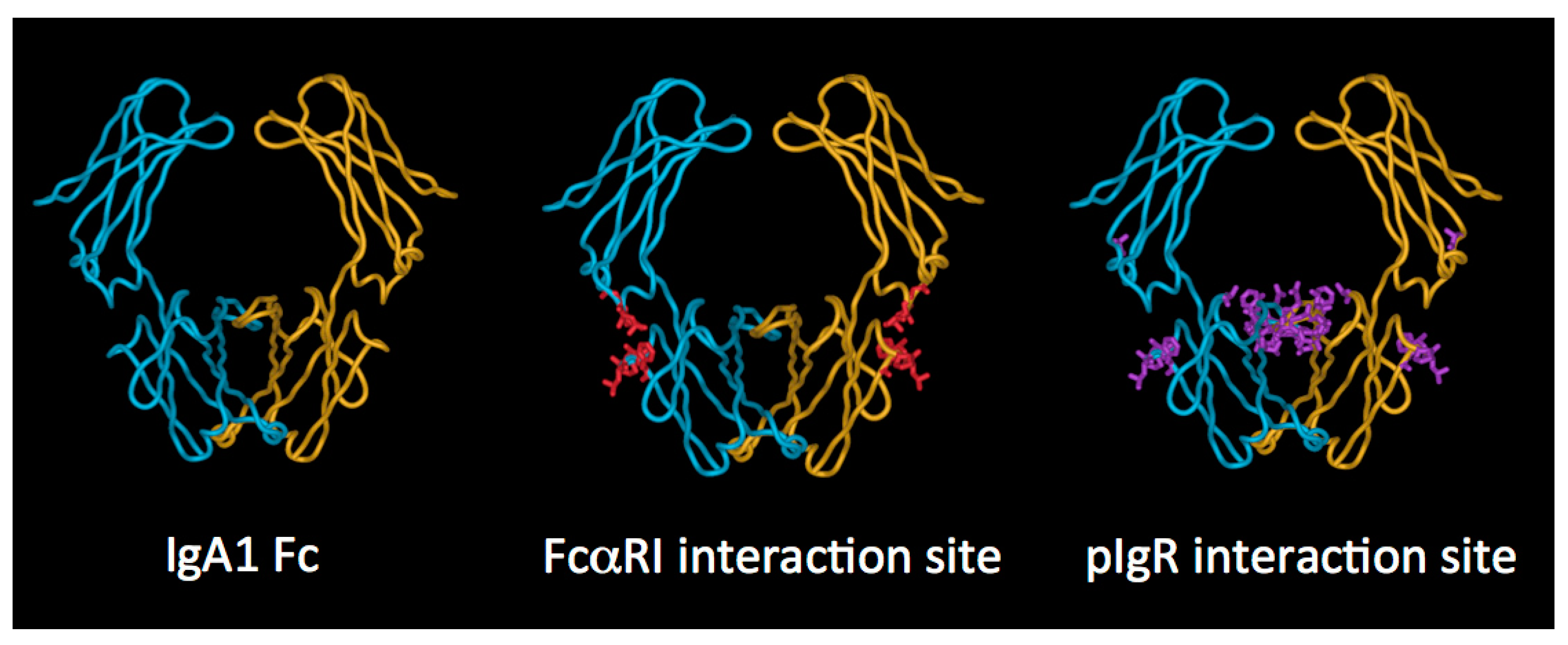
2.3. IgA Fc Region
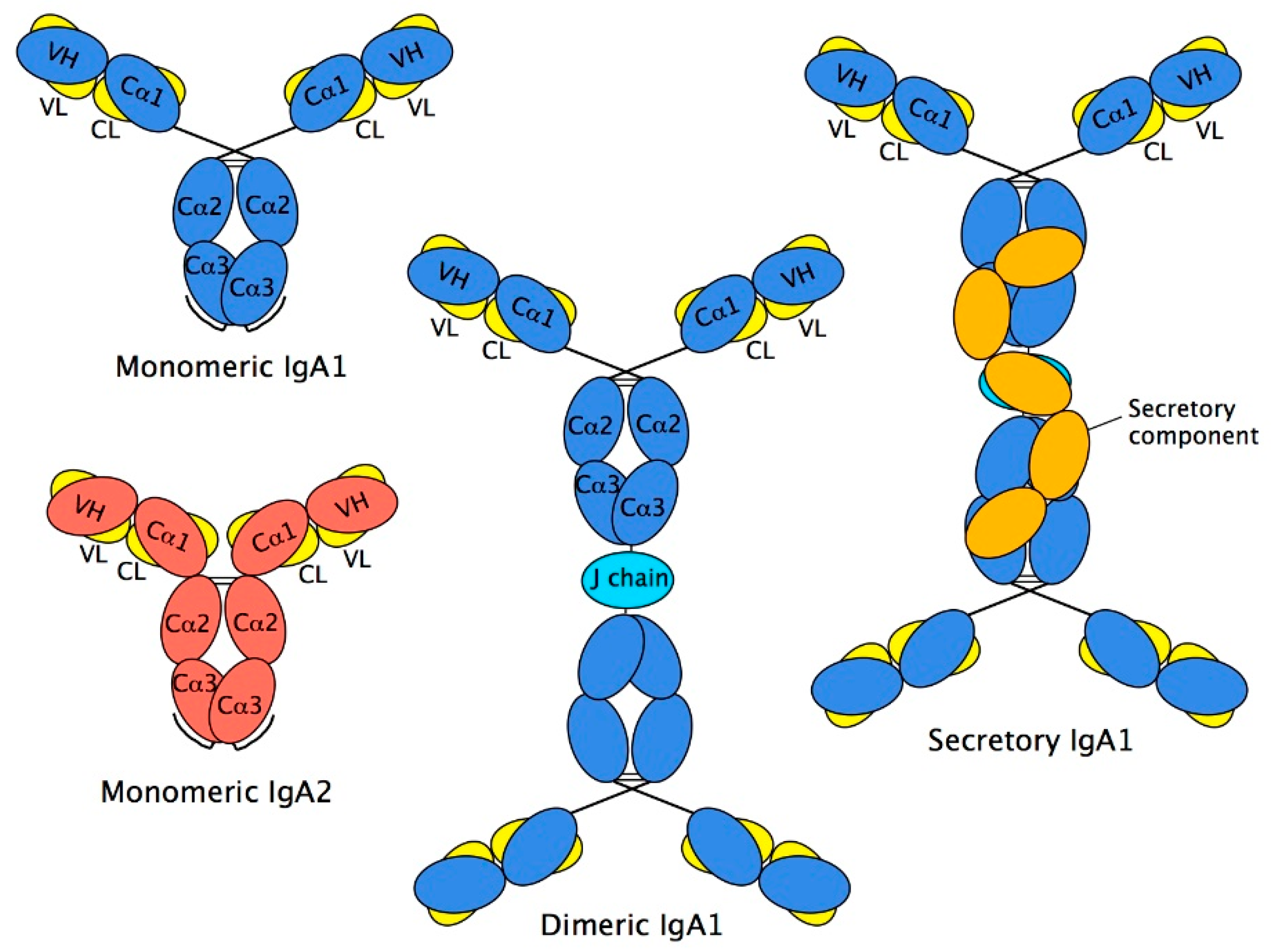
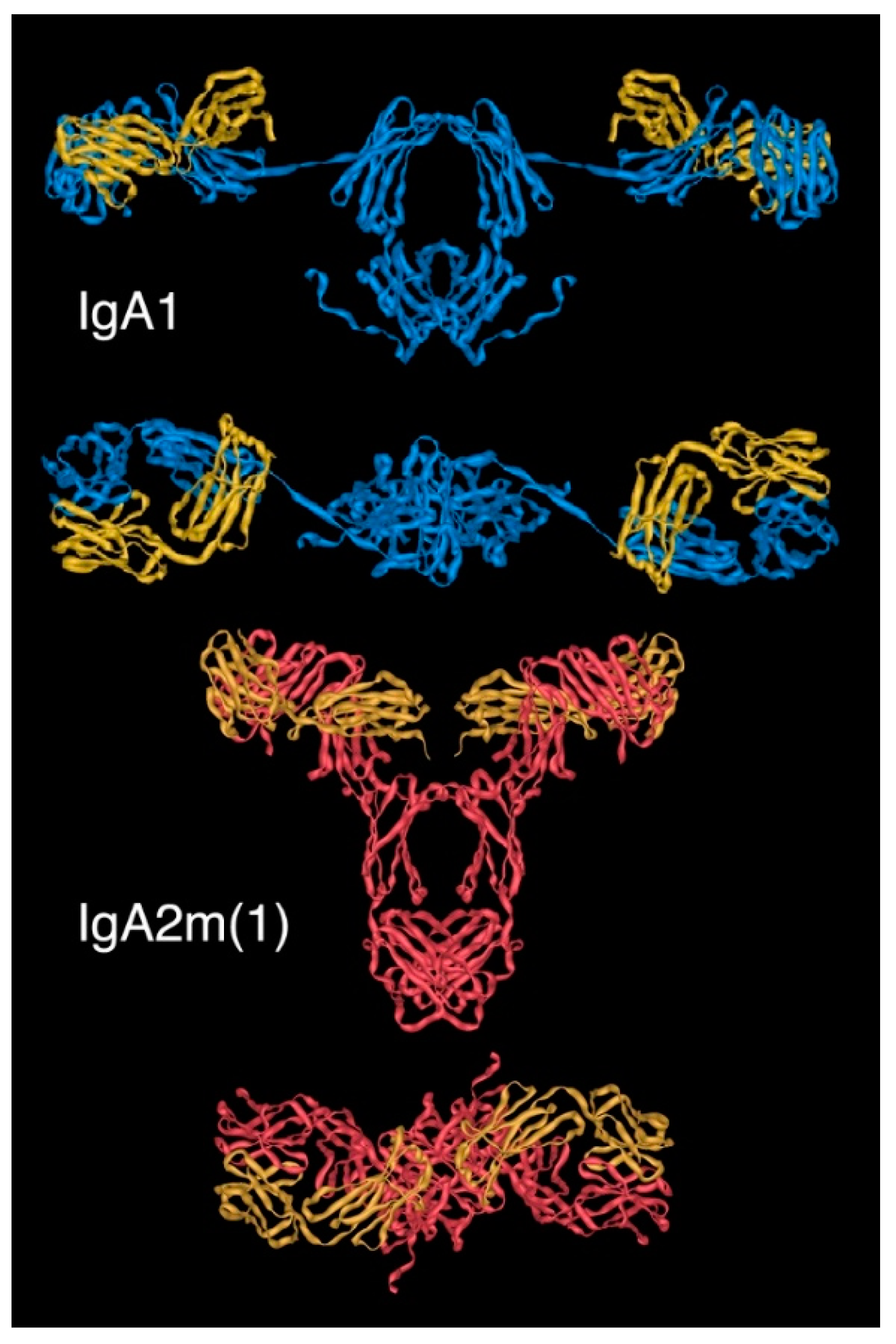
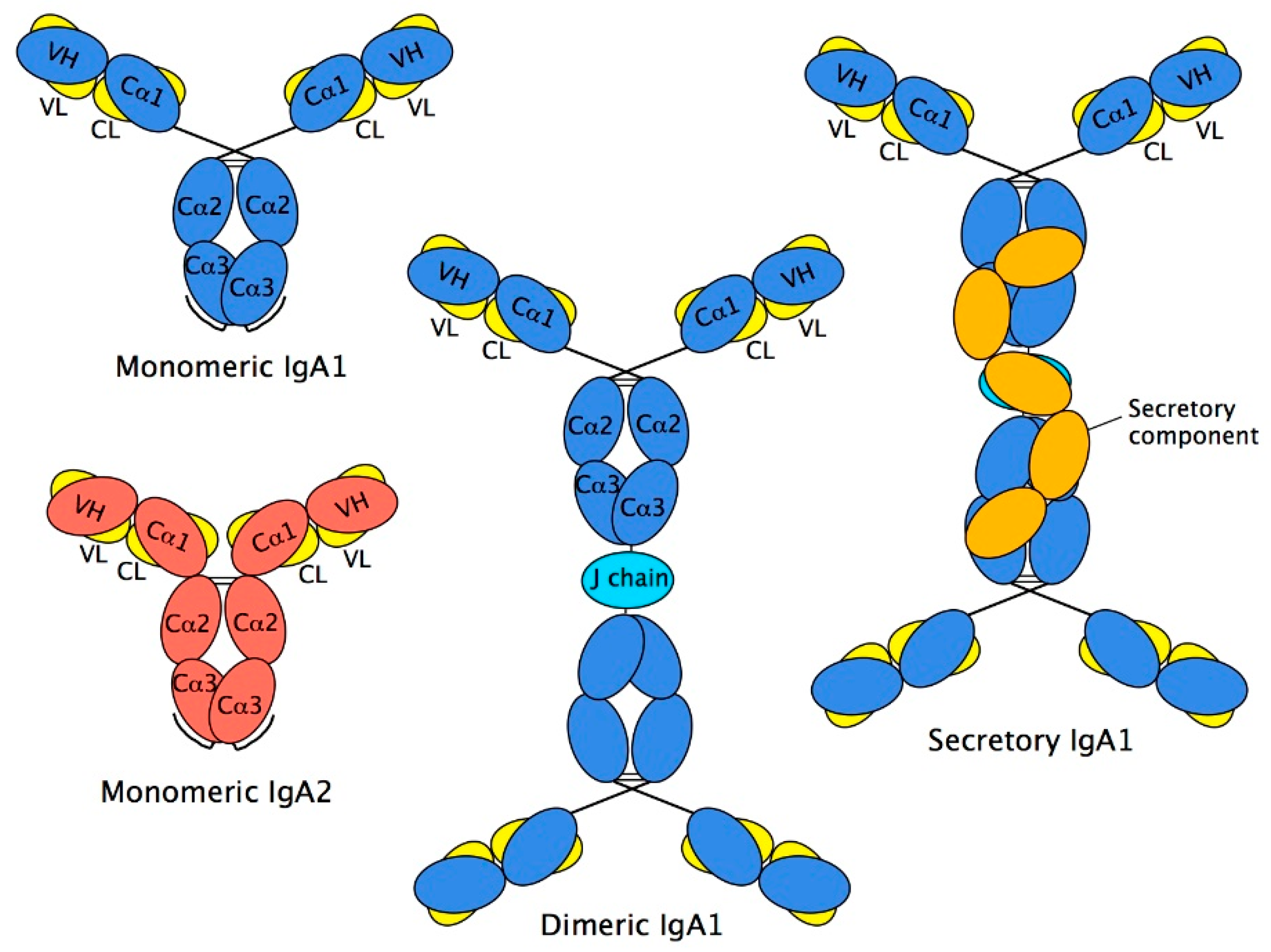
2.4. Structure of Monomeric IgA
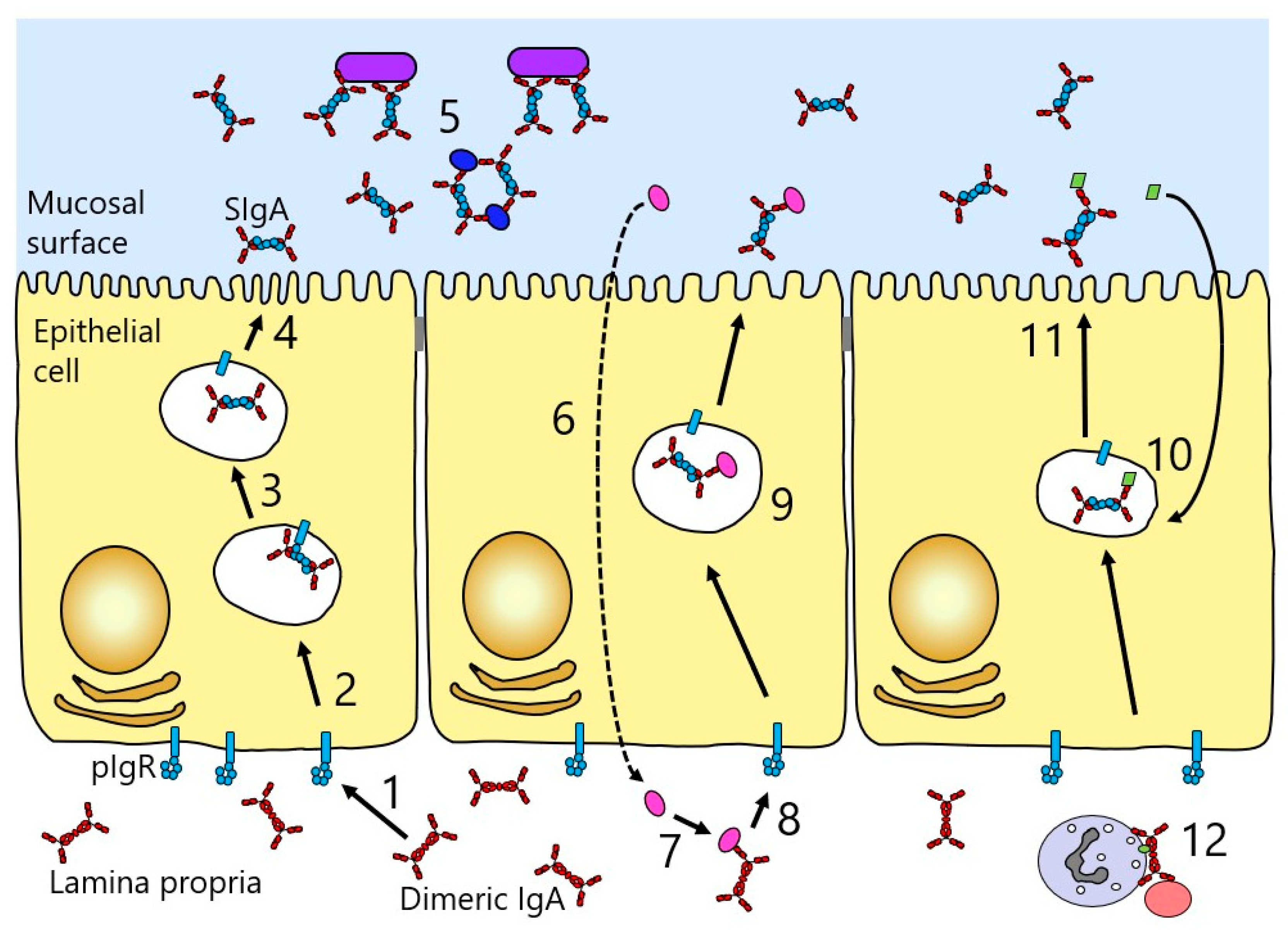
2.5. Dimeric IgA
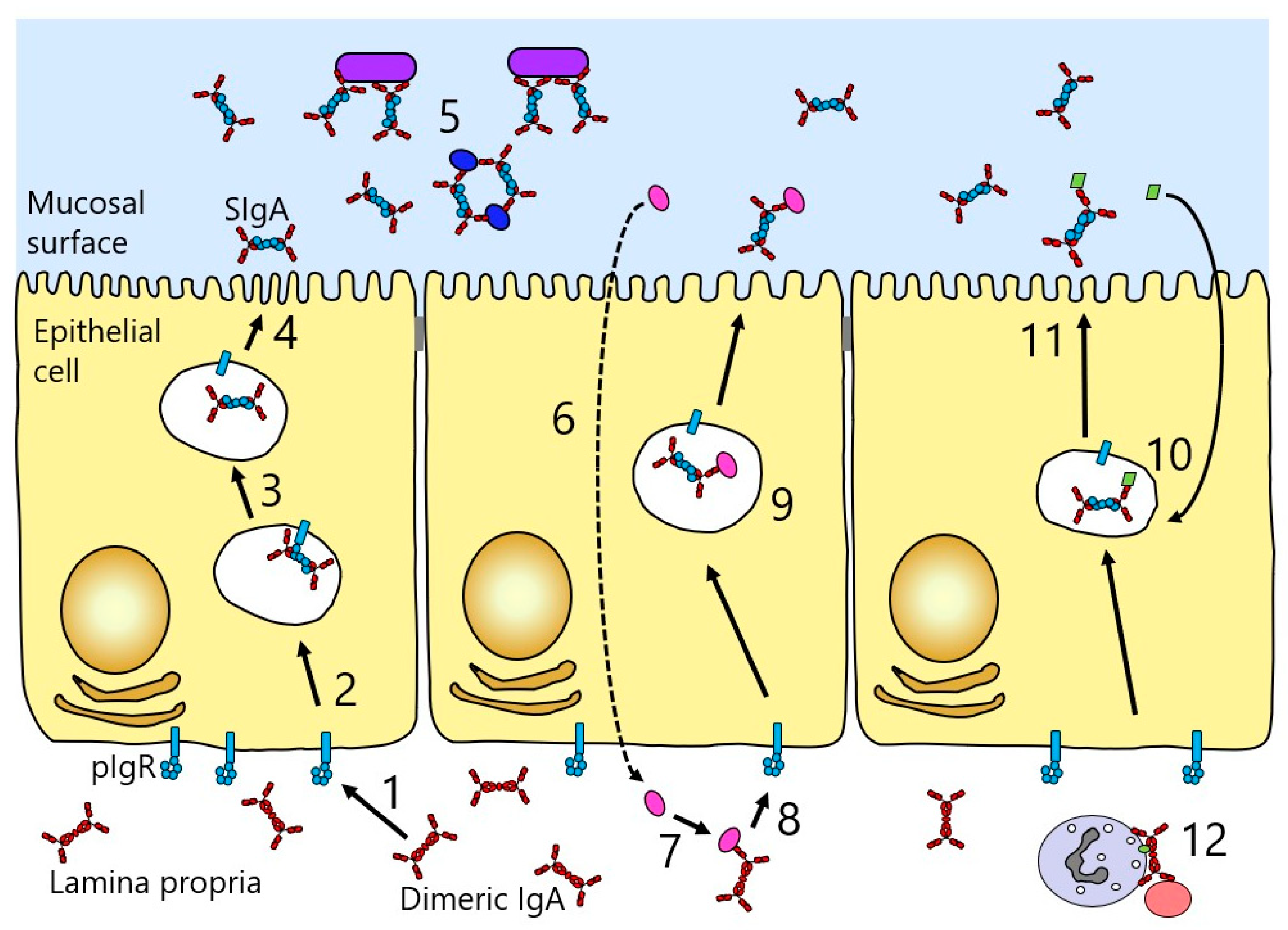
2.6. Secretory IgA
3. IgA Function
3.1. Neutralisation
3.2. Complement Activation
3.3. Interaction of the IgA Fc Region with Host Receptors
3.4. FcαRI
4. Circumvention of IgA Function by Pathogens
4.1. Bacterial IgA Binding Proteins
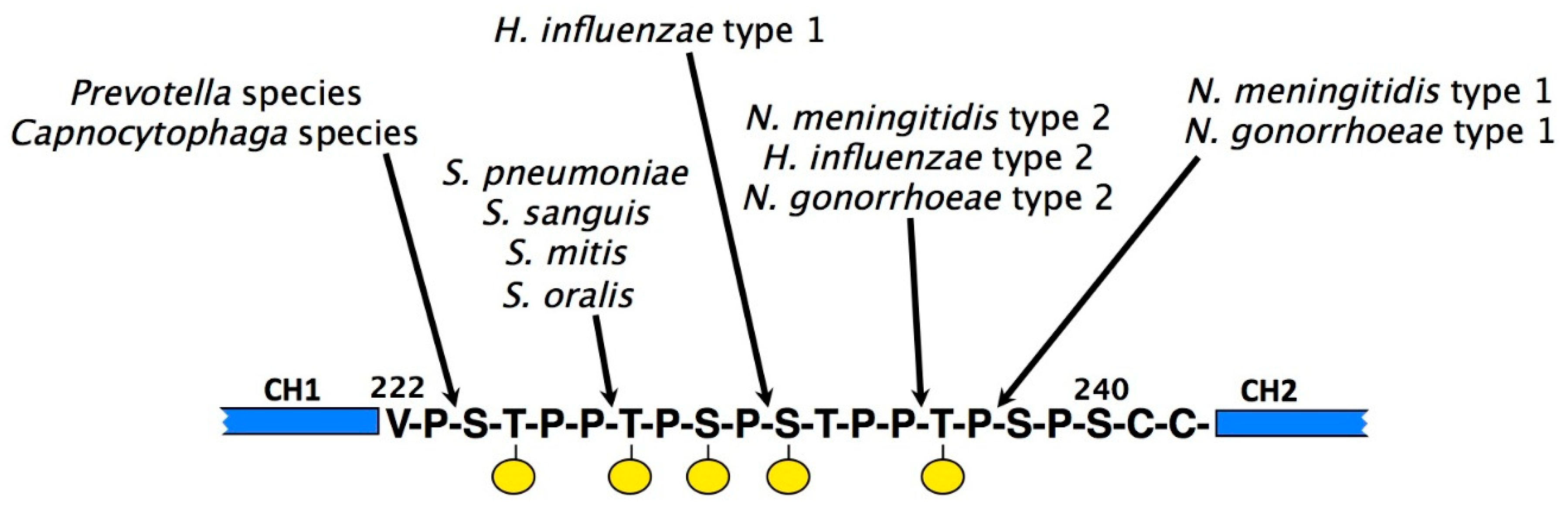
4.2. Bacterial Proteases That Target IgA
5. IgA Developability
5.1. Advantages of IgA-Based Therapeutics
5.2. Constraints of Using IgA Therapeutically and Efforts to Resolve These
6. Current landscape of IgA-Based Therapeutics
6.1. Comparisons of IgG and IgA mAbs in Cancer Therapy
6.2. IgA mAbs in Treating or Preventing Infections
6.3. FcαRI Blocking Agents
7. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mestecky, J.; Russell, M.W.; Jackson, S.; Brown, T.A. The human IgA system: A reassessment. Clin. Immunol. Immunopathol. 1986, 40, 105–114. [Google Scholar] [CrossRef]
- Conley, M.E.; Delacroix, D.L. Intravascular and mucosal immunoglobulin A: Two separate but related systems of immune defense? Ann. Intern. Med. 1987, 106, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Childers, N.K.; Bruce, M.G.; McGhee, J.R. Molecular mechanisms of immunoglobulin A defense. Annu. Rev. Immunol. 1989, 43, 503–536. [Google Scholar] [CrossRef] [PubMed]
- Chintalacharuvu, K.R.; Raines, M.; Morrison, S.L. Divergence of human alpha-chain constant region gene sequences. A novel recombinant alpha2 gene. J. Immunol. 1994, 152, 5299–5304. [Google Scholar]
- Kawamura, S.; Saitou, N.; Ueda, S. Concerted evolution of the primate immunoglobulin α-gene through gene conversion. J. Biol. Chem. 1992, 267, 7359–7367. [Google Scholar]
- Pinheiro, A.; de Sousa-Pereira, P.; Strive, T.; Knight, K.L.; Woof, J.M.; Esteves, P.J.; Abrantes, J. Identification of a new European rabbit IgA with a serine-rich hinge region. PLoS ONE 2018, 13, e0201567. [Google Scholar] [CrossRef] [Green Version]
- Snoeck, V.; Peters, I.R.; Cox, E. The IgA system: A comparison of structure and function in different species. Vet. Res. 2006, 37, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Putnam, F.W.; Yu-Sheng, V.L.; Low, T.L.K. Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease digestion, Fab and Fc fragment and the complete amino acid sequence of the α1 heavy chain. J. Biol. Chem. 1979, 254, 2865–2874. [Google Scholar]
- Tomana, M.; Niedermeier, W.; Mestecky, J.; Skvaril, F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry 1976, 13, 325–328. [Google Scholar] [CrossRef]
- Maurer, M.A.; Meyer, L.; Bianchi, M.; Turner, H.L.; Le, N.P.L.; Steck, M.; Wyrzucki, A.; Orlowski, V.; Ward, A.B.; Crispin, M.; et al. Glycosylation of human IgA directly inhibits influenza A and other sialic-acid-binding viruses. Cell Rep. 2018, 23, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Field, M.C.; Amatayakul-Chantler, S.; Rademacher, T.W.; Rudd, P.M.; Dwek, R.A. Structural analysis of the N-glycans from human immunoglobulin A1: Comparison of normal human serum immunoglobulin A1 from that isolated from patients with rheumatoid arthritis. Biochem. J. 1994, 299, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattu, T.S.; Pleass, R.P.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royle, L.; Roos, A.; Harvey, D.J.; Wormald, M.R.; van Gijlswijk-Jannsen, D.; El Redwan, R.M.; Wilson, I.A.; Daha, M.R.; Dwek, R.A.; Rudd, P.M. Secretory IgA N- and O-linked glycans provide a link between the innnate and adaptive immune systems. J. Biol. Chem. 2003, 278, 20140–20153. [Google Scholar] [CrossRef] [PubMed]
- Satow, Y.; Cohen, G.H.; Padlan, E.A.; Davies, D.R. Phosphocholine binding immunoglobulin Fab McPC603. An X-ray diffraction study at 2.7 Å. J. Mol. Biol. 1986, 190, 593–604. [Google Scholar] [CrossRef]
- Suh, S.W.; Bhat, T.N.; Navia, M.A.; Cohen, G.H.; Rao, D.N.; Rudikoff, S.; Davies, D.R. The galactan-binding immunoglobulin Fab J539: An X-ray diffraction study at 2.6-A resolution. Proteins 1986, 1, 74–80. [Google Scholar] [CrossRef]
- Correa, A.; Trajtenberg, F.; Obal, G.; Pritsch, O.; Dighiero, G.; Oppezzo, P.; Buschiazzo, A. Structure of a human IgA1 Fab fragment at 1.55 Å resolution: Potential effect of the constant domains on antigen-affinity modulation. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Janda, A.; Bowen, A.; Greenspan, N.S.; Casadevall, A. Ig constant region effects on variable region structure and function. Front. Microbiol. 2016, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Herr, A.B.; Ballister, E.R.; Bjorkman, P.J. Insights into IgA-mediated immune responses from the crystal structures of human FcαRI and its complex with IgA1-Fc. Nature 2003, 423, 614–620. [Google Scholar] [CrossRef]
- Ramsland, P.A.; Willoughby, N.; Trist, H.M.; Farrugia, W.; Hogarth, P.M.; Fraser, J.D.; Wines, B.D. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc. Natl. Acad. Sci. USA 2007, 10, 15051–15056. [Google Scholar] [CrossRef] [Green Version]
- Göritzer, K.; Turupcu, A.; Maresch, D.; Novak, J.; Altmann, F.; Oostenbrink, C.; Obinger, C.; Strasser, R. Distinct Fcα receptor N-glycans modulate the binding affinity to immunoglobulin A (IgA) antibodies. J. Biol. Chem. 2019, 294, 13995–14008. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, A.; Munn, E.; Richardson, N. The three-dimensional conformation of γM and γA globulin molecules. Ann. N. Y. Acad. Sci. 1971, 190, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Munn, E.A.; Feinstein, A.; Munro, A.J. Electron microscope examination of free IgA molecules and of their complexes with antigen. Nature 1971, 231, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.H.; Strelets, L.; Brekke, O.H.; Sandlie, I.; Michaelsen, T.E. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: A role for flexibility and geometry. J. Immunol. 1998, 161, 4083–4090. [Google Scholar] [PubMed]
- Boehm, M.K.; Woof, J.M.; Kerr, M.A.; Perkins, S.J. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: A study by X-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 1999, 286, 1421–1447. [Google Scholar] [CrossRef] [PubMed]
