Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid

 ,
,  , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Study Design
2.3. COX-mediated Effect of Acetylsalicylic Acid: TXA2 Metabolite Measurement
2.4. LC-QTOF-MS Metabolic Fingerprinting
2.4.1. Sample Preparation
2.4.2. Ultra-High-Performance LC-QTOF-MS Method Sample Analysis
2.4.3. Performance Evaluation
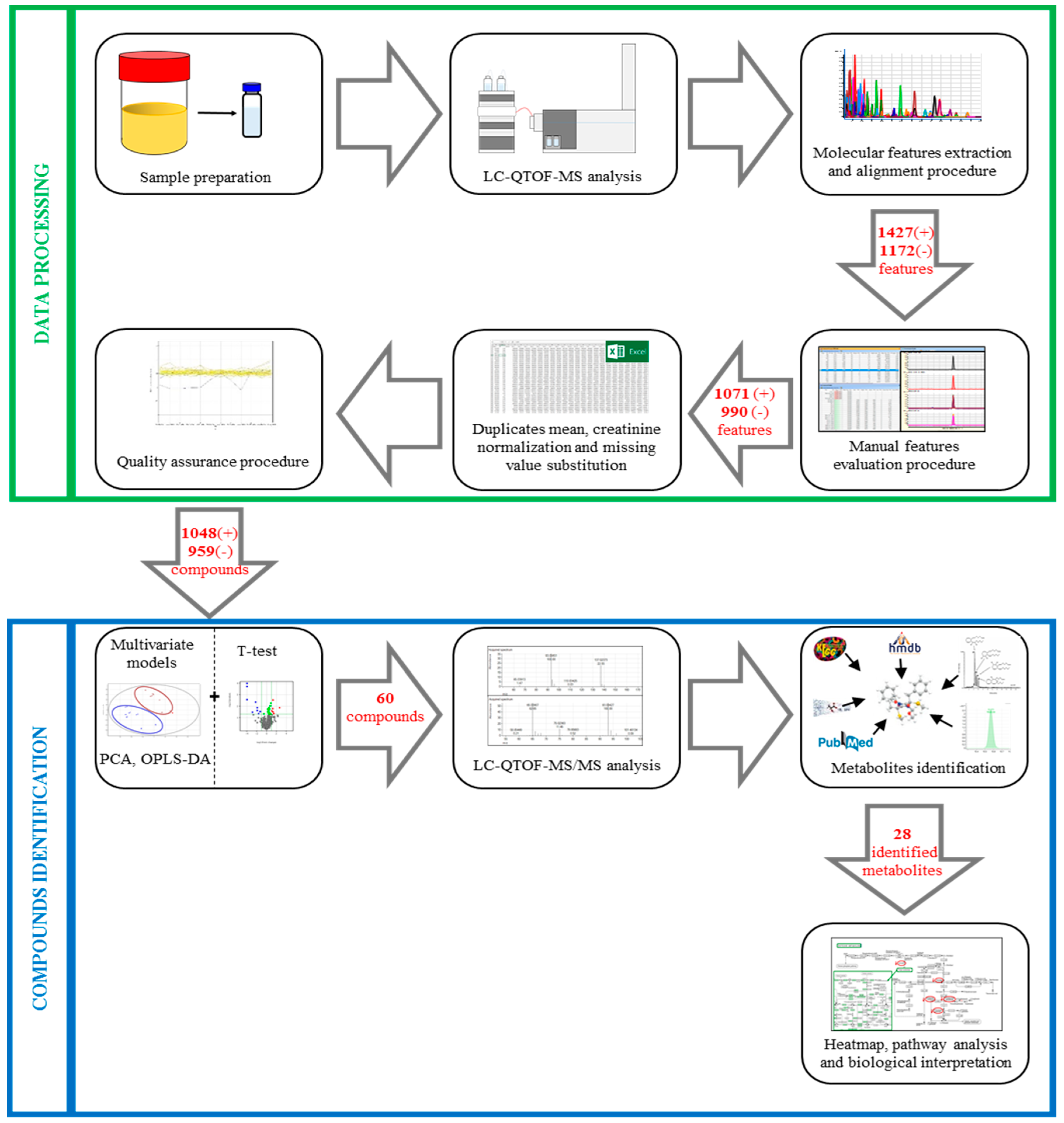
2.4.4. Data Processing
2.4.5. Compound Identification
2.4.6. Ultra-High-Performance LC-QTOF-MS/MS Method Sample Analysis
2.5. Statistics
3. Results
3.1. TXA2 Metabolite Measurement
3.2. LC-QTOF-MS Metabolic Fingerprint
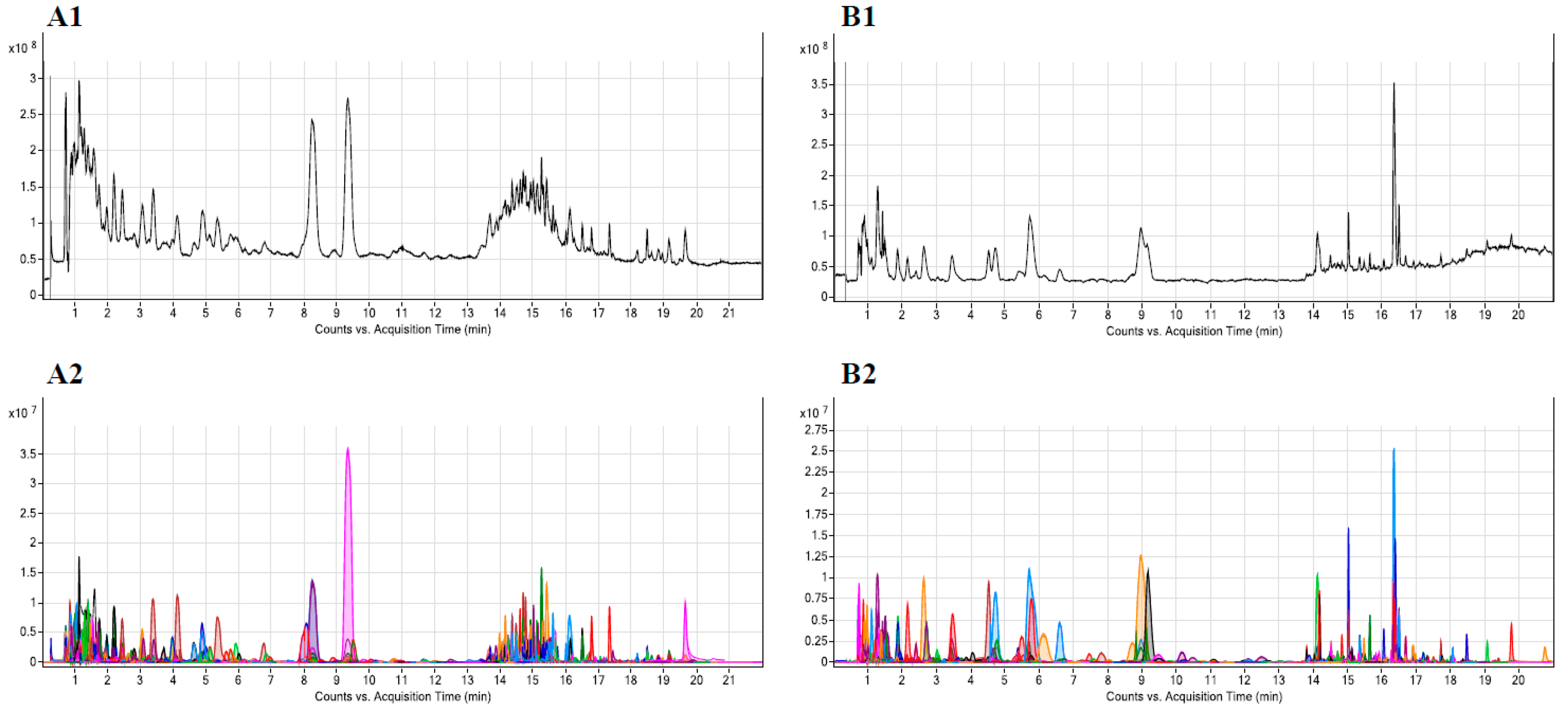
3.2.1. LC-QTOF-MS Sample Analysis
3.2.2. Performance Evaluation
3.2.3. Data Processing
3.3. Compound Identification
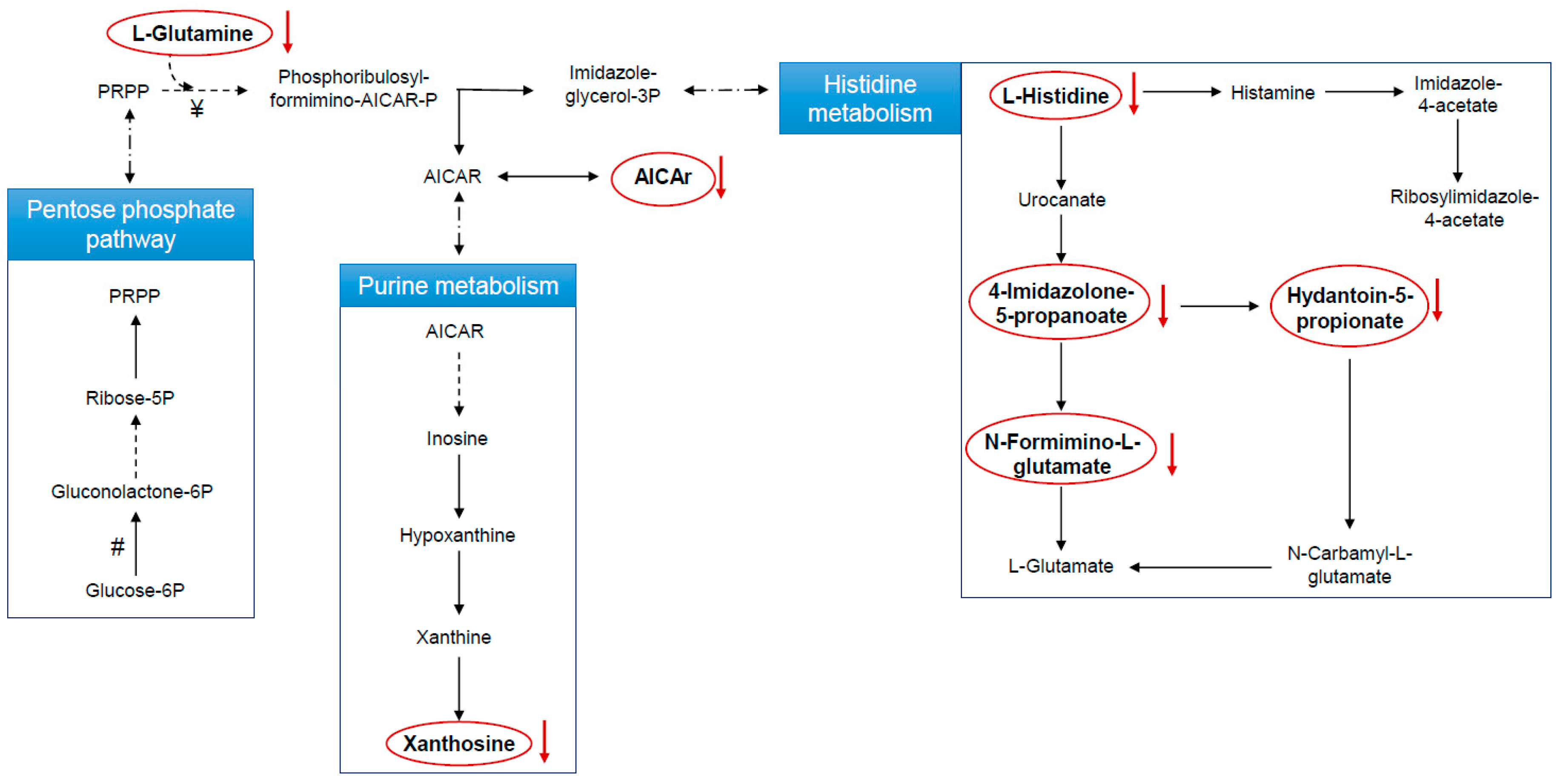
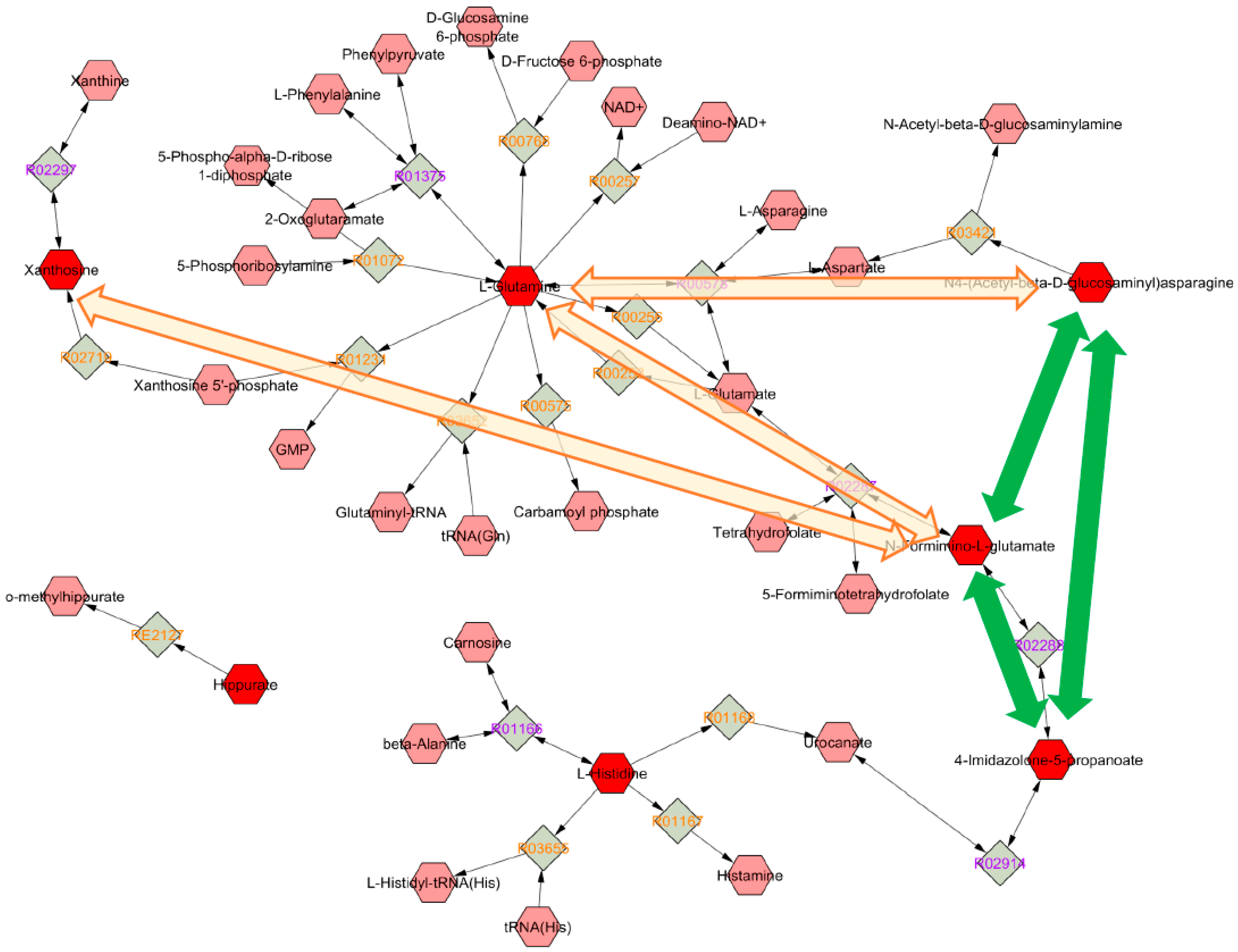
Metabolic Changes Induced by ASA
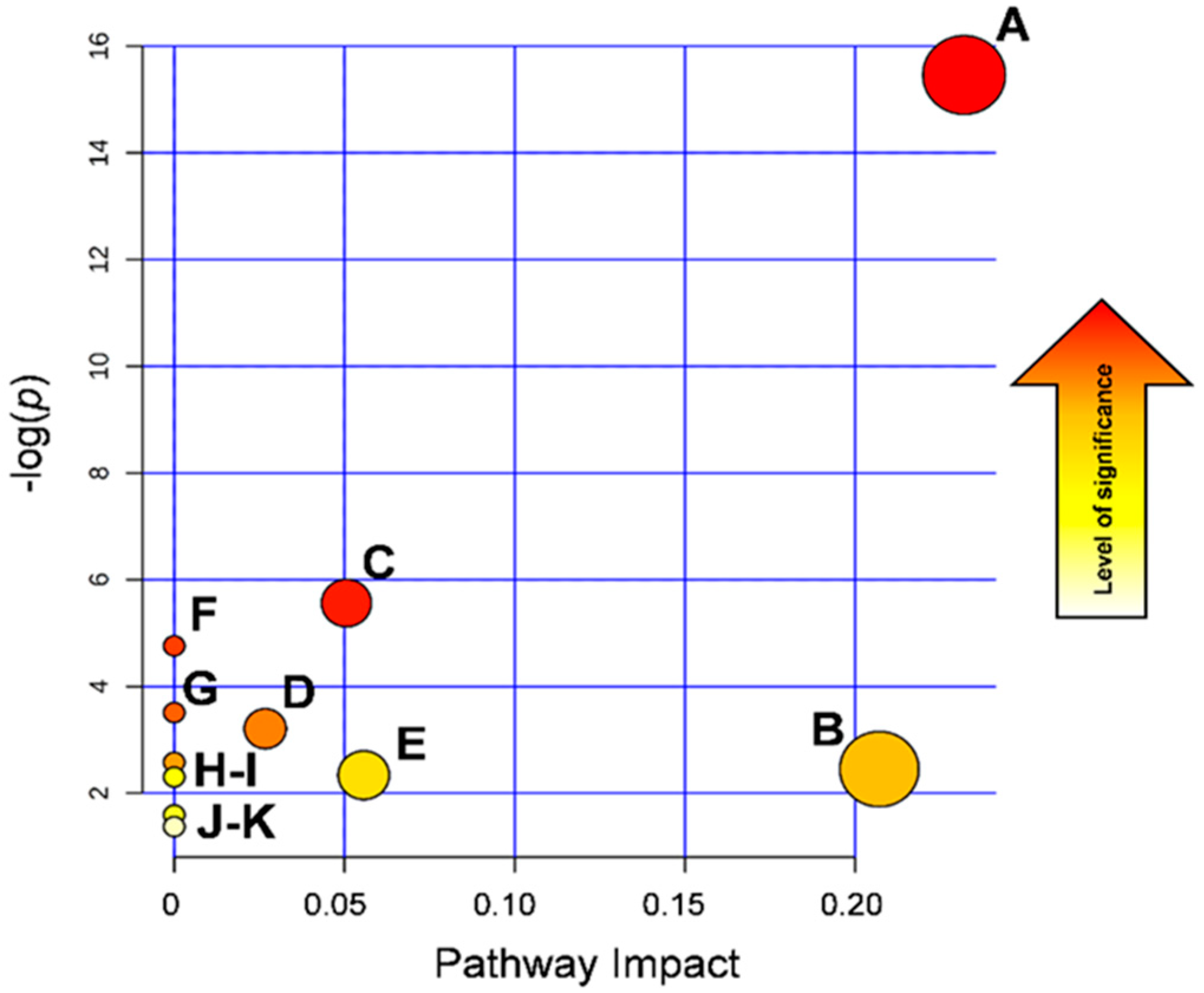
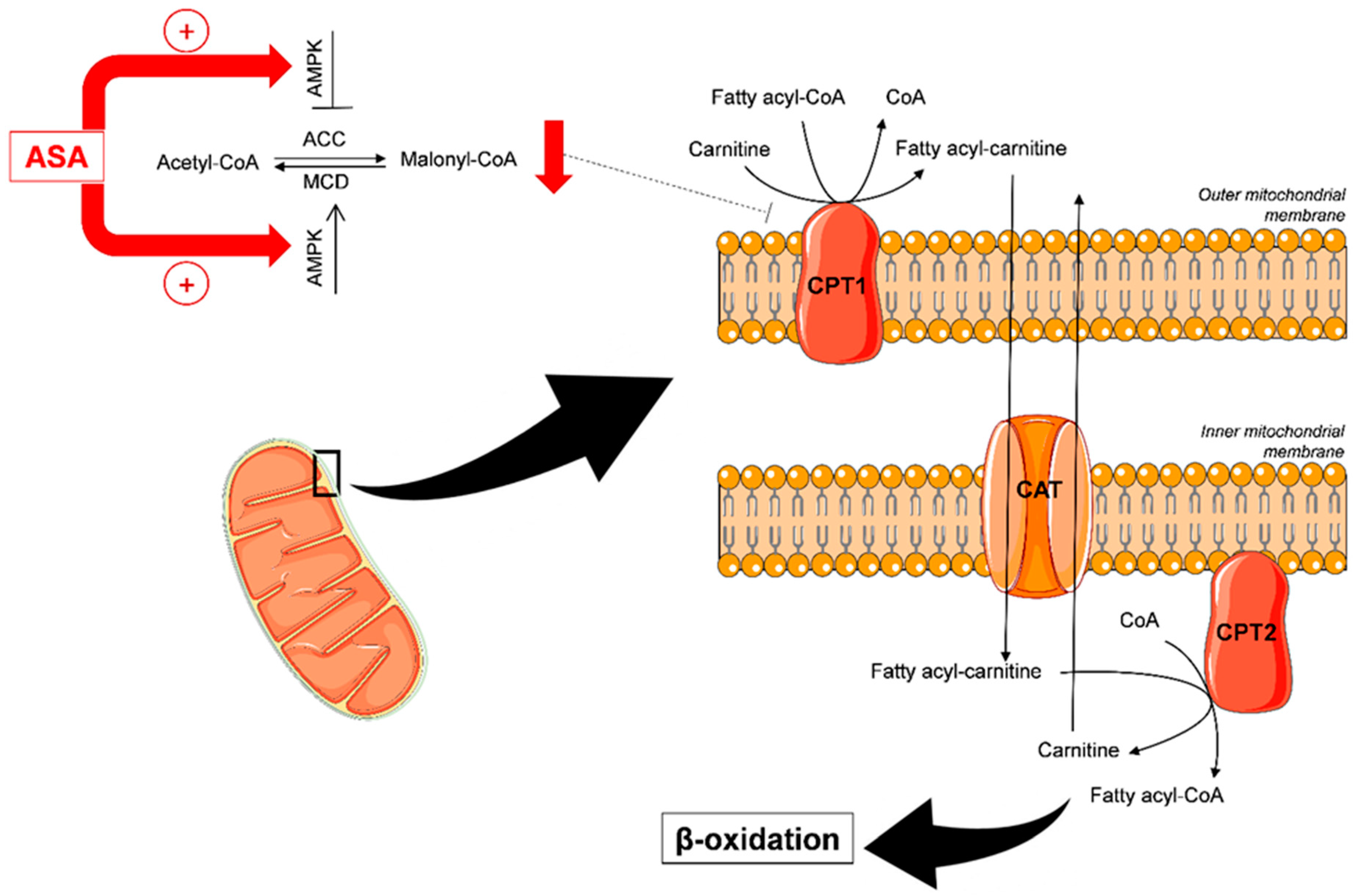
3.4. Biological Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
Reagents, Reference Materials, and Apparatus
References
- Patrono, C.; Morais, J.; Baigent, C.; Collet, J.P.; Fitzgerald, D.; Halvorsen, S.; Rocca, B.; Siegbahn, A.; Storey, R.F.; Vilahur, G. Antiplatelet Agents for the Treatment and Prevention of Coronary Atherothrombosis. J. Am. Coll. Cardiol. 2017, 70, 1760–1776. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Ornelas, A.; Zacharias-Millward, N.; Menter, D.G.; Davis, J.S.; Lichtenberger, L.; Hawke, D.; Hawk, E.; Vilar, E.; Bhattacharya, P.; Millward, S. Beyond COX-1: The effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Rev. 2017. