Results and Discussion
So far there have been no reports of the transesterification reaction of 3,5-diester-γ-pyrone. Nevertheless, in our laboratory some crown ethers and podands have been prepared by in good yield by the transesterification reaction of 2,6-diester-γ-pyrone with triethylene glycol in the presence of NaOMe as a catalyst [
13]. A similar reaction was carried out with pyrone-3,5-dicarboxylate (
6) which was unsuccessful and starting material remained intact.
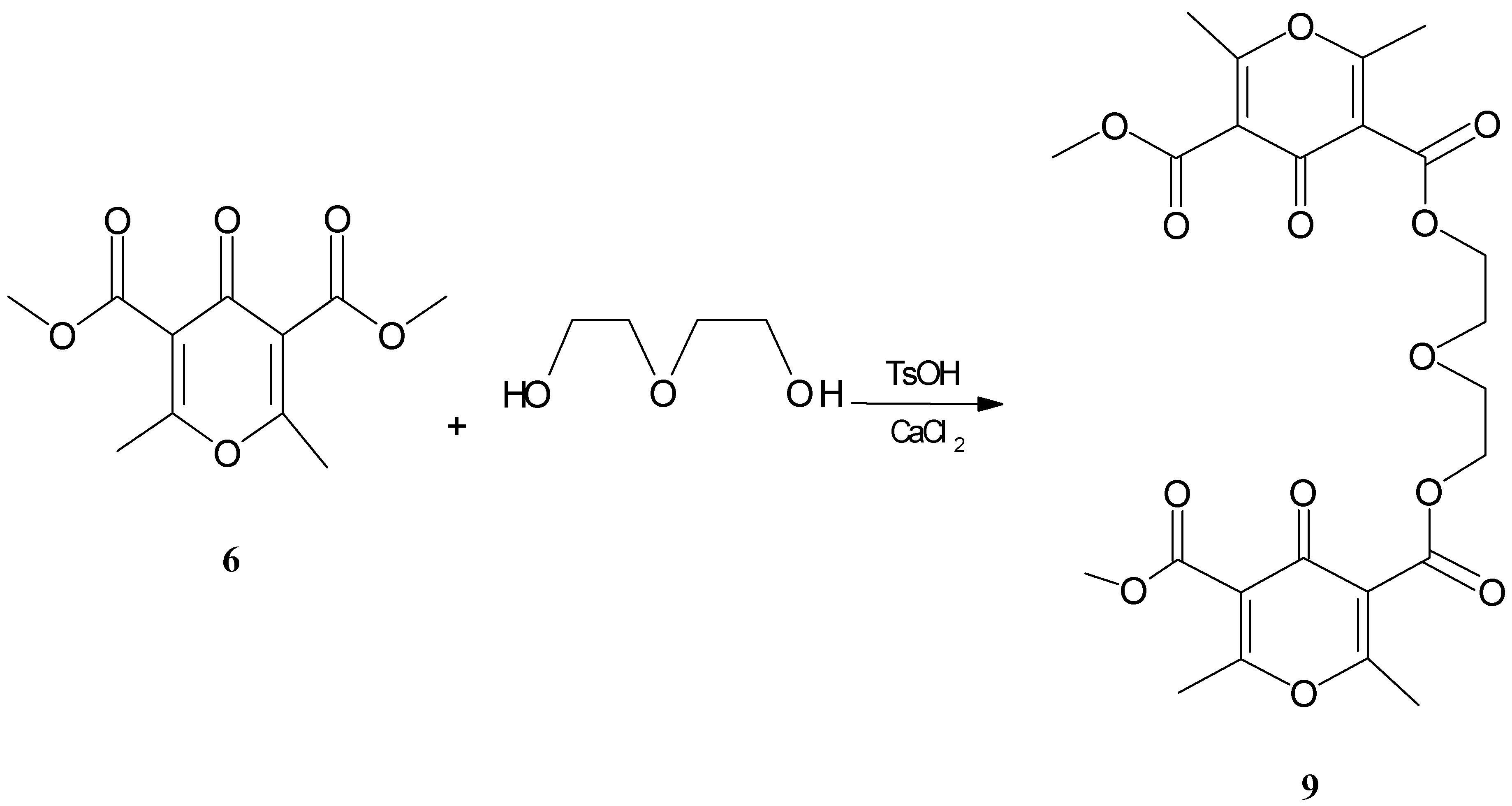
Consequently, we used acidic conditions for the transesterification reaction. Thus, diester (
6) reacted at 150
0C with diethylene glycol in the presence of TsOH as a catalyst and CaCl
2 as a template to give compound (
9) in 6% yield (
Scheme 1).