- Furtado, P.B.; Whitty, P.W.; Robertson, A.; Eaton, J.T.; Almogren, A.; Kerr, M.A.; Woof, J.M.; Perkins, S.J. Solution structure determination of monomeric human IgA2 by X-ray and neutron scattering, analytical ultracentrifugation and constrained modelling: A comparison with monomeric human IgA1. J. Mol. Biol. 2004, 338, 921–941. [Google Scholar] [CrossRef] [PubMed]
- Hui, G.K.; Wright, D.W.; Vennard, O.L.; Rayner, L.E.; Pang, M.; Yeo, S.C.; Gor, J.; Molyneux, K.; Barratt, J.; Perkins, S.J. The solution structures of native and patient monomeric human IgA1 reveal asymmetric extended structures: Implications for function and IgAN disease. Biochem. J. 2015, 471, 167–185. [Google Scholar] [CrossRef] [Green Version]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 2000, 52, 240–248. [Google Scholar] [CrossRef]
- Xiong, E.; Li, Y.; Min, Q.; Cui, C.; Liu, J.; Hong, R.; Lai, N.; Wang, Y.; Sun, J.; Matsumoto, R.; et al. MZB1 promotes the secretion of J-chain-containing dimeric IgA and is critical for the suppression of gut inflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 13480–13489. [Google Scholar] [CrossRef] [Green Version]
- Bastian, A.; Kratzin, H.; Eckart, K.; Hilschmann, N. Intra- and inter-chain disulphide bridges of the human J chain in secretory immunoglobulin A. Biol. Chem. Hoppe Seyler 1992, 373, 1255–1263. [Google Scholar] [CrossRef]
- Frutiger, S.; Hughes, G.J.; Paquet, N.; Luthy, R.; Jaton, J.C. Disulfide bond assignment in human J chain and its covalent pairing with immunoglobulin M. Biochemistry 1992, 31, 12643–12647. [Google Scholar] [CrossRef]
- Cann, G.M.; Zaritsky, A.; Koshland, M.E. Primary structure of the immunoglobulin J chain from the mouse. Proc. Natl. Acad. Sci. USA 1982, 79, 6656–6660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin, J.D.; Pleass, R.J.; Owens, R.J.; Woof, J.M. Mutagenesis of the human IgA1 heavy chain tailpiece that prevents dimer assembly. J. Immunol. 1996, 157, 156–159. [Google Scholar] [PubMed]
- Krugmann, S.; Pleass, R.J.; Atkin, J.D.; Woof, J.M. Structural requirements for assembly of dimeric IgA probed by site-directed mutagenesis of J chain and a cysteine residue of the α chain CH2 domain. J. Immunol. 1997, 159, 244–249. [Google Scholar] [PubMed]
- Bonner, A.; Furtado, P.B.; Almogren, A.; Kerr, M.A.; Perkins, S.J. Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J. Immunol. 2008, 180, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Woof, J.M.; Mestecky, J. Mucosal Immunoglobulins. In Mucosal Immunology, 4th ed.; Mestecky, J., Strober, W., Russell, M.W., Kelsall, B.L., Cheroutre, H., Lambrecht, B.N., Eds.; Academic Press: Oxford, UK, 2015; pp. 287–324. [Google Scholar]
- Natvig, I.B.; Johansen, F.E.; Nordeng, T.W.; Haraldsen, G.; Brandtzaeg, P. Mechanism for enhanced external transfer of dimeric IgA over pentameric IgM: Studies of diffusion, binding to the human polymeric Ig receptor, and epithelial transcytosis. J. Immunol. 1997, 159, 4330–4340. [Google Scholar] [PubMed]
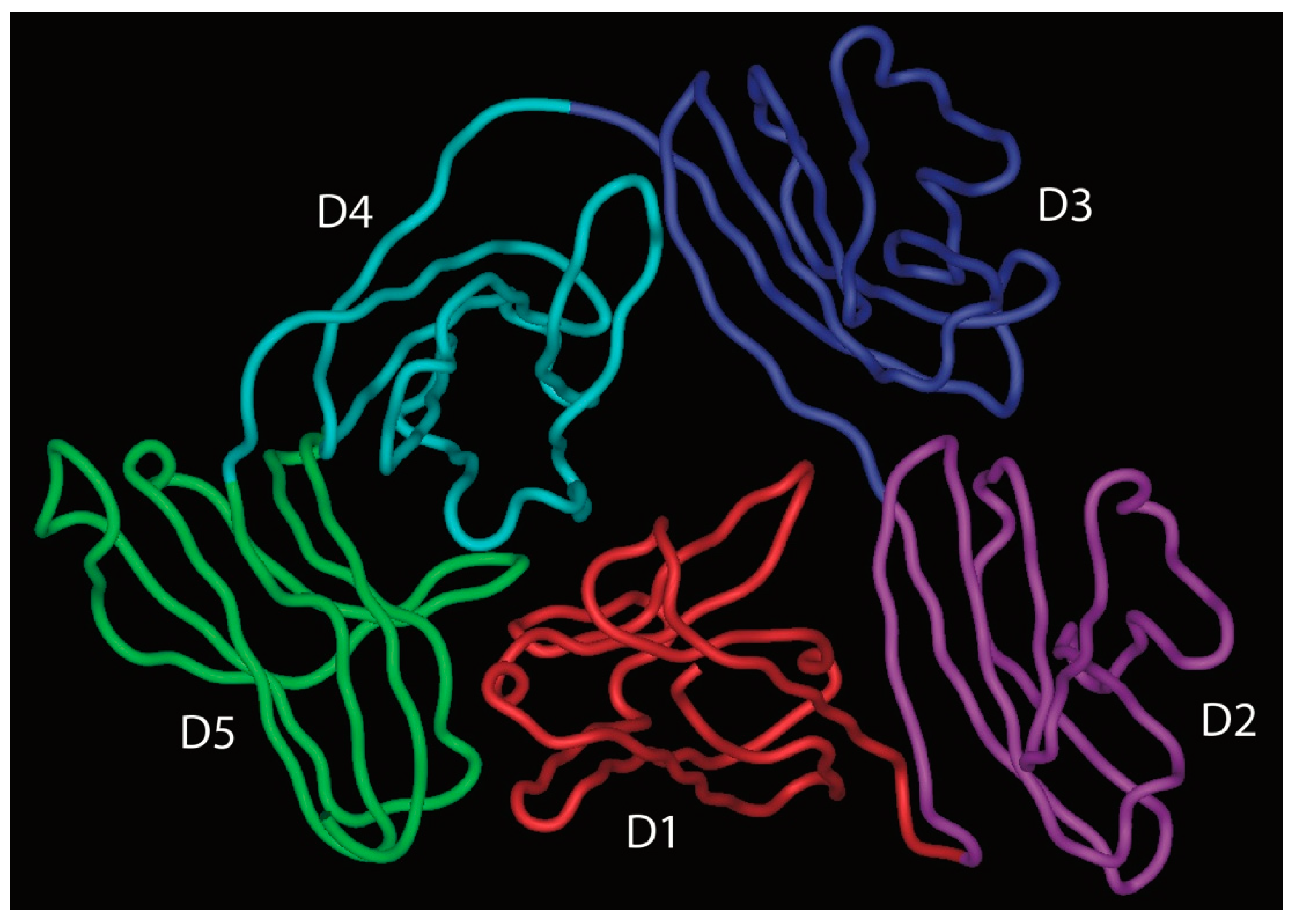
- Kaetzel, C.S. The polymeric immunoglobulin receptor: Bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 2005, 206, 83–99. [Google Scholar] [CrossRef]
- Bakos, M.A.; Kurosky, A.; Cwerwinski, E.W.; Goldblum, R.M. A conserved binding site on the receptor for polymeric Ig is homologous to CDR1 of Ig V kappa domains. J. Immunol. 1993, 151, 1346–1352. [Google Scholar]
- Coyne, R.S.; Siebrecht, M.; Peitsch, M.C.; Casanova, J.E. Mutational analysis of polymeric immunoglobulin receptor/ligand interactions. Evidence for the involvement of multiple complementarity determining region (CDR)-like loops in receptor domain I. J. Biol. Chem. 1994, 269, 31620–31625. [Google Scholar]
- Hamburger, A.E.; West, A.P.; Bjorkman, P.J. Crystal structure of a polymeric immunoglobulin binding fragment of the human polymeric immunoglobulin receptor. Structure 2004, 12, 1925–1935. [Google Scholar] [CrossRef] [Green Version]
- Hexham, J.M.; White, K.D.; Carayannopoulos, L.N.; Mandecki, W.; Brisette, R.; Yang, Y.S.; Capra, J.D. A human immunoglobulin (Ig) A Cα3 domain motif directs polymeric Ig receptor-mediated secretion. J. Exp. Med. 1999, 189, 747–752. [Google Scholar] [CrossRef]
- Braathen, R.; Sorensen, V.; Brandtzaeg, P.; Sandlie, I.; Johansen, F.E. The carboxyl-terminal domains of IgA and IgM direct isotype-specific polymerization and interaction with the polymeric immunoglobulin receptor. J. Biol. Chem. 2002, 277, 42755–42762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Pleass, R.J.; Batten, M.R.; Atkin, J.D.; Woof, J.M. Structural requirements for the interaction of human IgA with the human polymeric Ig receptor. J. Immunol. 2005, 175, 6694–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallgren-Gebauer, E.; Gebauer, W.; Bastian, A.; Kratzin, H.; Eiffert, H.; Zimmerman, B.; Karas, M.; Hilschmann, N. The covalent linkage of the secretory component to IgA. Adv. Exp. Med. Biol. 1995, 371A, 625–628. [Google Scholar] [PubMed]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. The J chain is essential for polymeric Ig receptor-mediated epithelial transport of IgA. J. Immunol. 2001, 167, 5185–5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmueller, B.M.; Huey-Tubman, K.E.; López, C.J.; Yang, Z.; Hubbell, W.L.; Bjorkman, P.J. The structure and dynamics of secretory component and its interactions with polymeric immunoglobulins. Elife 2016, 5, e10640. [Google Scholar] [CrossRef]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef]
- Mantis, N.J.; McGuinness, C.R.; Sonuyi, O.; Edwards, G.; Farrant, S.A. Immunoglobulin A antibodies against ricin A and B subunits protect epithelial cells from ricin intoxication. Infect. Immun. 2006, 74, 3455–3462. [Google Scholar] [CrossRef] [Green Version]
- Wold, A.; Mestecky, J.; Tomana, M.; Kobata, A.; Ohbayashi, H.; Endo, T.; Edén, C.S. Secretory immunoglobulin A carries oligosaccharide receptors for Escherichia coli type 1 fimbrial lectin. Infect. Immun. 1990, 58, 3073–3077. [Google Scholar]
- Schroten, H.; Stapper, C.; Plogmann, R.; Köhler, H.; Hacker, J.; Hanisch, F.G. Fab-independent antiadhesion effects of secretory immunoglobulin A on S-fimbriated Escherichia coli are mediated by sialyloligosaccharides. Infect. Immun. 1998, 66, 3971–3973. [Google Scholar]
- Ruhl, S.; Sandberg, A.L.; Cole, M.F.; Cisar, J.O. Recognition of immunoglobulin A1 by oral actinomyces and streptococcal lectins. Infect. Immun. 1996, 64, 5421–5424. [Google Scholar]