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Peng, Y.; Duan, W.; Tian, Y.; Zhang, J.; Hu, T.; Cai, Y.; Feng, Y.; Li, G. Aspirin regulates hepatocellular lipid metabolism by activating AMPK signaling pathway. J. Toxicol. Sci. 2015, 40, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Kamble, P.; Litvinov, D.; Aluganti Narasimhulu, C.; Jiang, X.; Parthasarathy, S. Aspirin may influence cellular energy status. Eur. J. Pharmacol. 2015, 749, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.L.; Huang, P.H.; Kao, C.L.; Leu, H.B.; Cheng, Y.H.; Liao, Y.W.; Yang, Y.P.; Chien, Y.; Wang, C.Y.; Hsiao, C.Y.; et al. Aspirin attenuates vinorelbine-induced endothelial inflammation via modulating SIRT1/AMPK axis. Biochem. Pharmacol. 2014, 88, 189–200. [Google Scholar] [CrossRef]
- Everett, J.R. Pharmacometabonomics in humans: A new tool for personalized medicine. Pharmacogenomics 2015, 16, 737–754. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Wilson, I.D.; Lindon, J.C. Pharmacometabonomics as an effector for personalized medicine. Pharmacogenomics 2011, 12, 103–111. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [Green Version]
- Squellerio, I.; Porro, B.; Songia, P.; Veglia, F.; Caruso, D.; Tremoli, E.; Cavalca, V. Liquid chromatography-tandem mass spectrometry for simultaneous measurement of thromboxane B2 and 12(S)-hydroxyeicosatetraenoic acid in serum. J. Pharm. Biomed. Anal. 2014, 96, 256–262. [Google Scholar] [CrossRef]