In contrast with the transesterification reaction of dimethyl chelidonate [
13], reaction of 2,6-dimethyl-4-oxo-4
H-pyran-3,5-dicarboxylate (
6) with triethylene glycol or tetraethylene glycol produced a mixture of higher molecular weight products which could not be identified. Therefore we decided to use ethylene glycol monoalkyl ethers instead of ethylene glycols to prevent this polymerization reaction. Thus, treatment of ester (
7) with excess ethylene glycol monomethyl ether in the presence of TsOH led to compounds (
10) and (
11) in 29 and 16.4% yields respectively (
Scheme 2).
In the second part of this work, we used nucleophilic substitution reactions of 3,5-bis(bromomethyl) pyrone for the synthesis of new podands. For instance, 3,5-bis(bromomethyl)-4
H-pyran-4-one (
8) was prepared according to the lit. [
12] and treated with methoxy ethanol or ethoxyethoxy ethanol in the presence of an excess of sodium hydride to give podands (
12) and (
13) in 62.5 and 53% yields respectively (
Scheme 3).
Compounds (
14,
15) were prepared by the reaction of catechol or salicylaldehyde with (
10) in DMF and in the presence of excess Et
3N at room temperature, in 32 and 13% yields respectively (
Scheme 4).
The data obtained from mass, IR, 1H and 13C NMR spectra and elemental analyses are fully consistent with the proposed structures.
Experimental
General
Melting points were determined with an Electrothermal Instrument model 9100 and are uncorrected. Infrared (IR) spectra were run on a Shimadzu IR 435 Spectrophotometer as KBr disks or as smears between salt plates. The
1H NMR spectra were recorded on a Varian-EM 390 spectrometer. The
13C NMR spectra were recorded on a FT-NMR Brucker 80 MHz spectrometer. Chemical shifts were reported in values in ppm with TMS as the internal standard. Elemental analyses were performed on a Heareus, CHN-O-RAPID analyzer. The following compounds were prepared using literature methods: (
6) [
10,
11], (
7) [
1] and (
8) [
12].
Bis[2-(3-carbonyloxy-5-methoxycarbonyl-2,6-dimethyl-4-oxo-4H-pyran) ethyl)]ether (9)
A mixture of diethylene glycol (0.22g, 2.07 mmol), TsOH (0.036g, 0.207 mmol) and CaCl2 (0.0155 g, 0.207 mmol), was heated at 130-140oC and pyrone (6) (0.5 g, 2.07 mmol) was added slowly over 1.5 hr. The reaction mixture was heated for 2 hr, cooled to room temperature, then 15 ml water was added and the mixture was extracted with 2×30 ml CHCl3, washed with water and dried over MgSO4.The solvent was evaporated and resulting material was purified by dry column chromatography on silicagel using ethyl acetate as eluent. Compound (9) was obtained in 6% yield. Mp.119-120oC.
1H NMR (CDCl3] δ: 2.29 (12H, s) 3.79 (4H, t, J = 5 Hz), 3.89 (10H, s) 4.44 (4H, J = 5 Hz).
13C NMR (CDCl3) δ: 19.00, 19.05, 53.07, 64.89, 64.15, 121.63, 121.75, 164.25, 164.99, 166.17, 166.31, 172.08.
IR (KBr) cm-1: 1730, 1655, 1615.
Ms: m/z 522(M +): Analyses: Found: C 55.17, H 5.02. Calc. for C24H26O13: C 55.16, H 5.02.
Reaction of compound (6) with ethylene glycol monomethyl ether
A mixture of pyrone (6, 0.25 g, 0.93 mmol), TsOH (0.02 g, 0.093 mmol) and ethylene glycol monomethyl ether (8 mL) was refluxed for 76 hr. Alcohol was removed under vacuum and the resulting crude material was purified by dry column chromatography on silicagel using ethyl acetatepetroleum ether 2:3 as eluent. Compounds (10) and (11) were obtained 29% (mp:70-71oC) and 16.4% (mp:109-110oC) yields respectively.
2,6-Dimethyl-4H-pyran-4-one-3,5-dicarboxylic acid, 5-methyl, 3-(2-methoxyethyl)ester (10)
1H NMR (CDCl3) δ: 1.35 (3H, t, J = 7 Hz), 2.37 (6H, s), 3.38 (3H, s), 3.67 (2H, t, J = 5Hz), 4.35 (2H, q, J = 7 Hz), 4.44 (2H, t, J = 5Hz).