- Biesbrock, A.R.; Reddy, M.S.; Levine, M.J. Interaction of a salivary mucin-secretory immunoglobulin A complex with mucosal pathogens. Infect. Immun. 1991, 59, 3492–3497. [Google Scholar] [PubMed]
- Xu, F.; Newby, J.M.; Schiller, J.L.; Schroeder, H.A.; Wessler, T.; Chen, A.; Forest, M.G.; Lai, S.K. Modeling barrier properties of intestinal mucus reinforced with IgG and secretory IgA against motile bacteria. ACS Infect. Dis. 2019, 5, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Tenovuo, J.; Moldoveanu, Z.; Mestecky, J.; Pruitt, K.M.; Rahemtulla, B.M. Interaction of specific and innate factors of immunity: IgA enhances the antimicrobial effect of the lactoperoxidase system against Streptococcus mutans. J. Immunol. 1982, 128, 726–731. [Google Scholar] [PubMed]
- Wright, A.; Lamm, M.E.; Huang, Y.T. Excretion of human immunodeficiency virus type 1 through polarized epithelium by immunoglobulin A. J. Virol. 2008, 82, 11526–11535. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.K.; Blanchard, T.G.; Levine, A.D.; Emancipator, S.N.; Lamm, M.E. A mucosal IgA-mediated excretory immune system in vivo. J. Immunol. 2001, 166, 3688–3692. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.I.; Pedron, T.; Tournebize, R.; Olivo-Marin, J.C.; Sansonetti, P.J.; Phalipon, A. Anti-inflammatory role for intracellular dimeric immunoglobulin a by neutralization of lipopolysaccharide in epithelial cells. Immunity 2003, 18, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Mazanec, M.B.; Kaetzel, C.S.; Lamm, M.E.; Fletcher, D.; Nedrud, J.G. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc. Natl. Acad. Sci. USA 1992, 89, 6901–6905. [Google Scholar] [CrossRef] [Green Version]
- Mazanec, M.B.; Coudret, C.L.; Fletcher, D.R. Intracellular neutralization of influenza virus by immunoglobulin A anti-hemagglutinin monoclonal antibodies. J. Virol. 1995, 69, 1339–1343. [Google Scholar]
- Zhou, D.; Zhang, Y.; Li, Q.; Chen, Y.; He, B.; Yang, J.; Tu, H.; Lei, L.; Yan, H. Matrix protein-specific IgA antibody inhibits measles virus replication by intracellular neutralization. J. Virol. 2011, 85, 11090–11097. [Google Scholar] [CrossRef] [Green Version]
- Feng, N.; Lawton, J.A.; Gilbert, J.; Kuklin, N.; Vo, P.; Prasad, B.V.; Greenberg, H.B. Inhibition of rotavirus replication by a non-neutralizing, rotavirus VP6-specific IgA mAb. J. Clin. Investig. 2002, 109, 1203–1213. [Google Scholar] [CrossRef]
- Corthésy, B.; Benureau, Y.; Perrier, C.; Fourgeux, C.; Parez, N.; Greenberg, H.; Schwartz-Cornil, I. Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J. Virol. 2006, 80, 10692–10699. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.T.; Wright, A.; Gao, X.; Kulick, L.; Yan, H.; Lamm, M.E. Intraepithelial cell neutralization of HIV-1 replication by IgA. J. Immunol. 2005, 174, 4828–4835. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Yan, H.; Lamm, M.E.; Huang, Y.T. Immunoglobulin A antibodies against internal HIV-1 proteins neutralize HIV-1 replication inside epithelial cells. Virology 2006, 356, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.W.; Siadat-Pajouh, M.; Krishnaney, A.A.; Greenberg, H.B. Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science 1996, 272, 104–107. [Google Scholar] [CrossRef]
- Schwartz-Cornil, I.; Benureau, Y.; Greenberg, H.; Hendrickson, B.A.; Cohen, J. Heterologous protection induced by the inner capsid proteins of rotavirus requires transcytosis of mucosal immunoglobulins. J. Virol. 2002, 76, 8110–8117. [Google Scholar] [CrossRef] [Green Version]
- Lohse, S.; Loew, S.; Kretschmer, A.; Jansen, J.H.M.; Meyer, S.; Ten Broeke, T.; Rosner, T.; Dechant, M.; Derer, S.; Klausz, K.; et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br. J. Haematol. 2018, 181, 413–417. [Google Scholar] [CrossRef]
- Roos, A.; Bouwman, L.H.; van Gijlswijk-Janssen, D.J.; Faber-Krol, M.C.; Stahl, G.L.; Daha, M.R. Human IgA activates the complement system via the mannan-binding lectin pathway. J. Immunol. 2001, 167, 2861–2868. [Google Scholar] [CrossRef] [Green Version]
- Ghumra, A.; Shi, J.; Mcintosh, R.S.; Rasmussen, I.B.; Braathen, R.; Johansen, F.E.; Sandlie, I.; Mongini, P.K.; Areschoug, T.; Lindahl, G.; et al. Structural requirements for the interaction of human IgM and IgA with the human Fcα/µ receptor. Eur. J. Immunol. 2009, 39, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Matysiak-Budnik, T.; Moura, I.C.; Arcos-Fajardo, M.; Lebreton, C.; Menard, S.; Candalh, C.; Ben-Khalifa, K.; Dugave, C.; Tamouza, H.; van Niel, G.; et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 2008, 205, 143–154. [Google Scholar] [CrossRef]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013, 11, e1001658. [Google Scholar] [CrossRef]
- Baumann, J.; Park, C.G.; Mantis, N.J. Recognition of secretory IgA by DC-SIGN: Implications for immune surveillance in the intestine. Immunol. Lett. 2010, 131, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.J.; Fuchs, A.; Colonna, M. Cutting edge: Human FcRL4 and FcRL5 are receptors for IgA and IgG. J. Immunol. 2012, 188, 4741–4745. [Google Scholar] [CrossRef] [Green Version]
- Rifai, A.; Fadden, K.; Morrison, S.L.; Chintalacharuvu, K.R. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J. Exp. Med. 2000, 191, 2171–2182. [Google Scholar] [CrossRef] [Green Version]
- Molyneux, K.; Wimbury, D.; Pawluczyk, I.; Muto, M.; Bhachu, J.; Mertens, P.R.; Feehally, J.; Barratt, J. beta1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. 2017, 92, 1458–1468. [Google Scholar] [CrossRef] [Green Version]
- Lamkhioued, B.; Gounni, A.S.; Gruart, V.; Pierce, A.; Capron, A.; Capron, M. Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur. J. Immunol. 1995, 25, 117–125. [Google Scholar] [CrossRef]
- Bruhns, P.; Jönsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [Green Version]
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc receptor involvement in inflammatory diseases: From mechanisms to new therapeutic tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef] [Green Version]
- Breedveld, A.; van Egmond, M. IgA and FcαRI: Pathological roles and therapeutic opportunities. Front. Immunol. 2019, 10, 553. [Google Scholar] [CrossRef]
- van der Steen, L.; Tuk, C.W.; Bakema, J.E.; Kooij, G.; Reijerkerk, A.; Vidarsson, G.; Bouma, G.; Kraal, G.; de Vries, H.E.; Beelen, R.H.; et al. Immunoglobulin A: FcαRI interactions induce neutrophil migration through release of leukotriene B4. Gastroenterology 2009, 137, e1–e3. [Google Scholar] [CrossRef]