- Cavalca, V.; Minardi, F.; Scurati, S.; Guidugli, F.; Squellerio, I.; Veglia, F.; Dainese, L.; Guarino, A.; Tremoli, E.; Caruso, D. Simultaneous quantification of 8-iso-prostaglandin-F(2alpha) and 11-dehydro thromboxane B(2) in human urine by liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2010, 397, 168–174. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]
- Godzien, J.; Alonso-Herranz, V.; Barbas, C.; Armitage, E.G. Controlling the quality of metabolomics data: New strategies to get the best out of the QC sample. Metabolomics 2015, 11, 518–528. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Gika, H.G.; Theodoridis, G.A.; Wingate, J.E.; Wilson, I.D. Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: Application to human urine. J. Proteome Res. 2007, 6, 3291–3303. [Google Scholar] [CrossRef]
- Naz, S.; Vallejo, M.; Garcia, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef]
- Patrignani, P.; Filabozzi, P.; Patrono, C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Investig. 1982, 69, 1366–1372. [Google Scholar] [CrossRef]
- Van Ryn, J.; Kink-Eiband, M.; Kuritsch, I.; Feifel, U.; Hanft, G.; Wallenstein, G.; Trummlitz, G.; Pairet, M. Meloxicam does not affect the antiplatelet effect of aspirin in healthy male and female volunteers. J. Clin. Pharmacol. 2004, 44, 777–784. [Google Scholar] [CrossRef]
- Frelinger, A.L., 3rd; Furman, M.I.; Linden, M.D.; Li, Y.; Fox, M.L.; Barnard, M.R.; Michelson, A.D. Residual arachidonic acid-induced platelet activation via an adenosine diphosphate-dependent but cyclooxygenase-1- and cyclooxygenase-2-independent pathway: A 700-patient study of aspirin resistance. Circulation 2006, 113, 2888–2896. [Google Scholar] [CrossRef] [Green Version]
