Opipramol Inhibits Lipolysis in Human Adipocytes without Altering Glucose Uptake and Differently from Antipsychotic and Antidepressant Drugs with Adverse Effects on Body Weight Control

 ,
,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Subjects and Preparation of Adipose Cells
2.3. Lipolysis Assays
2.4. Oxidation of Radiolabeled Tyramine and Benzylamine by Adipose Tissue Homogenates
2.5. Hydrogen Peroxide Production by Adipose Tissue Homogenates
2.6. Glucose Transport Assays
2.7. Data Presentation and Statistical analyses
3. Results and Discussion
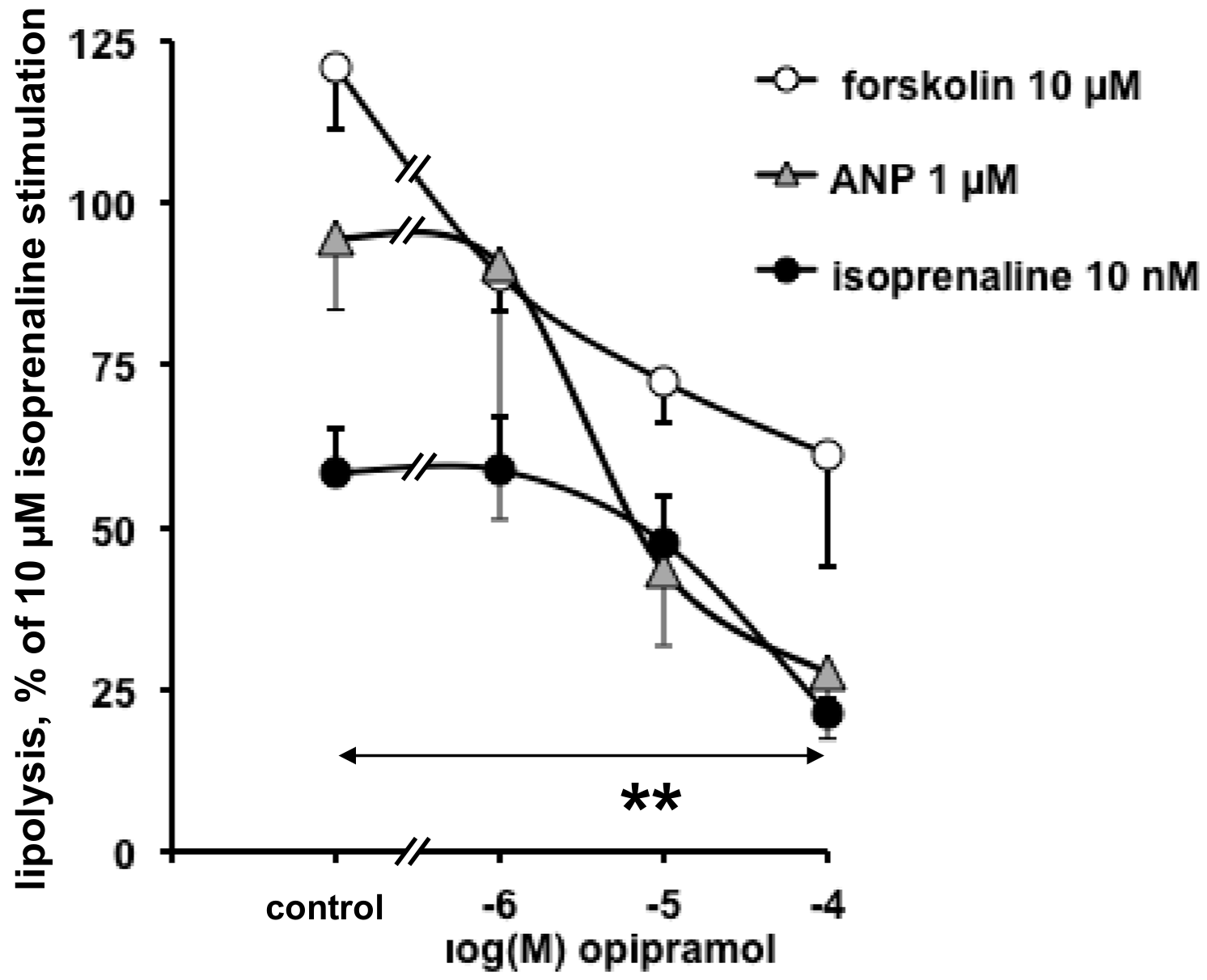