- Aleyd, E.; van Hout, M.W.; Ganzevles, S.H.; Hoeben, K.A.; Everts, V.; Bakema, J.E.; van Egmond, M. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcα receptor I. J. Immunol. 2014, 192, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Aleyd, E.; Heineke, M.H.; van Egmond, M. The era of the immunoglobulin A Fc receptor FcαRI; its function and potential as target in disease. Immunol. Rev. 2015, 268, 123–138. [Google Scholar] [CrossRef]
- Morton, H.C.; Schiel, A.E.; Janssen, S.W.; van de Winkel, J.G. Alternatively spliced forms of the human myeloid Fc alpha receptor (CD89) in neutrophils. Immunogenetics 1996, 43, 246–247. [Google Scholar] [CrossRef]
- Pleass, R.J.; Andrews, P.D.; Kerr, M.A.; Woof, J.M. Alternative splicing of the human IgA Fc receptor CD89 in neutrophils and eosinophils. Biochem. J. 1996, 318, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Ji, C.; Xie, F.; Langefeld, C.D.; Qian, K.; Gibson, A.W.; Edberg, J.C.; Kimberly, R.P. FcαRI (CD89) alleles determine the proinflammatory potential of serum IgA. J. Immunol. 2007, 178, 3973–3982. [Google Scholar] [CrossRef] [Green Version]
- Woof, J.M.; Burton, D.R. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol. 2004, 4, 89–99. [Google Scholar] [CrossRef]
- van Spriel, A.B.; Leusen, J.H.; Vilé, H.; van de Winkel, J.G. Mac-1 (CD11b/CD18) as accessory molecule for FcαR (CD89) binding of IgA. J. Immunol. 2002, 169, 3831–3836. [Google Scholar] [CrossRef] [Green Version]
- Carayannopoulos, L.; Hexham, J.M.; Capra, J.D. Localization of the binding site for the monocyte immunoglobulin (Ig) A-Fc receptor (CD89) to the domain boundary between Cα2 and Cα3 in human IgA1. J. Exp. Med. 1996, 183, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Pleass, R.J.; Dunlop, J.I.; Anderson, C.M.; Woof, J.M. Identification of residues in the CH2/CH3 domain interface of IgA essential for interaction with the human Fcα receptor (FcαR) CD89. J. Biol. Chem. 1999, 274, 23508–23514. [Google Scholar] [CrossRef] [Green Version]
- Pleass, R.J.; Dehal, P.K.; Lewis, M.J.; Woof, J.M. Limited role of charge matching in the interaction of human immunoglobulin A with the immunoglobulin A Fc receptor (FcαRI) CD89. Immunology 2003, 109, 331–335. [Google Scholar] [CrossRef]
- Wines, B.D.; Hulett, M.D.; Jamieson, G.P.; Trist, H.M.; Spratt, J.M.; Hogarth, P.M. Identification of residues in the first domain of human Fcα receptor essential for interaction with IgA. J. Immunol. 1999, 162, 2146–2153. [Google Scholar]
- Wines, B.D.; Sardjono, C.T.; Trist, H.H.; Lay, C.S.; Hogarth, P.M. The interaction of FcαRI with IgA and its implications for ligand binding by immunoreceptors of the leukocyte receptor cluster. J. Immunol. 2001, 166, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.M.; Wall, S.B.; Takahashi, K.; Novak, J.; Renfrow, M.B.; Herr, A.B. Analysis of IgA1 N-glycosylation and its contribution to FcαRI binding. Biochemistry 2008, 47, 11285–11299. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhao, Q.; Zhu, L.; Zhang, W. Deglycosylation of FcαR at N58 increases its binding to IgA. Glycobiology 2010, 20, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Posgai, M.T.; Tonddast-Navaei, S.; Jayasinghe, M.; Ibrahim, G.M.; Stan, G.; Herr, A.B. FcαRI binding at the IgA1 CH2–CH3 interface induces long-range conformational changes that are transmitted to the hinge region. Proc. Natl. Acad. Sci. USA 2018, 115, E8882–E8891. [Google Scholar] [CrossRef] [Green Version]
- Heineke, M.H.; van der Steen, L.P.E.; Korthouwer, R.M.; Hage, J.J.; Langedijk, J.P.M.; Benschop, J.J.; Bakema, J.E.; Slootstra, J.W.; van Egmond, M. Peptide mimetics of immunoglobulin A (IgA) and FcαRI block IgA-induced human neutrophil activation and migration. Eur. J. Immunol. 2017, 47, 1835–1845. [Google Scholar] [CrossRef] [Green Version]
- van der Steen, L.P.; Bakema, J.E.; Sesarman, A.; Florea, F.; Tuk, C.W.; Kirtschig, G.; Hage, J.J.; Sitaru, C.; van Egmond, M. Blocking Fcα receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J. Immunol. 2012, 189, 1594–1601. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Marjon, K.D.; Mold, C.; Marnell, L.; Du Clos, T.W.; Sun, P. Pentraxins and IgA share a binding hot-spot on FcαRI. Protein Sci. 2014, 23, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Abi-Rached, L.; Dorighi, K.; Norman, P.J.; Yawata, M.; Parham, P. Episodes of natural selection shaped the interactions of IgA-Fc with FcαRI and bacterial decoy proteins. J. Immunol. 2007, 178, 7943–7954. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, A.; Woof, J.M.; Abi-Rached, L.; Parham, P.; Esteves, P.J. Computational analyses of an evolutionary arms race between mammalian immunity mediated by immunoglobulin A and its subversion by bacterial pathogens. PLoS ONE 2013, 8, e73934. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, S.; Tillig, M.P.; Wolff, S.; Vaerman, J.P.; Chhatwal, G.S. Species-specific binding of human secretory component to SpsA protein of Streptococcus pneumoniae via a hexapeptide motif. Mol. Microbiol. 2000, 36, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.R.; Mostov, K.E.; Lamm, M.E.; Nanno, M.; Shimida, S.; Ohwaki, M.; Tuomanen, E. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 2000, 102, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Elm, C.; Braathen, R.; Bergmann, S.; Frank, R.; Vaerman, J.P.; Kaetzel, C.S.; Chhatwal, G.S.; Johansen, F.E.; Hammerschmidt, S. Ectodomains 3 and 4 of human polymeric immunoglobulin receptor (hpIgR) mediate invasion of Streptococcus pneumoniae into the epithelium. J. Biol. Chem. 2004, 279, 6296–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmat, T.M.; Agarwal, V.; Räth, S.; Hildebrandt, J.P.; Hammerschmidt, S. Streptococcus pneumoniae infection of host epithelial cells via polymeric immunoglobulin receptor transiently induces calcium release from intracellular stores. J. Biol. Chem. 2011, 286, 17861–17869. [Google Scholar] [CrossRef] [Green Version]
- Frithz, E.; Héden, L.O.; Lindahl, G. Extensive sequence homology between IgA receptor and M proteins in Streptococcus pyogenes. Mol. Microbiol. 1989, 3, 1111–1119. [Google Scholar] [CrossRef]
- Stenberg, L.; O’Toole, P.W.; Mestecky, J.; Lindahl, G. Molecular characterization of protein Sir, a streptococcal cell surface protein that binds both immunoglobulin A and immunoglobulin G. J. Biol. Chem. 1994, 2691, 3458–13464. [Google Scholar]
