Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. ER Proteostasis and Alpha 1-Antitrypsin
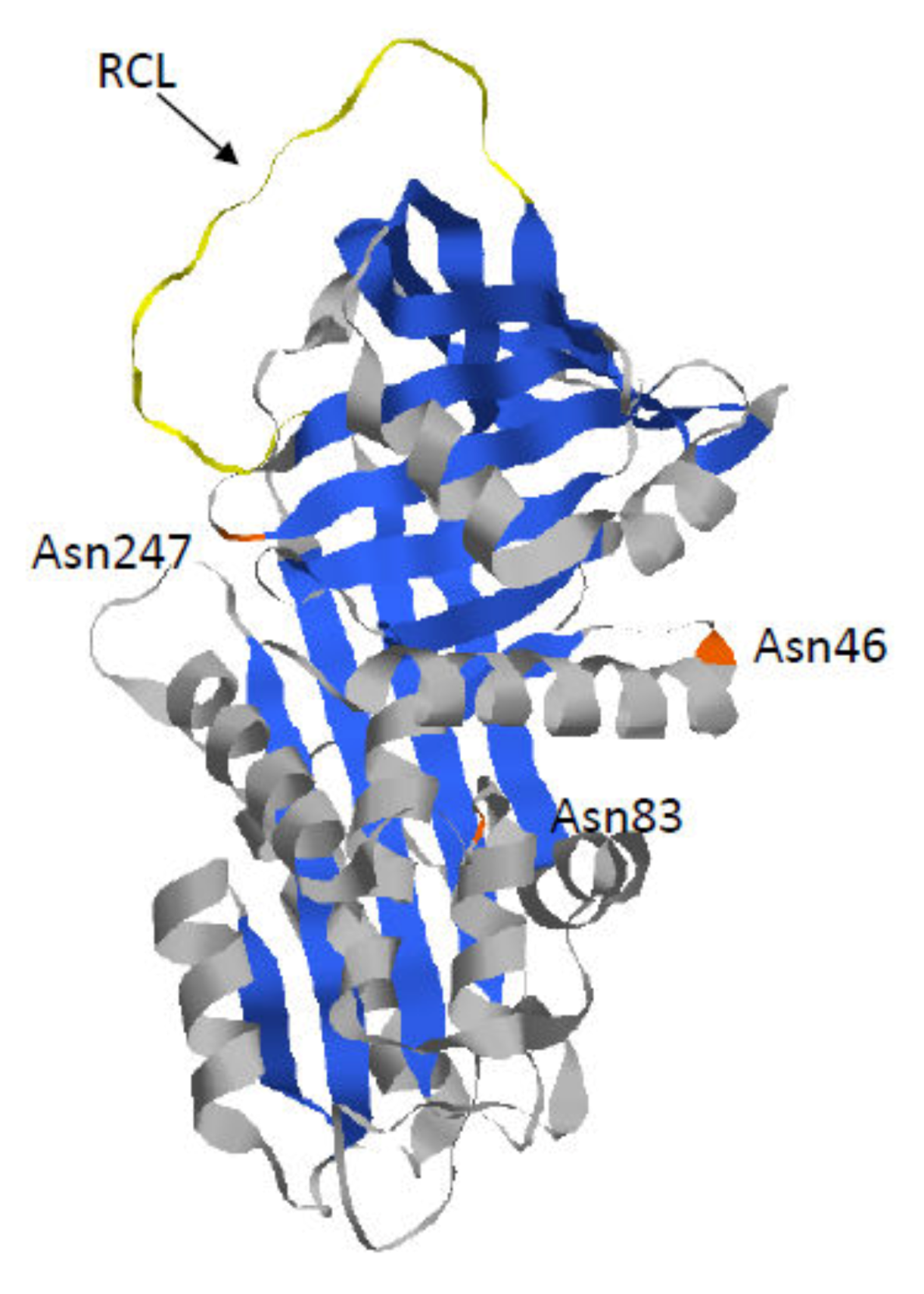
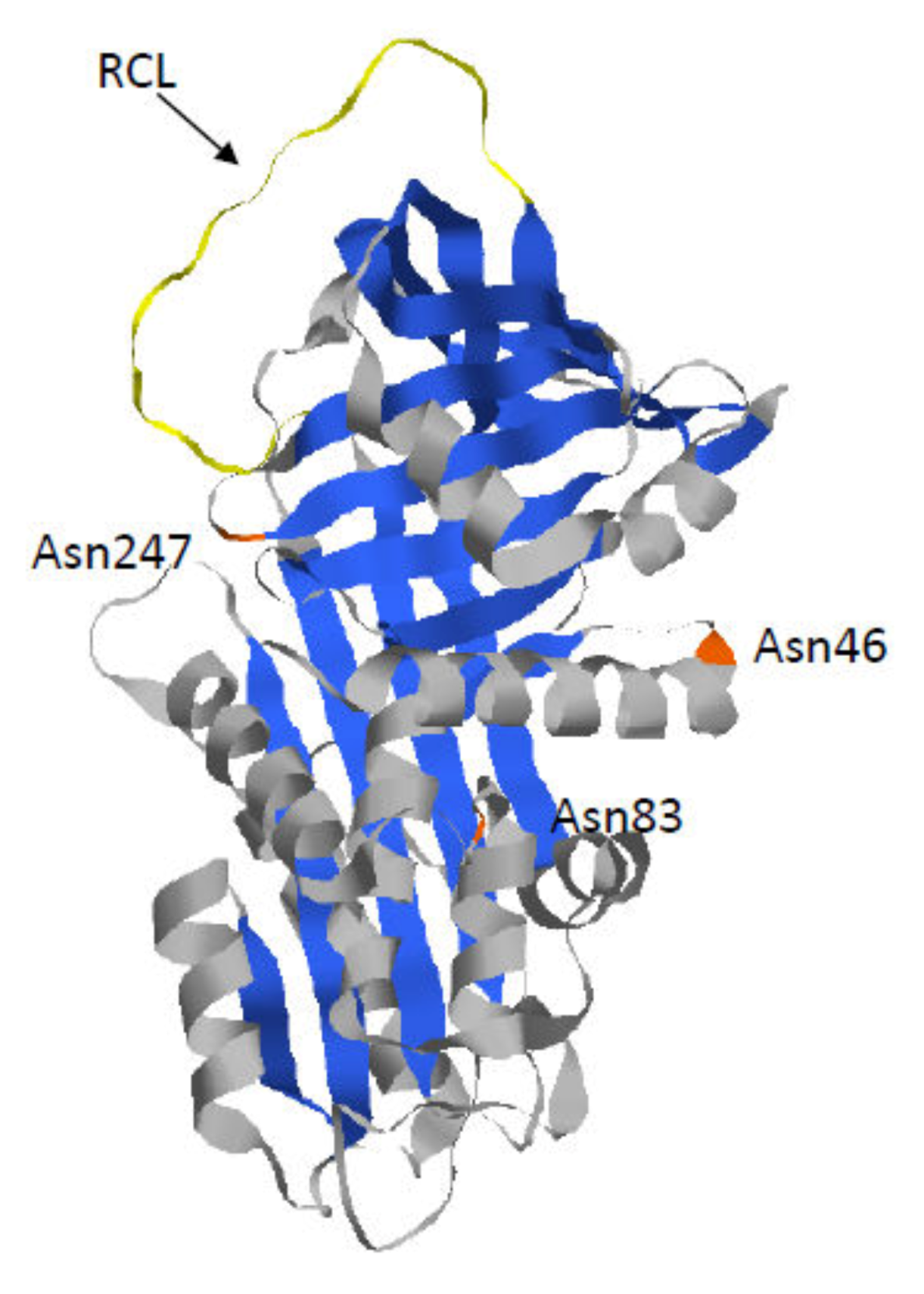
1.1. Alpha 1-Antitrypsin
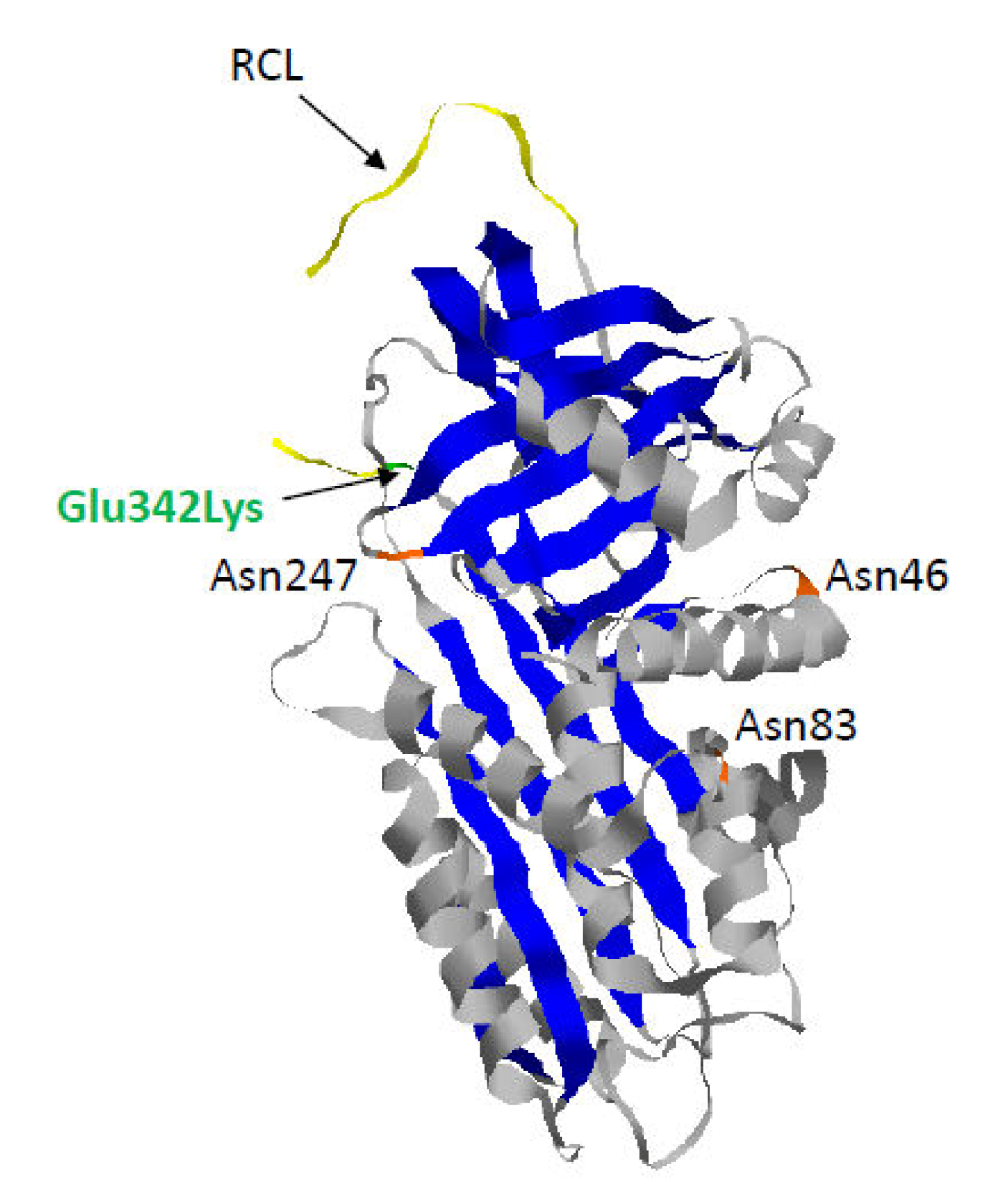
1.2. Z-AAT: The Most Common and Severe Variant
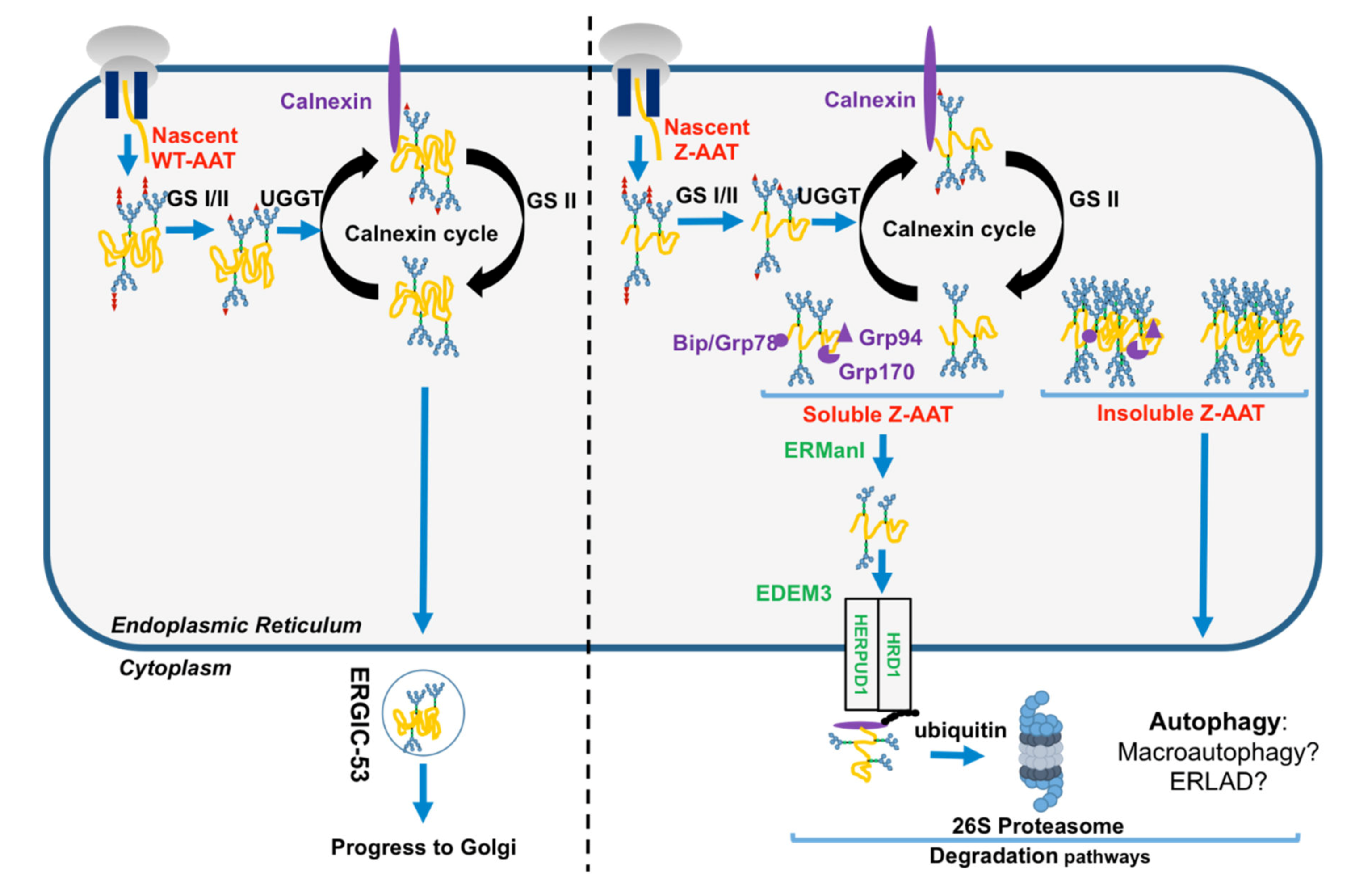
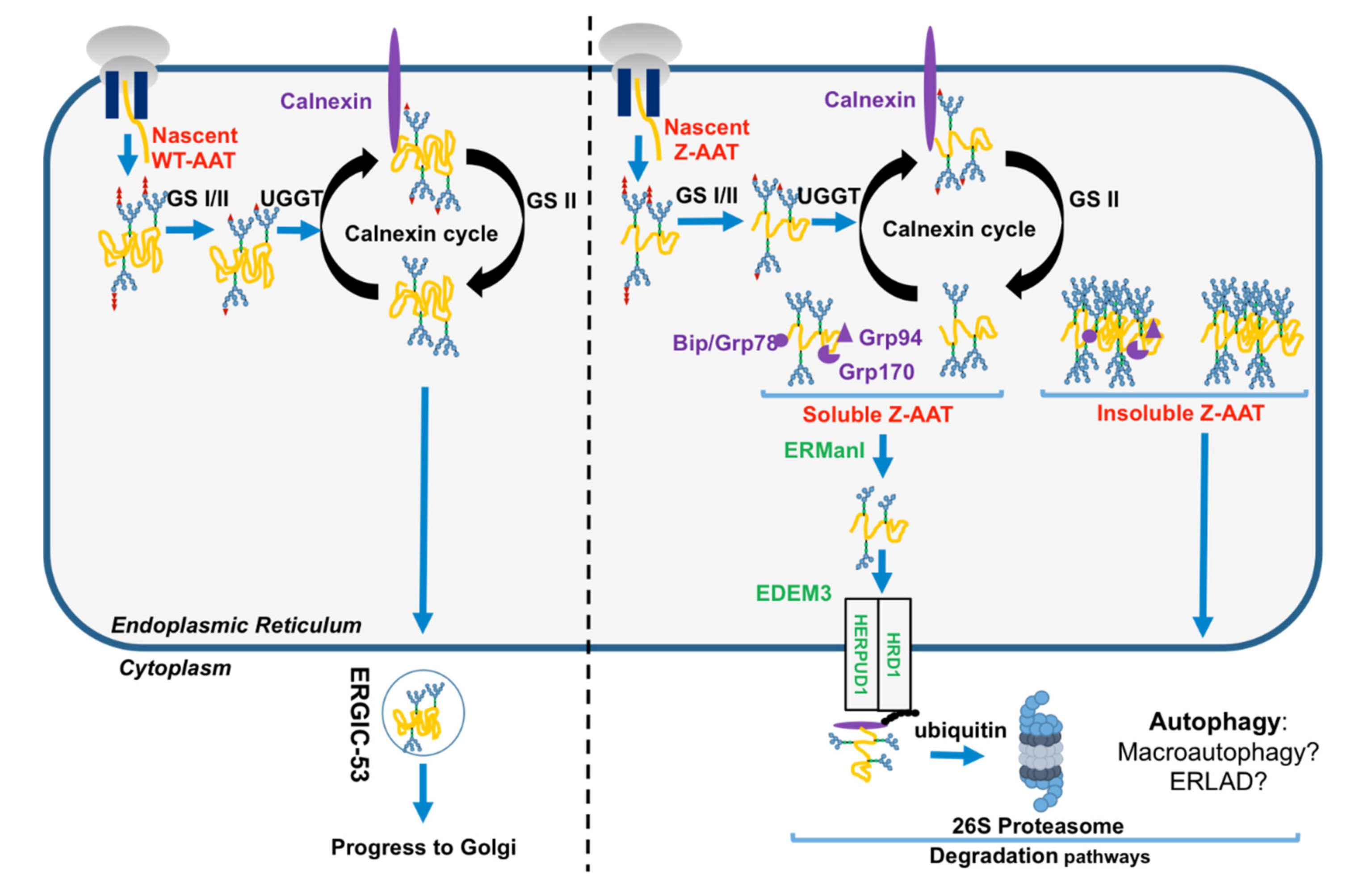
1.3. The Folded AAT vs. the Unfolded Z-AAT in the Secretory Pathway
2. Disposal of the Z Mutant
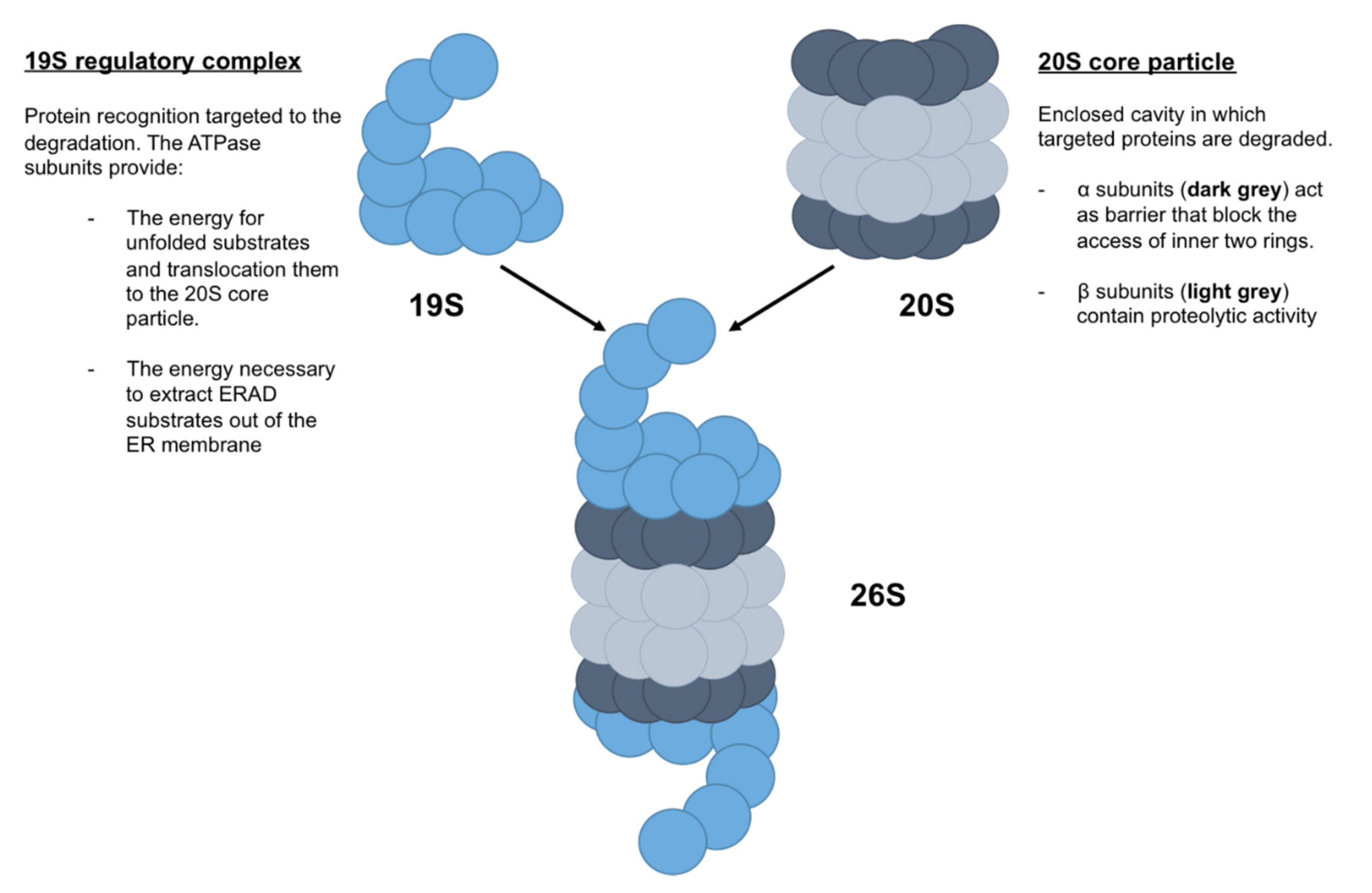
2.1. Z-AAT and Proteasome
2.2. Z-AAT and Autophagy
2.3. Autophagy and Proteasome Crosstalk
3. Proteostasis Imbalance and AATD-Mediated Liver Toxicity
4. Proteostasis Modulators in Correction of AATD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAT | Alpha 1-antitrypsin |
| AATD | Alpha 1-antitrypsin deficiency |
| PN | Proteostasis network |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| NSPs | Neutrophil serine proteases |
| RCL | Reactive center loop |
| PAS + D | Periodic acid–Schiff-positive diastase |
| GS1 and GS2 | Glucosidase I and II |
| CNX | Calnexin |
| ERGIC53 | Endoplasmic reticulum–Golgi intermediate compartment |
| ERp57 | Calnexin–endoplasmic reticulum protein 57 |
| GRP94 | Glucose-regulated protein 94 |
| UGGT1 | Uridine diphosphate (UDP)-glucose:glycoprotein glucosyltransferase 1 |
| ERManI | ER α-mannosidase I |
| EDEM | ER degradation-enhancing α-mannosidase-like proteins |
| HRD1 | HMG-CoA reductase degradation 1 homolog |
| HERPUD1 | Homocysteine inducible ER protein with ubiquitin-like domain 1 |
| UPS | Ubiquitin–proteasome system |
| DUBs enzyme | Deubiquitinilating enzyme |
| CBZ | Carbamazepine |
| TFEB | Transcription factor EB master gene |
| ERLAD | ER-to-lysosome-associated degradation |
| UPR | Unfolded protein response |
| CF | Cystic fibrosis |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| 4-PBA | Sodium 4-phenylbutyrate |