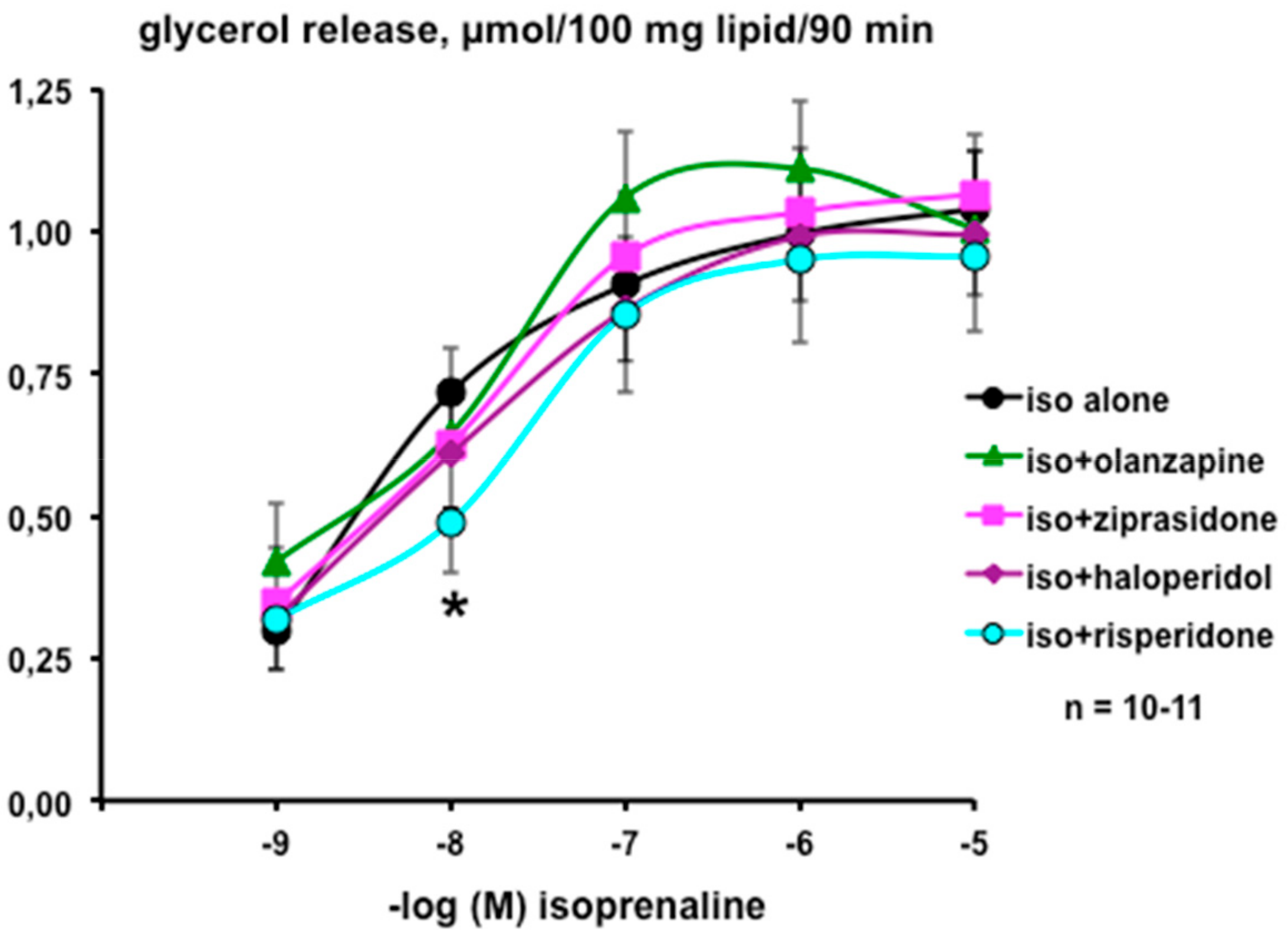
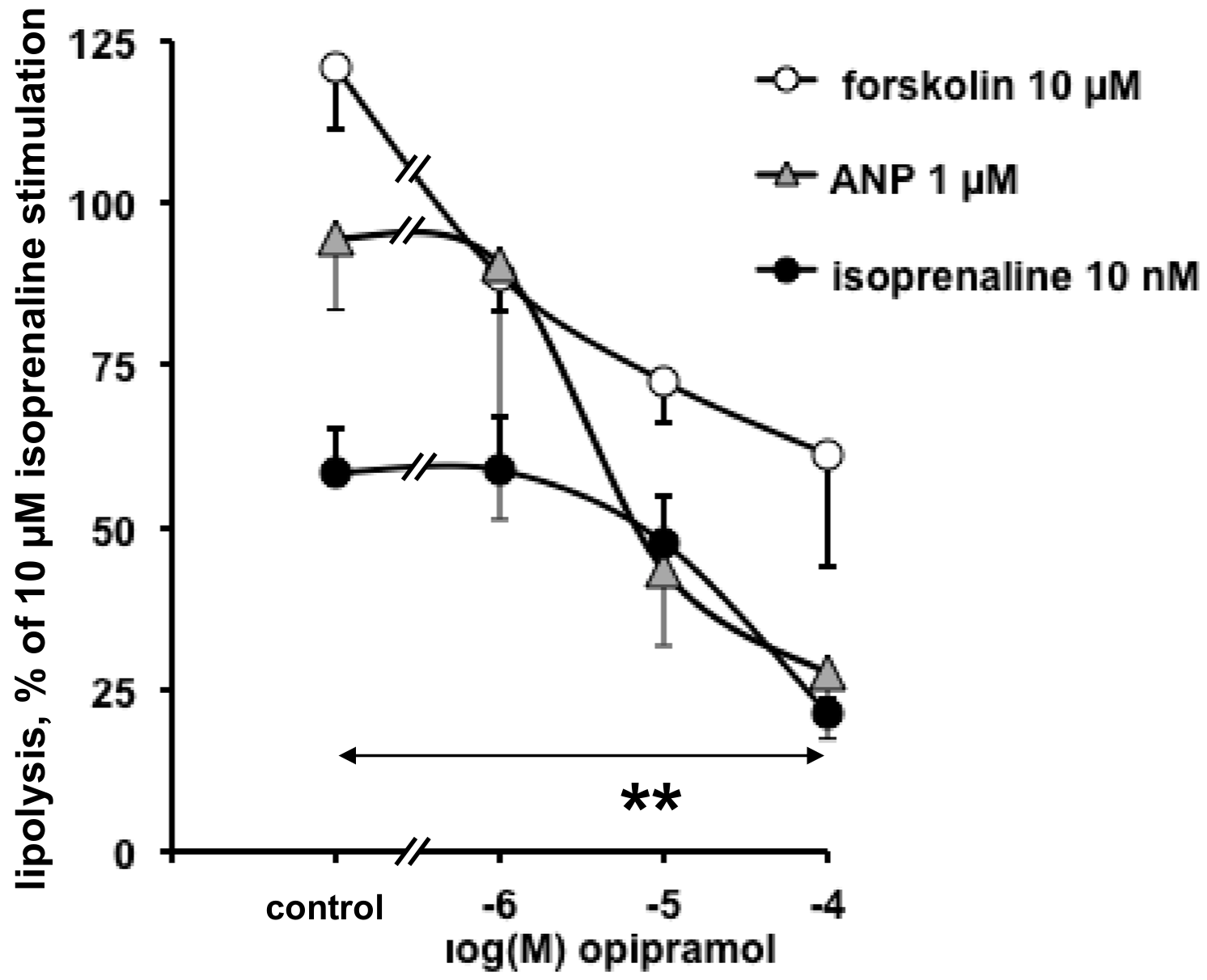
3.1. In Vitro Evaluation of the Direct Influence of Antipsychotics on the Lipolytic Responses of Human Adipose Cells