- Héden, L.O.; Frithz, E.; Lindahl, G. Molecular characterization of an IgA receptor from group B streptococci: Sequence of the gene, identification of a proline-rich region with unique structure and isolation of N-terminal fragments with IgA-binding capacity. Eur. J. Immunol. 1991, 21, 1481–1490. [Google Scholar] [CrossRef]
- Pleass, R.J.; Areschoug, T.; Lindahl, G.; Woof, J.M. Streptococcal IgA-binding proteins bind in the Cα2-Cα3 interdomain region and inhibit binding of IgA to human CD89. J. Biol. Chem. 2001, 276, 8197–8204. [Google Scholar] [CrossRef] [Green Version]
- Wines, B.D.; Willoughby, N.; Fraser, J.D.; Hogarth, P.M. A competitive mechanism for staphylococcal toxin SSL7 inhibiting the leukocyte IgA receptor, FcαRI, is revealed by SSL7 binding at the Cα2/Cα3 interface of IgA. J. Biol. Chem. 2006, 281, 1389–1393. [Google Scholar] [CrossRef] [Green Version]
- Mistry, D.; Stockley, R.A. IgA1 protease. Int. J. Biochem. Cell Biol. 2006, 38, 1244–1248. [Google Scholar] [CrossRef]
- Janoff, E.N.; Rubins, J.B.; Fasching, C.; Charboneau, D.; Rahkola, J.T.; Plaut, A.G.; Weiser, J.N. Pneumococcal IgA1 protease subverts specific protection by human IgA1. Mucosal Immunol. 2014, 7, 249–256. [Google Scholar] [CrossRef]
- Senior, B.W.; Dunlop, J.I.; Batten, M.R.; Kilian, M.; Woof, J.M. Cleavage of a recombinant human immunoglobulin A2 (IgA2)-IgA1 hybrid antibody by certain bacterial IgA1 proteases. Infect. Immun. 2000, 68, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batten, M.R.; Senior, B.W.; Kilian, M.; Woof, J.M. Amino acid sequence requirements in the hinge of human immunoglobulin A1 (IgA1) for cleavage by streptococcal IgA1 proteases. Infect. Immun. 2003, 71, 1462–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, B.W.; Woof, J.M. The influences of hinge length and composition on the susceptibility of human IgA to cleavage by diverse bacterial IgA1 proteases. J. Immunol. 2005, 174, 7792–7799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintalacharuvu, K.R.; Chuang, P.D.; Dragoman, A.; Fernandez, C.Z.; Qiu, J.; Plaut, A.G.; Trinh, K.R.; Gala, F.A.; Morrison, S.L. Cleavage of the human immunoglobulin A1 (IgA1) hinge region by IgA1 proteases requires structures in the Fc region of IgA. Infect. Immun. 2003, 71, 2563–2570. [Google Scholar] [CrossRef] [Green Version]
- Senior, B.W.; Woof, J.M. Sites in the CH3 domain of human IgA1 that influence sensitivity to bacterial IgA1 proteases. J. Immunol. 2006, 177, 3913–3919. [Google Scholar] [CrossRef]
- Johnson, T.A.; Qiu, J.; Plaut, A.G.; Holyoak, T. Active-site gating regulates substrate selectivity in a chymotrypsin-like serine protease: The structure of Haemophilus influenzae immunoglobulin A1 protease. J. Mol. Biol. 2009, 389, 559–574. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.; Wood, S.G.; Lynch, M.; Plaut, A.G. Substrate analogue inhibitors of the IgA1 proteinases from Neisseria gonorrhoeae. J. Med. Chem. 1988, 31, 1647–1651. [Google Scholar] [CrossRef]
- Bachovchin, W.W.; Plaut, A.G.; Flentke, G.R.; Lynch, M.; Kettner, C.A. Inhibition of IgA1 proteinases from Neisseria gonorrhoeae and Haemophilus influenzae by peptide prolyl boronic acids. J. Biol. Chem. 1990, 265, 3738–3743. [Google Scholar]
- Shehaj, L.; Choudary, S.K.; Makwana, K.M.; Gallo, M.C.; Murphy, T.F.; Kritzer, J.A. Small-molecule inhibitors of Haemophilus influenzae IgA1 protease. ACS Infect. Dis. 2019, 5, 1129–1138. [Google Scholar] [CrossRef]
- Lamm, M.E.; Emancipator, S.N.; Robinson, J.K.; Yamashita, M.; Fujioka, H.; Qiu, J.; Plaut, A.G. Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: Potential therapy for IgA nephropathy. Am. J. Pathol. 2008, 172, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, X.; Shen, H.; Mao, N.; Wang, H.; Cui, L.; Cheng, Y.; Fan, J. Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA Nephropathy. Sci. Rep. 2016, 6, 30964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- van Splunter, M.; van Hoffen, E.; Floris-Vollenbroek, E.G.; Timmerman, H.; de Bos, E.L.; Meijer, B.; Ulfman, L.H.; Witteman, B.; Wells, J.M.; Brugman, S.; et al. Oral cholera vaccination promotes homing of IgA+ memory B cells to the large intestine and the respiratory tract. Mucosal Immunol. 2018, 11, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; King, J.D.; Kataoka, K.; Kobayashi, R.; Gilbert, R.S.; Oishi, K.; Hollingshead, S.K.; Briles, D.E.; Fujihashi, K. Secretory-IgA antibodies play an important role in the immunity to Streptococcus pneumoniae. J. Immunol. 2010, 185, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planque, S.; Salas, M.; Mitsuda, Y.; Sienczyk, M.; Escobar, M.A.; Mooney, J.P.; Morris, M.K.; Nishiyama, Y.; Ghosh, D.; Kumar, A.; et al. Neutralization of genetically diverse HIV-1 strains by IgA antibodies to the gp120-CD4-binding site from long-term survivors of HIV infection. AIDS 2010, 24, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.; Hwang, K.K.; Liu, P.; Dennison, S.M.; Tay, M.Z.; Shen, X.; Pollara, J.; Lucas, J.T.; Parks, R.; Rerks-Ngarm, S.; et al. HIV-1-Specific IgA monoclonal antibodies from an HIV-1 vaccinee mediate galactosylceramide blocking and phagocytosis. J. Virol. 2018, 92, e01552-17. [Google Scholar] [CrossRef] [Green Version]
- Herremans, T.M.; Reimerink, J.H.; Buisman, A.M.; Kimman, T.G.; Koopmans, M.P. Induction of mucosal immunity by inactivated poliovirus vaccine is dependent on previous mucosal contact with live virus. J. Immunol. 1999, 162, 5011–5018. [Google Scholar]
- Silvey, K.J.; Hutchings, A.B.; Vajdy, M.; Petzke, M.M.; Neutra, M.R. Role of immunoglobulin A in protection against reovirus entry into murine Peyer’s patches. J. Virol. 2001, 75, 10870–10879. [Google Scholar] [CrossRef] [Green Version]
- Tamura, S.; Funato, H.; Hirabayashi, Y.; Suzuki, Y.; Nagamine, T.; Aizawa, C.; Kurata, T. Cross-protection against influenza A virus infection by passively transferred respiratory tract IgA antibodies to different hemagglutinin molecules. Eur. J. Immunol. 1991, 21, 1337–1344. [Google Scholar] [CrossRef]
- Bakema, J.E.; van Egmond, M. Immunoglobulin A: A next generation of therapeutic antibodies? MAbs 2011, 3, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Leusen, J.H. IgA as a therapeutic antibody. Mol. Immunol. 2015, 68, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Lombana, T.N.; Rajan, S.; Zorn, J.A.; Mandikian, D.; Chen, E.C.; Estevez, A.; Yip, V.; Bravo, D.D.; Phung, W.; Farahi, F.; et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. MAbs 2019, 11, 1122–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledford, H. Rush to protect lucrative antibody patents kicks into gear. Nature 2018, 557, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.C. The role of IgA and IgA Fc receptors as anti-inflammatory agents. J. Clin. Immunol. 2010, 30, S61–S64. [Google Scholar] [CrossRef] [PubMed]