| SAHA | Suberoylanilide hydroxamic acid |
References
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [PubMed]
- Bouchecareilh, M.; Balch, W.E. Proteostasis: A new therapeutic paradigm for pulmonary disease. Proc. Am. Thorac. Soc. 2011, 8, 189–195. [Google Scholar] [PubMed] [Green Version]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. alpha1-Antitrypsin deficiency. Nat. Rev. Dis Primers 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Esra, K.S.; Nathalie, D.-S.; Alain, L.; Marion, B. Overview of Alpha-1 Antitrypsin Deficiency-Mediated Liver Disease. EMJ 2019, 7, 65–79. [Google Scholar]
- Laurell, C.B. Electrophoretic Microheterogeneity of Serum Alpha-1-Antitrypsin. Scand. J. Clin Lab. Invest. 1965, 17, 271–274. [Google Scholar] [CrossRef]
- Ruiz, M.; Lacaille, F.; Berthiller, J.; Joly, P.; Dumortier, J.; Aumar, M.; Bridoux-Henno, L.; Jacquemin, E.; Lamireau, T.; Broué, P.; et al. Liver disease related to alpha1-antitrypsin deficiency in French children: The DEFI-ALPHA cohort. Liver Int. 2019, 39, 1136–1146. [Google Scholar] [CrossRef]
- Chu, A.S.; Chopra, K.B.; Perlmutter, D.H. Is severe progressive liver disease caused by alpha-1-antitrypsin deficiency more common in children or adults? Liver Transpl. 2016, 22, 886–894. [Google Scholar] [CrossRef]
- Bouchecareilh, M.; Balch, W.E. Proteostasis, an emerging therapeutic paradigm for managing inflammatory airway stress disease. Curr. Mol. Med. 2012, 12, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Bouchecareilh, M.; Conkright, J.J.; Balch, W.E. Proteostasis strategies for restoring alpha1-antitrypsin deficiency. Proc Am. Thorac Soc. 2010, 7, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim. Biophys. Acta. 2001, 1535, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharm. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [Green Version]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Dunlea, D.M.; Fee, L.T.; McEnery, T.; McElvaney, N.G.; Reeves, E.P. The impact of alpha-1 antitrypsin augmentation therapy on neutrophil-driven respiratory disease in deficient individuals. J. Inflamm. Res. 2018, 11, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignaud, H.; Cullin, C.; Bouchecareilh, M. Alpha-1 antitrypsin deficiency: A model of alteration of protein homeostasis or proteostasis. Rev. Mal. Respir. 2015, 32, 1059–1071. [Google Scholar] [CrossRef]
- Kalsheker, N.; Morley, S.; Morgan, K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem. Soc. Trans. 2002, 30, 93–98. [Google Scholar] [CrossRef]
- Andersen, O.J.; Risor, M.W.; Poulsen, E.C.; Nielsen, N.C.; Miao, Y.; Enghild, J.J.; Schiøtt, B. Reactive Center Loop Insertion in alpha-1-Antitrypsin Captured by Accelerated Molecular Dynamics Simulation. Biochemistry 2017, 56, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Belorgey, D.; Hagglof, P.; Karlsson-Li, S.; Lomas, D.A. Protein misfolding and the serpinopathies. Prion 2007, 1, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.S.; Yu, M.H. Kinetic dissection of alpha 1-antitrypsin inhibition mechanism. J. Biol Chem. 2002, 277, 11629–11635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zheng, Y.; Zhang, F.; Wei, Z.; Wang, Y.; Carrell, R.W.; Read, R.J.; Chen, G.-Q.; Zhou, A. Molecular Mechanism of Z alpha1-Antitrypsin Deficiency. J. Biol. Chem. 2016, 291, 15674–15686. [Google Scholar] [CrossRef] [Green Version]
- Ekeowa, U.I.; Freeke, J.; Miranda, E.; Gooptu, B.; Bush, M.F.; Perez, J.; Teckman, J.; Robinson, C.V.; Lomas, D.A. Defining the mechanism of polymerization in the serpinopathies. Proc. Natl. Acad. Sci. USA 2010, 107, 17146–17151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, M.; Sendall, T.J.; Pearce, M.C.; Whisstock, J.C.; Huntington, J.A. Molecular