- Santilli, F.; Rocca, B.; De Cristofaro, R.; Lattanzio, S.; Pietrangelo, L.; Habib, A.; Pettinella, C.; Recchiuti, A.; Ferrante, E.; Ciabattoni, G.; et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: Implications for aspirin “resistance”. J. Am. Coll. Cardiol. 2009, 53, 667–677. [Google Scholar] [CrossRef]
- Su, Y.F.; Yang, S.H.; Lee, Y.H.; Wu, B.C.; Huang, S.C.; Liu, C.M.; Chen, S.L.; Pan, Y.F.; Chou, S.S.; Chou, M.Y.; et al. Aspirin-induced inhibition of adipogenesis was p53-dependent and associated with inactivation of pentose phosphate pathway. Eur. J. Pharmacol. 2014, 738, 101–110. [Google Scholar] [CrossRef]
- Lewis, J.P.; Yerges-Armstrong, L.M.; Ellero-Simatos, S.; Georgiades, A.; Kaddurah-Daouk, R.; Hankemeier, T. Integration of pharmacometabolomic and pharmacogenomic approaches reveals novel insights into antiplatelet therapy. Clin. Pharmacol. Ther. 2013, 94, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.P.; McNicol, A.; Brandes, L.J.; Becker, A.B.; Gerrard, J.M. A role for intracellular histamine in collagen-induced platelet aggregation. Blood 1990, 75, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.M.; Segura, J.A.; Campos-Sandoval, J.A.; Lobo, C.; Alonso, L.; Alonso, F.J.; Marquez, J. Glutamine homeostasis and mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 2051–2061. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, V.H.; Merhi, F.; Djavaheri-Mergny, M.; Duran, R.V. Glutaminolysis and autophagy in cancer. Autophagy 2015, 11, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Ellero-Simatos, S.; Beitelshees, A.L.; Lewis, J.P.; Yerges-Armstrong, L.M.; Georgiades, A.; Dane, A.; Harms, A.C.; Strassburg, K.; Guled, F.; Hendriks, M.M.; et al. Oxylipid Profile of Low-Dose Aspirin Exposure: A Pharmacometabolomics Study. J. Am. Heart Assoc. 2015, 4, e002203. [Google Scholar] [CrossRef] [Green Version]
- Cory, J.G.; Cory, A.H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: Asparaginase treatment in childhood acute lymphoblastic leukemia. In Vivo 2006, 20, 587–589. [Google Scholar]
- Rothwell, P.M.; Algra, A.; Chen, Z.; Diener, H.C.; Norrving, B.; Mehta, Z. Effects of aspirin on risk and severity of early recurrent stroke after transient ischaemic attack and ischaemic stroke: Time-course analysis of randomised trials. Lancet 2016, 388, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Tatham, M.H.; Cole, C.; Scullion, P.; Wilkie, R.; Westwood, N.J.; Stark, L.A.; Hay, R.T. A Proteomic Approach to Analyze the Aspirin-mediated Lysine Acetylome. Mol. Cell. Proteom. MCP 2017, 16, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Kappler, M.; Pabst, U.; Rot, S.; Taubert, H.; Wichmann, H.; Schubert, J.; Bache, M.; Weinholdt, C.; Immel, U.D.; Grosse, I.; et al. Normoxic accumulation of HIF1alpha is associated with glutaminolysis. Clin. Oral Investig. 2017, 21, 211–224. [Google Scholar] [CrossRef]