- Challacombe, S.J.; Russell, M.W. Estimations of the intravascular half-lives of normal rhesus monkey IgG, IgA, and IgM. Immunology 1979, 36, 331–338. [Google Scholar]
- Strober, W.; Wochner, R.D.; Barlow, M.H.; McFarlin, D.E.; Waldmann, T.A. Immunoglobulin metabolism in ataxia telangiectasia. J. Clin. Investig. 1968, 47, 1905–1915. [Google Scholar] [CrossRef]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.; van Tetering, G.; Dechant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef]
- Lohse, S.; Meyer, S.; Meulenbroek, L.A.; Jansen, J.H.; Nederend, M.; Kretschmer, A.; Klausz, K.; Moginger, U.; Derer, S.; Rosner, T.; et al. An anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. 2016, 76, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Rouwendal, G.J.; van der Lee, M.M.; Meyer, S.; Reiding, K.R.; Schouten, J.; de Roo, G.; Egging, D.F.; Leusen, J.H.; Boross, P.; Wuhrer, M.; et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs 2016, 8, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Nederend, M.; Jansen, J.H.; Reiding, K.R.; Jacobino, S.R.; Meeldijk, J.; Bovenschen, N.; Wuhrer, M.; Valerius, T.; Ubink, R.; et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs 2016, 8, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Beyer, T.; Lohse, S.; Berger, S.; Peipp, M.; Valerius, T.; Dechant, M. Serum-free production and purification of chimeric IgA antibodies. J. Immunol. Methods 2009, 346, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Hart, F.; Danielczyk, A.; Goletz, S. Human cell line-derived monoclonal IgA antibodies for cancer immunotherapy. Bioengineering 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vink, T.; Oudshoorn-Dickmann, M.; Roza, M.; Reitsma, J.J.; de Jong, R.N. A simple, robust and highly efficient transient expression system for producing antibodies. Methods 2014, 65, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker, M.; Maresch, D.; Strasser, R. Glyco-engineering for the production of recombinant IgA1 with distinct mucin-type O-glycans in plants. Bioengineered 2016, 7, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker, M.; Tschofen, M.; Maresch, D.; König, J.; Juarez, P.; Orzaez, D.; Altmann, F.; Steinkellner, H.; Strasser, R. Transient glyco-engineering to produce recombinant IgA1 with defined N- and O-glycans in plants. Front. Plant Sci. 2016, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Westerhof, L.B.; Wilbers, R.H.; van Raaij, D.R.; Nguyen, D.L.; Goverse, A.; Henquet, M.G.; Hokke, C.H.; Bosch, D.; Bakker, J.; Schots, A. Monomeric IgA can be produced in planta as efficient as IgG, yet receives different N-glycans. Plant Biotechnol. J. 2014, 12, 1333–1342. [Google Scholar] [CrossRef]
- Yoo, E.M.; Yu, L.J.; Wims, L.A.; Goldberg, D.; Morrison, S.L. Differences in N-glycan structures found on recombinant IgA1 and IgA2 produced in murine myeloma and CHO cell lines. MAbs 2010, 2, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Göritzer, K.; Maresch, D.; Altmann, F.; Obinger, C.; Strasser, R. Exploring site-specific N-glycosylation of HEK293 and plant-produced human IgA isotypes. J. Proteome Res. 2017, 16, 2560–2570. [Google Scholar] [CrossRef]
- Sandin, C.; Linse, S.; Areschoug, T.; Woof, J.M.; Reinholdt, J.; Lindahl, G. Isolation and detection of human IgA using a streptococcal IgA-binding peptide. J. Immunol. 2002, 169, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, S.; Depicker, A.; Schepens, B.; Saelens, X.; Juarez, P. A two-amino acid mutation in murine IgA enables downstream processing and purification on staphylococcal superantigen-like protein 7. J. Biotechnol. 2019, 294, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Ohzono, S.; Park, M.; Sakamoto, K.; Tsukamoto, S.; Sugita, R.; Ishitobi, H.; Mori, T.; Ito, O.; Sorajo, K.; et al. Human IgA-binding peptides selected from random peptide libraries: Affinity maturation and application in IgA purification. J. Biol. Chem. 2012, 287, 43126–43136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintalacharuvu, K.R.; Yu, L.J.; Bhola, N.; Kobayashi, K.; Fernandez, C.Z.; Morrison, S.L. Cysteine residues required for the attachment of the light chain in human IgA2. J. Immunol. 2002, 169, 5072–5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintalacharuvu, K.R.; Morrison, S.L. Production of secretory immunoglobulin A by a single mammalian cell. Proc. Natl. Acad Sci. USA 1997, 94, 6364–6368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Sano, K.; Suzuki, T.; Ainai, A.; Taga, Y.; Ueno, T.; Tabata, K.; Saito, K.; Wada, Y.; Ohara, Y.; et al. IgA tetramerization improves target breadth but not peak potency of functionality of anti-influenza virus broadly neutralizing antibody. PLoS Pathog. 2019, 15, e1007427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Egmond, M.; van Vuuren, A.J.; Morton, H.C.; van Spriel, A.B.; Shen, L.; Hofhuis, F.M.; Saito, T.; Mayadas, T.N.; Verbeek, J.S.; van de Winkel, J.G. Human immunoglobulin A receptor (FcαRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood 1999, 93, 4387–4394. [Google Scholar] [CrossRef]
- Launay, P.; Grossetete, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcα receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Duchez, S.; Amin, R.; Cogné, N.; Delpy, L.; Sirac, C.; Pascal, V.; Corthésy, B.; Cogné, M. Premature replacement of mu with alpha immunoglobulin chains impairs lymphopoiesis and mucosal homing but promotes plasma cell maturation. Proc. Natl. Acad Sci. USA 2010, 107, 3064–3069. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [Google Scholar]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, A.; Hafsi, S.; Militello, L.; Russo, A.E.; Soua, Z.; Mazzarino, M.C.; Stivala, F.; Libra, M. Understanding rituximab function and resistance: Implications for tailored therapy. Front. Biosci. 2011, 16, 770–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyiadzis, M.; Foon, K.A. Approved monoclonal antibodies for cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 1151–1158. [Google Scholar] [CrossRef]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glennie, M.J.; French, R.R.; Cragg, M.S.; Taylor, R.P. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 2007, 44, 3823–3837. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef]