basis of alpha1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011, 12, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Dolmer, K.; Gettins, P.G. How the serpin alpha1-proteinase inhibitor folds. J. Biol. Chem. 2012, 287, 12425–12432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomas, D.A.; Hurst, J.R.; Gooptu, B. Update on alpha-1 antitrypsin deficiency: New therapies. J. Hepatol. 2016, 65, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Ghouse, R.; Chu, A.; Wang, Y.; Perlmutter, D.H. Mysteries of alpha1-antitrypsin deficiency: Emerging therapeutic strategies for a challenging disease. Dis. Model. Mech. 2014, 7, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Gooptu, B.; Dickens, J.A.; Lomas, D.A. The molecular and cellular pathology of alpha(1)-antitrypsin deficiency. Trends Mol. Med. 2014, 20, 116–127. [Google Scholar] [CrossRef]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Bouchecareilh, M.; Hutt, D.M.; Szajner, P.; Flotte, T.R.; Balch, W.E. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency. J. Biol. Chem. 2012, 287, 38265–38278. [Google Scholar] [CrossRef] [Green Version]
- Nyfeler, B.; Reiterer, V.; Wendeler, M.W.; Stefan, E.; Zhang, B.; Michnick, S.W.; Hauri, H.-P. Identification of ERGIC-53 as an intracellular transport receptor of alpha1-antitrypsin. J. Cell Biol. 2008, 180, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, P.; Vignaud, H.; Di Martino, J.; Ruiz, M.; Garin, R.; Restier, L.; Belmalih, A.; Marchal, C.; Cullin, C.; Arveiler, B.; et al. ERAD defects and the HFE-H63D variant are associated with increased risk of liver damages in Alpha 1-Antitrypsin Deficiency. PLoS ONE 2017, 12, e0179369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I. Association of the SEL1L protein transmembrane domain with HRD1 ubiquitin ligase regulates ERAD-L. Febs J. 2016, 283, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct Mol Biol. 2011, 18, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Q.; Shen, Y.; Sun, A.; Zhu, X.; Fang, S.; Shen, Y. The ubiquitin ligase Hrd1 promotes degradation of the Z variant alpha 1-antitrypsin and increases its solubility. Mol. Cell Biochem. 2011, 346, 137–145. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, J.; Zhu, N.; Ding, Q.; Zhang, X.; Yu, J.; Qiang, W.; Zhang, Z.; Ma, Y.; Huang, D.; et al. Ubiquitin ligase SYVN1/HRD1 facilitates degradation of the SERPINA1 Z variant/alpha-1-antitrypsin Z variant via SQSTM1/p62-dependent selective autophagy. Autophagy 2017, 13, 686–702. [Google Scholar] [CrossRef] [Green Version]
- Qu, D.; Teckman, J.H.; Omura, S.; Perlmutter, D.H. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol Chem. 1996, 271, 22791–22795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in aging, disease and death: The true identity of a cell death impostor. Cell Death Differ. 2009, 16, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol Med. (Maywood) 2010, 235, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Pastore, N.; Ballabio, A.; Brunetti-Pierri, N. Autophagy master regulator TFEB induces clearance of toxic SERPINA1/alpha-1-antitrypsin polymers. Autophagy 2013, 9, 1094–1096. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Fasana, E.; Bergmann, T.J.; Raimondi, A.; Loi, M.; Solda, T.; Galli, C.; D’Antuono, R.; Morone, D.; Danieli, A.; et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018, 37, e99259. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, D.A.; Knight, M.K.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- Haddock, C.J.; Blomenkamp, K.; Gautam, M.; James, J.; Mielcarska, J.; Gogol, E.; Teckman, J.; Skowyra, D. PiZ mouse liver accumulates polyubiquitin conjugates that associate with catalytically active 26S proteasomes. PLoS ONE 2014, 9, e106371. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell. 2006, 17, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.; Huang, L.; McPherson, J.; Muzny, D.; Rouhani, F.; Brantly, M.; Gibbs, R.; Sifers, R.N. Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology 2009, 50, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Whitman, I.; Molmenti, E.; Moore, K.; Hippenmeyer, P.; Perlmutter, D.H. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc. Natl. Acad. Sci. USA 1994, 91, 9014–9018. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; Perlmutter, D.H. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J. Biol. Chem. 1996, 271, 13215–13220. [Google Scholar] [CrossRef] [Green Version]
- Aad, G.; Abbott, B.; Abdallah, J.; Abdel Khalek, S.; Abdinov, O.; Aben, R.; Abi, B.; Abolins, M.; Abouzeid, O.S.; Abramowicz, H.; et al. Observation of an excited Bc(+/-) meson state with the ATLAS detector. Phys Rev. Lett. 2014, 113, 212004. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef] [Green Version]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Balch, W.E.; Roth, D.M.; Hutt, D.M. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb. Perspect Biol. 2011, 3, a004499. [Google Scholar] [CrossRef] [Green Version]
- Paterson, S.L.; Barry, P.J.; Horsley, A.R. Tezacaftor and ivacaftor for the treatment of cystic fibrosis. Expert Rev. Respir Med. 2020, 14, 15–30. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef] [Green Version]
- Devlin, G.L.; Parfrey, H.; Tew, D.J.; Lomas, D.A.; Bottomley, S.P. Prevention of polymerization of M and Z alpha1-Antitrypsin (alpha1-AT) with trimethylamine N-oxide. Implications for the treatment of alpha1-at deficiency. Am. J. Respir. Cell Mol. Biol. 2001, 24, 727–732. [Google Scholar] [CrossRef]
- Lomas, D.A. New Therapeutic Targets for Alpha-1 Antitrypsin Deficiency. Chronic. Obs. Pulm. Dis. 2018, 5, 233–243. [Google Scholar] [CrossRef]