- Liu, Y.X.; Feng, J.Y.; Sun, M.M.; Liu, B.W.; Yang, G.; Bu, Y.N.; Zhao, M.; Wang, T.J.; Zhang, W.Y.; Yuan, H.F.; et al. Aspirin inhibits the proliferation of hepatoma cells through controlling GLUT1-mediated glucose metabolism. Acta Pharmacol. Sin. 2019, 40, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Kappler, M.; Pabst, U.; Weinholdt, C.; Taubert, H.; Rot, S.; Kaune, T.; Kotrba, J.; Porsch, M.; Guttler, A.; Bache, M.; et al. Causes and Consequences of a Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism. Int. J. Mol. Sci. 2019, 20, 4742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27 (Suppl. 2), S43–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Uppala, R.; Dudiak, B.; Beck, M.E.; Bharathi, S.S.; Zhang, Y.; Stolz, D.B.; Goetzman, E.S. Aspirin increases mitochondrial fatty acid oxidation. Biochem. Biophys. Res. Commun. 2017, 482, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Glasgow, J.F.; Middleton, B.; Moore, R.; Gray, A.; Hill, J. The mechanism of inhibition of beta-oxidation by aspirin metabolites in skin fibroblasts from Reye’s syndrome patients and controls. Biochim. Biophys. Acta 1999, 1454, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Wang, S.; Osame, M. Aspirin induces short-chain free fatty acid accumulation in rats. Eur. J. Pharmacol. 1998, 349, 49–52. [Google Scholar] [CrossRef]
- Ma, N.; Karam, I.; Liu, X.W.; Kong, X.J.; Qin, Z.; Li, S.H.; Jiao, Z.H.; Dong, P.C.; Yang, Y.J.; Li, J.Y. UPLC-Q-TOF/MS-based urine and plasma metabonomics study on the ameliorative effects of aspirin eugenol ester in hyperlipidemia rats. Toxicol. Appl. Pharmacol. 2017, 332, 40–51. [Google Scholar] [CrossRef]