13C NMR (CDCl3) δ: 14.50,18.90, 18.95, 59.28, 62.24, 64.85, 70.52, 121.00, 164.36, 165.91, 172.11.
IR (KBr) cm-1:, 1720, 1665.
Ms: m/z 298 (M +); Analyses: Found: C 56.36, H 6.22. Calc. for C14H18O7: C 56.37, H 6.08.
2,6-Dimethyl-4H-pyran-4-one-3,5-dicarboxylic acid, bis(2-methoxyethyl)ester (11)
1H NMR (CDCl3) δ: 2.38 (6H, s), 3.39 (6H, s), 3.68 (4H, t, J = 4.7), 4.45 (4H, t, J = 4.7).
13C NMR (CDCl3 ) δ: 18.9, 59.3, 64.8, 70.5, 121.7, 164, 165, 172.
IR (KBr) cm-1: 1730, 1655.
Ms: m/z 328; Analyses: Found: C 54.75, H 6.18. Calc. for C15H20O8: C 54.87, H 6.14.
3,5-Bis(methoxyethoxymethyl)-2,6-diphenyl-4H-pyran-4-one (12)
A mixture of 2-methoxyethanol (0.18 mL, 2.3 mmol), sodium hydride (80% in mineral oil; 0.1 g, 3.45 mmol) and anhydrous THF (70 mL) was stirred at room temperature under nitrogen for 10 min and then refluxed for 60 min. After cooling again to r.t., pyrone (8, 0.5 g, 1.15 mmol) in THF (30 mL) was added over a period of 15 min. The resulting mixture was stirred at room temperature for 14 hr.
The aqueous phase was adjusted to pH 7 with dilute HCl and the solvent was removed under reduced pressure. The aqueous phase was extracted with 3×50 CHCl3. The combined CHCl3 extracts were dried over MgSO4 and solvent was removed under vacuum. The crude product was purified by dry column chromatography on silicagel using ethyl acetate as eluent. Compound (12) was obtained in 62.5% yield.
1H NMR (CDCl3) δ: 3.35 (6H, s), 3.40 (4H, m), 3.56(4H, m), (4.25 (4H, s), 7.25-7.75 (10H, m).
13C NMR (CDCl3) δ: 58.75, 63.75, 70, 72.5, 121.25, 128.75, 129, 131.25, 132.5, 165, 180.
IR (neat) cm -1: 1645, 1615.
Ms: m/z 424 (M + ); Analyses: Found: C 70.52, H 6.50. Calc. for C25H28O6 : C 70.74, H 6.25.
3,5-Bis(ethoxyethoxyethoxymethyl)-2,6-diphenyl-4H-pyran-4-one (13)
This compound was prepared according to the above procedure in 53% yield.
1H NMR (CDCl3)δ: 1.2(6H, t, J = 6.5), 3.5-3.8 (20H, m), 4.38 (4H, s), 7.25-7.8 (10H, m).
13C NMR (CDCl3) δ: 31.6, 59.0, 61.7, 63.9, 66.7, 70.1, 72.3, 121.5, 129.3, 129.64, 131.6, 132.6, 166.2, 180.2
IR (neat) cm -1: 3100, 2850, 1620.
Ms: m/z 540 (M+); Analyses: Found: C 68.47, H 7.16. Calc. for C31H40O8: C 68.87, H 7.46.
3,5-Bis[(2-hydroxyphenoxy)methyl]-2,6-diphenyl-4H-pyran-4-one (14)
A mixture of pyrone (10, 0.3 g, 0.69 mmol), catechol (0.15 g, 1.38 mmol), Et3N (0.58 mL, 4.14 mmol) and anhydrous DMF (4 mL) was stirred at room temperature under nitrogen for two days. Then water (15mL) was added, the pH was adjusted to 7 with dilute HCl and the precipitate formed was filtered off. The crude product was purified by dry column chromatography on silicagel using CHCl3 as eluent. Compound (14) was obtained in 32% yield ( mp: 110-111oC).
1H NMR (CDCl3) δ: 4.8 (4H, s), 6.5-7 (8H, m), 7.5-7.9 (10H, m), 8.28 (2H, s).
13C NMR (CDCl3) δ (ppm): 67.5, 117.5, 120, 120.1, 125, 129, 131.25, 132.5, 146.25, 150, 165, 180.0.
IR (KBr) cm -1: 3200, 1645.
Ms: m/z 492 (M +), Analyses: Found: C 75.35, H 4.94. Calc. for C31H24O6: C 75.60, H 4.91.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