- Burrows, J.A.; Willis, L.K.; Perlmutter, D.H. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc. Natl. Acad. Sci. USA 2000, 97, 1796–1801. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H. Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: A preliminary study. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 34–37. [Google Scholar] [CrossRef]
- Mallya, M.; Phillips, R.L.; Saldanha, S.A.; Gooptu, B.; Brown, S.C.; Termine, D.J.; Shirvani, A.M.; Wu, Y.; Sifers, R.N.; Abagyan, R.; et al. Small molecules block the polymerization of Z alpha1-antitrypsin and increase the clearance of intracellular aggregates. J. Med. Chem. 2007, 50, 5357–5363. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Stein, P.E.; Huntington, J.A.; Sivasothy, P.; Lomas, D.A.; Carrell, R.W. How small peptides block and reverse serpin polymerisation. J. Mol. Biol. 2004, 342, 931–941. [Google Scholar] [CrossRef]
- Sidhar, S.K.; Lomas, D.A.; Carrell, R.W.; Foreman, R.C. Mutations which impede loop/sheet polymerization enhance the secretion of human alpha 1-antitrypsin deficiency variants. J. Biol. Chem. 1995, 270, 8393–8396. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Pei, X.Y.; Dafforn, T.R.; Lomas, D.A. Topography of a 2.0 A structure of alpha1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, A.; Perez, J.; Tan, L.; Dickens, J.A.; Motamedi-Shad, N.; Irving, J.A.; Haq, I.; Ekeowa, U.; Marciniak, S.J.; Miranda, E.; et al. A single-chain variable fragment intrabody prevents intracellular polymerization of Z alpha1-antitrypsin while allowing its antiproteinase activity. Faseb. J. 2015, 29, 2667–2678. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, L.P.; Long, O.S.; Cobanoglu, M.C.; Benson, J.A.; Luke, C.J.; Miedel, M.T.; Hale, P.; Perlmutter, D.H.; Bahar, I.; Silverman, G.A.; et al. A genome-wide RNAi screen identifies potential drug targets in a C. elegans model of alpha1-antitrypsin deficiency. Hum. Mol. Genet. 2014, 23, 5123–5132. [Google Scholar] [CrossRef] [Green Version]