- Fiamoncini, J.; Lima, T.M.; Hirabara, S.M.; Ecker, J.; Gorjao, R.; Romanatto, T.; ELolimy, A.; Worsch, S.; Laumen, H.; Bader, B.; et al. Medium-chain dicarboxylic acylcarnitines as markers of n-3 PUFA-induced peroxisomal oxidation of fatty acids. Mol. Nutr. Food Res. 2015, 59, 1573–1583. [Google Scholar] [CrossRef]
- Fu, Y.; Zhen, J.; Lu, Z. Synergetic Neuroprotective Effect of Docosahexaenoic Acid and Aspirin in SH-Y5Y by Inhibiting miR-21 and Activating RXRalpha and PPARalpha. DNA Cell Biol. 2017, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Khamis, M.M.; Adamko, D.J.; El-Aneed, A. Mass spectrometric based approaches in urine metabolomics and biomarker discovery. Mass Spectrom. Rev. 2017, 36, 115–134. [Google Scholar] [CrossRef]
- Tsiropoulou, S.; McBride, M.; Padmanabhan, S. Urine Metabolomics in Hypertension Research. Methods Mol. Biol. 2017, 1527, 61–68. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Fold Change (T0 vs. T7) | VIP Score |
|---|---|---|
| 1,3,7-trimethyluric acid | −2.44 | 3.21 |
| aspartylglycosamine | −1.79 | 2.90 |
| aspartyl-isoleucine | −1.29 | 2.79 |
| tiglylcarnitine | −1.30 | 2.53 |
| 2-methylhippuric acid | −1.69 | 2.18 |
| nicotinuric acid | −1.26 | 2.13 |
| 2-isopropylmaleate | 3.79 | 2.11 |
| heptanoylcarnitine | −1.23 | 2.10 |
| 3-methylglutarylcarnitine | 2.56 | 1.98 |
| L-histidine | −1.56 | 1.96 |
| xanthosine | −1.33 | 1.93 |
| N-formimino-L-glutamate | −1.37 | 1.68 |
| hydantoin-5-propionate | −1.35 | 1.66 |
| corchoionoside B | −1.54 | 1.63 |
| 2-(2-phenylacetoxy)propionylglycine | 1.37 | 1.63 |
| prunasin | 3.14 | 1.62 |
| 4-imidazolone-5-propanoate | −1.34 | 1.50 |