- van der Bij, G.J.; Bogels, M.; Otten, M.A.; Oosterling, S.J.; Kuppen, P.J.; Meijer, S.; Beelen, R.H.; van Egmond, M. Experimentally induced liver metastases from colorectal cancer can be prevented by mononuclear phagocyte-mediated monoclonal antibody therapy. J. Hepatol. 2010, 53, 677–685. [Google Scholar] [CrossRef]
- Colombo, M.P.; Ferrari, G.; Stoppacciaro, A.; Parenza, M.; Rodolfo, M.; Mavilio, F.; Parmiani, G. Granulocyte colony-stimulating factor gene transfer suppresses tumorigenicity of a murine adenocarcinoma in vivo. J. Exp. Med. 1991, 173, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Bakema, J.E.; Ganzevles, S.H.; Fluitsma, D.M.; Schilham, M.W.; Beelen, R.H.; Valerius, T.; Lohse, S.; Glennie, M.J.; Medema, J.P.; van Egmond, M. Targeting FcαRI on polymorphonuclear cells induces tumor cell killing through autophagy. J. Immunol. 2011, 187, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Hernandez-Ilizaliturri, F.J.; Jupudy, V.; Ostberg, J.; Oflazoglu, E.; Huberman, A.; Repasky, E.; Czuczman, M.S. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin. Cancer Res. 2003, 9, 5866–5873. [Google Scholar] [PubMed]
- Pullarkat, V.; Deo, Y.; Link, J.; Spears, L.; Marty, V.; Curnow, R.; Groshen, S.; Gee, C.; Weber, J.S. A phase I study of a HER2/neu bispecific antibody with granulocyte-colony-stimulating factor in patients with metastatic breast cancer that overexpresses HER2/neu. Cancer Immunol. Immunother. 1999, 48, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.D.; Beelen, A.P.; Cole, B.F.; Wallace, P.K.; Fisher, J.L.; Waugh, M.G.; Kaufman, P.A.; Ernstoff, M.S. The pharmacokinetics of the bispecific antibody MDX-H210 when combined with interferon gamma-1b in a multiple-dose phase I study in patients with advanced cancer. Cancer Chemother. Pharmacol. 2002, 49, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Repp, R.; van Ojik, H.H.; Valerius, T.; Groenewegen, G.; Wieland, G.; Oetzel, C.; Stockmeyer, B.; Becker, W.; Eisenhut, M.; Steininger, H.; et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcγRI x anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br. J. Cancer 2003, 89, 2234–2243. [Google Scholar] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef]
- Otten, M.A.; Leusen, J.H.; Rudolph, E.; van der Linden, J.A.; Beelen, R.H.; van de Winkel, J.G.; van Egmond, M. FcR γ-chain dependent signaling in immature neutrophils is mediated by FcαRI, but not by FcγRI. J. Immunol. 2007, 179, 2918–2924. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rosner, T.; Valerius, T.; Leusen, J.H.W.; Ten Broeke, T. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- Huls, G.; Heijnen, I.A.; Cuomo, E.; van der Linden, J.; Boel, E.; van de Winkel, J.G.; Logtenberg, T. Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res. 1999, 59, 5778–5784. [Google Scholar]
- Dechant, M.; Beyer, T.; Schneider-Merck, T.; Weisner, W.; Peipp, M.; van de Winkel, J.G.; Valerius, T. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J. Immunol. 2007, 179, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, N.; van Egmond, M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur. J. Clin. Investig. 2018, 48, e12962. [Google Scholar] [CrossRef] [Green Version]
- Otten, M.A.; Rudolph, E.; Dechant, M.; Tuk, C.W.; Reijmers, R.M.; Beelen, R.H.; van de Winkel, J.G.; van Egmond, M. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J. Immunol. 2005, 174, 5472–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockmeyer, B.; Dechant, M.; van Egmond, M.; Tutt, A.L.; Sundarapandiyan, K.; Graziano, R.F.; Repp, R.; Kalden, J.R.; Gramatzki, M.; Glennie, M.J.; et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J. Immunol. 2000, 165, 5954–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guettinger, Y.; Barbin, K.; Peipp, M.; Bruenke, J.; Dechant, M.; Horner, H.; Thierschmidt, D.; Valerius, T.; Repp, R.; Fey, G.H.; et al. A recombinant bispecific single-chain fragment variable specific for HLA class II and FcαRI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J. Immunol. 2010, 184, 1210–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, V.; Laffleur, B.; Debin, A.; Cuvillier, A.; van Egmond, M.; Drocourt, D.; Imbertie, L.; Pangault, C.; Tarte, K.; Tiraby, G.; et al. Anti-CD20 IgA can protect mice against lymphoma development: Evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica 2012, 97, 1686–1694. [Google Scholar] [CrossRef]
- Wagner, E.K.; Maynard, J.A. Engineering therapeutic antibodies to combat infectious diseases. Curr. Opin. Chem. Eng. 2018, 19, 131–141. [Google Scholar] [CrossRef]
- Williams, A.; Reljic, R.; Naylor, I.; Clark, S.O.; Falero-Diaz, G.; Singh, M.; Challacombe, S.; Marsh, P.D.; Ivanyi, J. Passive protection with immunoglobulin A antibodies against tuberculous early infection of the lungs. Immunology 2004, 111, 328–333. [Google Scholar] [CrossRef]
- Balu, S.; Reljic, R.; Lewis, M.J.; Pleass, R.J.; McIntosh, R.; van Kooten, C.; van Egmond, M.; Challacombe, S.; Woof, J.M.; Ivanyi, J. A novel human IgA monoclonal antibody protects against tuberculosis. J. Immunol. 2011, 186, 3113–3119. [Google Scholar] [CrossRef] [Green Version]
- van Egmond, M.; van Garderen, E.; van Spriel, A.B.; Damen, C.A.; van Amersfoort, E.S.; van Zandbergen, G.; van Hattum, J.; Kuiper, J.; van de Winkel, J.G. FcαRI-positive liver Kupffer cells: Reappraisal of the function of immunoglobulin A in immunity. Nat. Med. 2000, 6, 680–685. [Google Scholar] [CrossRef]
- van Spriel, A.B.; van den Herik-Oudijk, I.E.; van Sorge, N.M.; Vile, H.A.; van Strijp, J.A.; van de Winkel, J.G. Effective phagocytosis and killing of Candida albicans via targeting FcγRI (CD64) or FcαRI (CD89) on neutrophils. J. Infect. Dis. 1999, 179, 661–669. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, W.; Vidarsson, G.; Vile, H.A.; van de Winkel, J.G.; Rodriguez, M.E. Pneumococcal capsular polysaccharide-specific IgA triggers efficient neutrophil effector functions via FcαRI (CD89). J. Infect. Dis. 2000, 182, 1139–1145. [Google Scholar] [CrossRef]