- Long, O.S.; Benson, J.A.; Kwak, J.H.; Luke, C.J.; Gosai, S.J.; O’Reilly, L.P.; Wang, Y.; Li, J.; Vetica, A.C.; Miedel, M.T.; et al. A C. elegans model of human alpha1-antitrypsin deficiency links components of the RNAi pathway to misfolded protein turnover. Hum. Mol Genet. 2014, 23, 5109–5122. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pak, S.C.; O’Reilly, L.P.; Benson, J.A.; Wang, Y.; Hidvegi, T.; Hale, P.; Dippold, C.; Ewing, M.; Silverman, G.A.; et al. Fluphenazine reduces proteotoxicity in C. elegans and mammalian models of alpha-1-antitrypsin deficiency. PLoS ONE 2014, 9, e87260. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cobanoglu, M.C.; Li, J.; Hidvegi, T.; Hale, P.; Ewing, M.; Chu, A.S.; Gong, Z.; Muzumdar, R.; Pak, S.C.; et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded alpha1-antitrypsin Z. PLoS ONE 2019, 14, e0209748. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karatas, E.; Bouchecareilh, M. Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int. J. Mol. Sci. 2020, 21, 1493. https://doi.org/10.3390/ijms21041493
Karatas E, Bouchecareilh M. Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. International Journal of Molecular Sciences. 2020; 21(4):1493. https://doi.org/10.3390/ijms21041493
Chicago/Turabian StyleKaratas, Esra, and Marion Bouchecareilh. 2020. "Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways" International Journal of Molecular Sciences 21, no. 4: 1493. https://doi.org/10.3390/ijms21041493
APA StyleKaratas, E., & Bouchecareilh, M. (2020). Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. International Journal of Molecular Sciences, 21(4), 1493. https://doi.org/10.3390/ijms21041493