| AICAr | −1.26 | 1.48 |
| isovalerylcarnitine | −1.35 | 1.43 |
| glycochenodeoxycholate 7-sulfate | −1.41 | 1.39 |
| L-glutamine | −1.29 | 1.30 |
| 1-malonylamino)cyclopropanecarboxylic acid | −1.26 | 1.22 |
| butyryl-L-carnitine | −1.31 | 1.15 |
| piperidine | −1.65 | 1.08 |
| benzeneacetamide-4-O-sulphate | −1.18 | 1.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Minno, A.; Porro, B.; Turnu, L.; Manega, C.M.; Eligini, S.; Barbieri, S.; Chiesa, M.; Poggio, P.; Squellerio, I.; Anesi, A.; et al. Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid. J. Clin. Med. 2020, 9, 51. https://doi.org/10.3390/jcm9010051
Di Minno A, Porro B, Turnu L, Manega CM, Eligini S, Barbieri S, Chiesa M, Poggio P, Squellerio I, Anesi A, et al. Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid. Journal of Clinical Medicine. 2020; 9(1):51. https://doi.org/10.3390/jcm9010051
Chicago/Turabian StyleDi Minno, Alessandro, Benedetta Porro, Linda Turnu, Chiara Maria Manega, Sonia Eligini, Simone Barbieri, Mattia Chiesa, Paolo Poggio, Isabella Squellerio, Andrea Anesi, and et al. 2020. "Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid" Journal of Clinical Medicine 9, no. 1: 51. https://doi.org/10.3390/jcm9010051
APA StyleDi Minno, A., Porro, B., Turnu, L., Manega, C. M., Eligini, S., Barbieri, S., Chiesa, M., Poggio, P., Squellerio, I., Anesi, A., Fiorelli, S., Caruso, D., Veglia, F., Cavalca, V., & Tremoli, E. (2020). Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid. Journal of Clinical Medicine, 9(1), 51. https://doi.org/10.3390/jcm9010051