- Hellwig, S.M.; van Spriel, A.B.; Schellekens, J.F.; Mooi, F.R.; van de Winkel, J.G. Immunoglobulin A-mediated protection against Bordetella pertussis infection. Infect. Immun. 2001, 69, 4846–4850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Yamamoto, K.; Sugita, N.; van Spriel, A.B.; Kaneko, S.; van de Winkel, J.G.; Yoshie, H. Effective in vitro clearance of Porphyromonas gingivalis by Fcα receptor I (CD89) on gingival crevicular neutrophils. Infect. Immun. 2001, 69, 2935–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidarsson, G.; van Der Pol, W.L.; van Den Elsen, J.M.; Vile, H.; Jansen, M.; Duijs, J.; Morton, H.C.; Boel, E.; Daha, M.R.; Corthésy, B.; et al. Activity of human IgG and IgA subclasses in immune defense against Neisseria meningitidis serogroup B. J. Immunol. 2001, 166, 6250–6256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioley, G.; Monnerat, J.; Lötscher, M.; Vonarburg, C.; Zuercher, A.; Corthésy, B. Plasma-derived polyreactive secretory-like IgA and IgM opsonizing Salmonella enterica Typhimurium reduces invasion and gut tissue inflammation through agglutination. Front. Immunol. 2017, 8, 1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corthésy, B.; Monnerat, J.; Lotscher, M.; Vonarburg, C.; Schaub, A.; Bioley, G. Oral passive immunization with plasma-derived polyreactive secretory-Like IgA/M partially protects mice against experimental salmonellosis. Front. Immunol. 2018, 9, 2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langereis, J.D.; van der Flier, M.; de Jonge, M.I. Limited innovations after more than 65 years of immunoglobulin replacement therapy: Potential of IgA- and IgM-enriched formulations to prevent bacterial respiratory tract infections. Front. Immunol. 2018, 9, 1925. [Google Scholar] [CrossRef]
- Clements, M.L.; Betts, R.F.; Tierney, E.L.; Murphy, B.R. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J. Clin. Microbiol. 1986, 24, 157–160. [Google Scholar]
- Wagner, D.K.; Clements, M.L.; Reimer, C.B.; Snyder, M.; Nelson, D.L.; Murphy, B.R. Analysis of immunoglobulin G antibody responses after administration of live and inactivated influenza A vaccine indicates that nasal wash immunoglobulin G is a transudate from serum. J. Clin. Microbiol. 1987, 25, 559–562. [Google Scholar]
- Renegar, K.B.; Small, P.A. Passive transfer of local immunity to influenza virus infection by IgA antibody. J. Immunol. 1991, 146, 1972–1978. [Google Scholar]
- Renegar, K.B.; Small, P.A.; Boykins, L.G.; Wright, P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004, 173, 1978–1986. [Google Scholar] [CrossRef]
- Watkins, J.D.; Sholukh, A.M.; Mukhtar, M.M.; Siddappa, N.B.; Lakhashe, S.K.; Kim, M.; Reinherz, E.L.; Gupta, S.; Forthal, D.N.; Sattentau, Q.J.; et al. Anti-HIV IgA isotypes: Differential virion capture and inhibition of transcytosis are linked to prevention of mucosal R5 SHIV transmission. AIDS 2013, 27, F13–F20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochereau, N.; Pavot, V.; Verrier, B.; Ensinas, A.; Genin, C.; Corthésy, B.; Paul, S. Secretory IgA as a vaccine carrier for delivery of HIV antigen to M cells. Eur. J. Immunol. 2015, 45, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Aleyd, E.; Al, M.; Tuk, C.W.; van der Laken, C.J.; van Egmond, M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcαRI. J. Immunol. 2016, 197, 4552–4559. [Google Scholar] [CrossRef] [PubMed]
- Ben Mkaddem, S.; Christou, I.; Rossato, E.; Berthelot, L.; Lehuen, A.; Monteiro, R.C. IgA, IgA receptors, and their anti-inflammatory properties. Curr. Top. Microbiol. Immunol. 2014, 382, 221–235. [Google Scholar]
- Pasquier, B.; Launay, P.; Kanamaru, Y.; Moura, I.C.; Pfirsch, S.; Ruffie, C.; Henin, D.; Benhamou, M.; Pretolani, M.; Blank, U.; et al. Identification of FcαRI as an inhibitory receptor that controls inflammation: Dual role of FcRγ ITAM. Immunity 2005, 22, 31–42. [Google Scholar]
- Rossato, E.; Ben Mkaddem, S.; Kanamaru, Y.; Hurtado-Nedelec, M.; Hayem, G.; Descatoire, V.; Vonarburg, C.; Miescher, S.; Zuercher, A.W.; Monteiro, R.C. Reversal of arthritis by human monomeric IgA through the receptor-mediated SH2 domain-containing phosphatase 1 inhibitory pathway. Arthritis Rheumatol. 2015, 67, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Kanamaru, Y.; Pfirsch, S.; Aloulou, M.; Vrtovsnik, F.; Essig, M.; Loirat, C.; Deschenes, G.; Guerin-Marchand, C.; Blank, U.; Monteiro, R.C.; et al. Inhibitory ITAM signaling by FcαRI-FcRγ chain controls multiple activating responses and prevents renal inflammation. J. Immunol. 2008, 180, 2669–2678. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kanamaru, Y.; Liu, C.; Suzuki, Y.; Tada, N.; Okumura, K.; Horikoshi, S.; Tomino, Y. Negative regulation of inflammatory responses by immunoglobulin A receptor (FcαRI) inhibits the development of Toll-like receptor-9 signalling-accelerated glomerulonephritis. Clin. Exp. Immunol. 2011, 166, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kanamaru, Y.; Watanabe, T.; Tada, N.; Horikoshi, S.; Suzuki, Y.; Liu, Z.; Tomino, Y. Targeted IgA Fc receptor I (FcαRI) therapy in the early intervention and treatment of pristane-induced lupus nephritis in mice. Clin. Exp. Immunol. 2015, 181, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic antibodies: What have we learnt from targeting CD20 and where are we going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonarburg, C.; Loetscher, M.; Spycher, M.O.; Kropf, A.; Illi, M.; Salmon, S.; Roberts, S.; Steinfuehrer, K.; Campbell, I.; Koernig, S.; et al. Topical application of nebulized human IgG, IgA and IgAM in the lungs of rats and non-human primates. Respir. Res. 2019, 20, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koernig, S.; Campbell, I.K.; Mackenzie-Kludas, C.; Schaub, A.; Loetscher, M.; Ching Ng, W.; Zehnder, R.; Pelczar, P.; Sanli, I.; Alhamdoosh, M.; et al. Topical application of human-derived Ig isotypes for the control of acute respiratory infection evaluated in a human CD89-expressing mouse model. Mucosal Immunol. 2019, 12, 1013–1024. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies 2019, 8, 57. https://doi.org/10.3390/antib8040057
de Sousa-Pereira P, Woof JM. IgA: Structure, Function, and Developability. Antibodies. 2019; 8(4):57. https://doi.org/10.3390/antib8040057
Chicago/Turabian Stylede Sousa-Pereira, Patrícia, and Jenny M. Woof. 2019. "IgA: Structure, Function, and Developability" Antibodies 8, no. 4: 57. https://doi.org/10.3390/antib8040057
APA Stylede Sousa-Pereira, P., & Woof, J. M. (2019). IgA: Structure, Function, and Developability. Antibodies, 8(4), 57. https://doi.org/10.3390/antib8040057