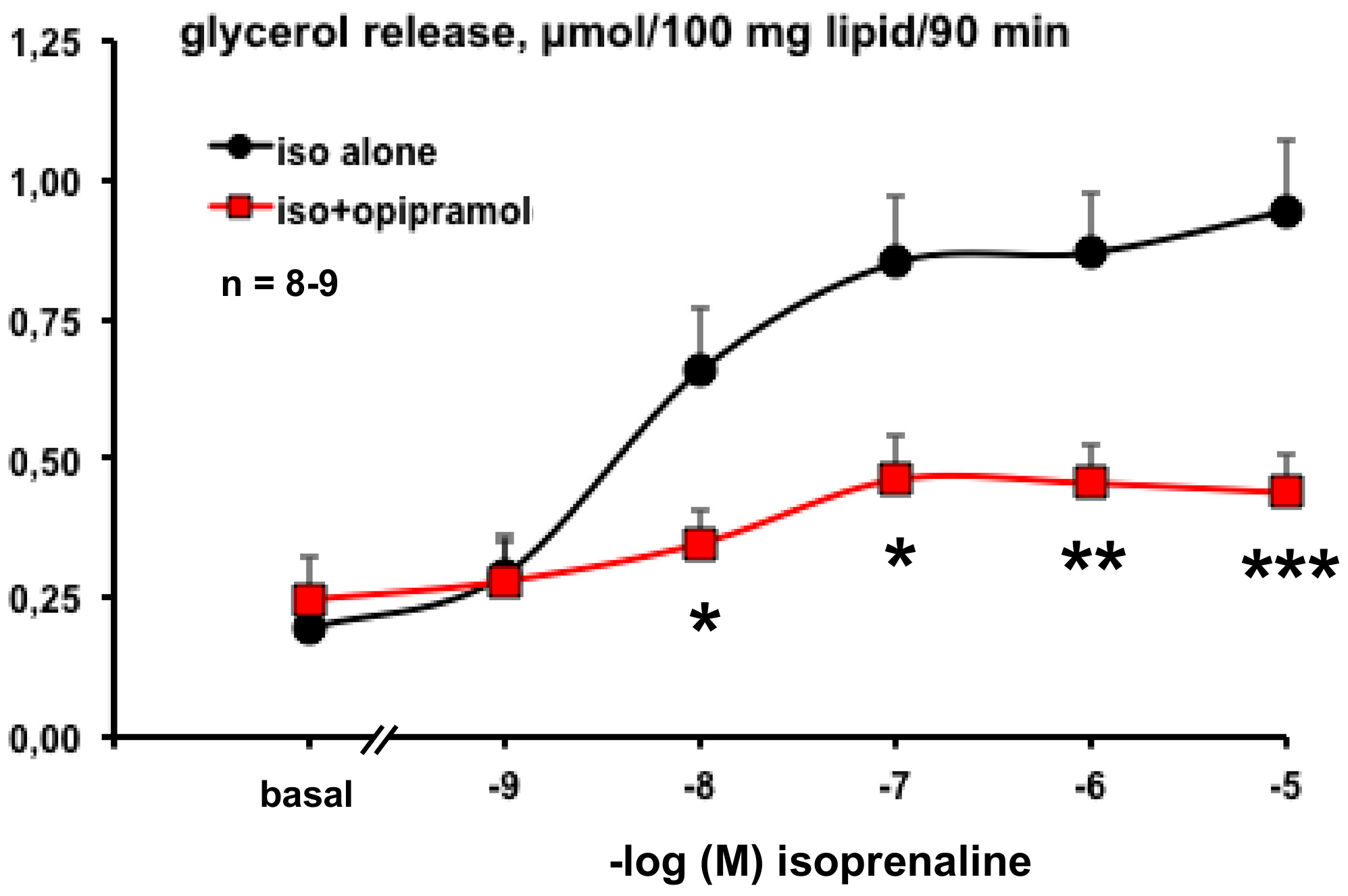
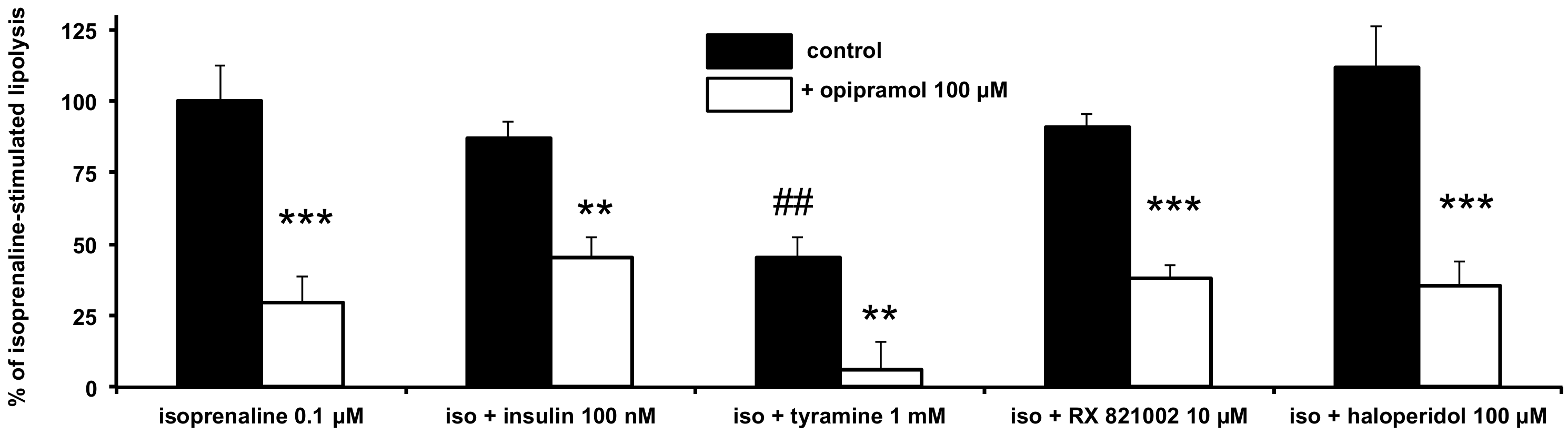
3.2. Influence of Opipramol on Basal and Stimulated Lipolytic Activity of Human Adipose Cells
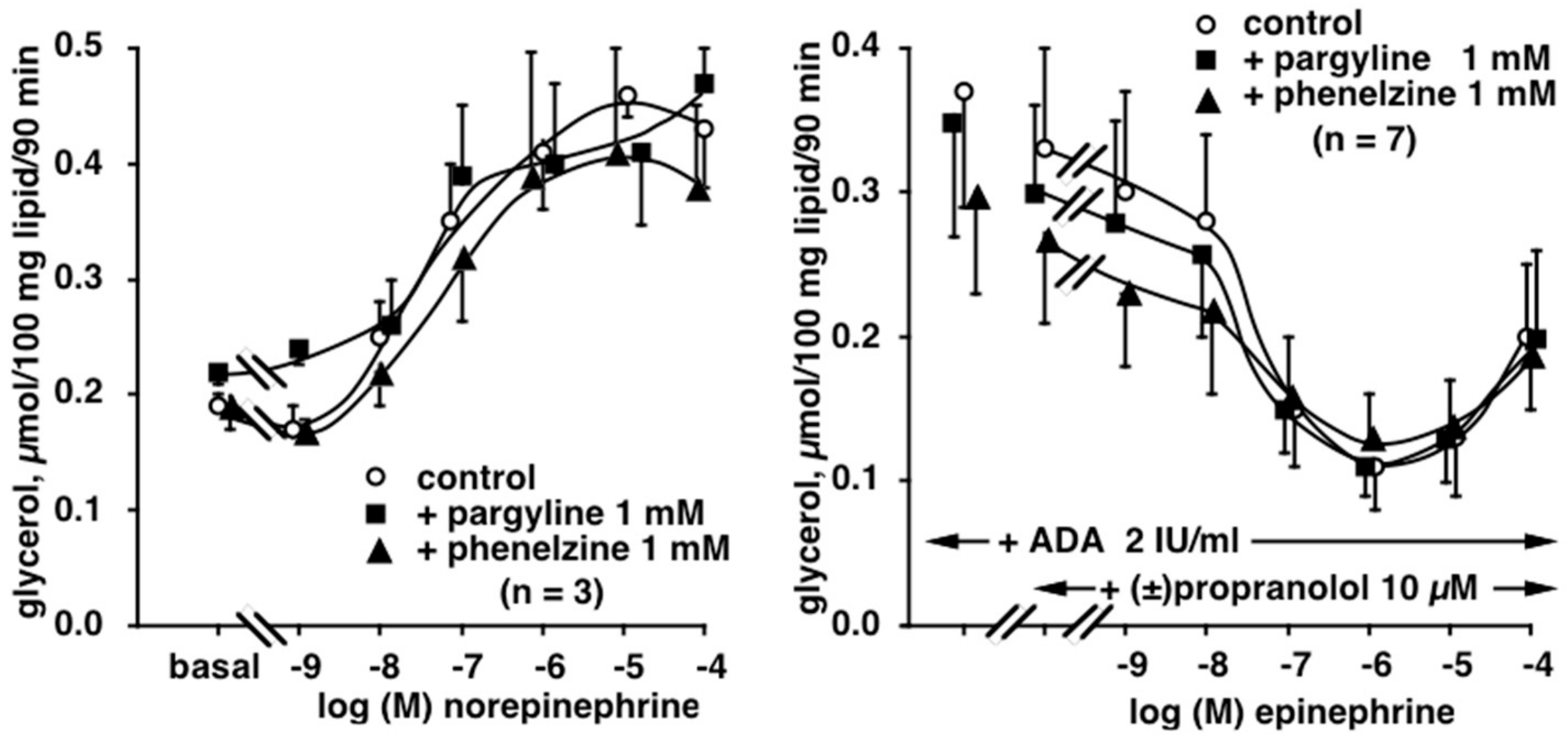
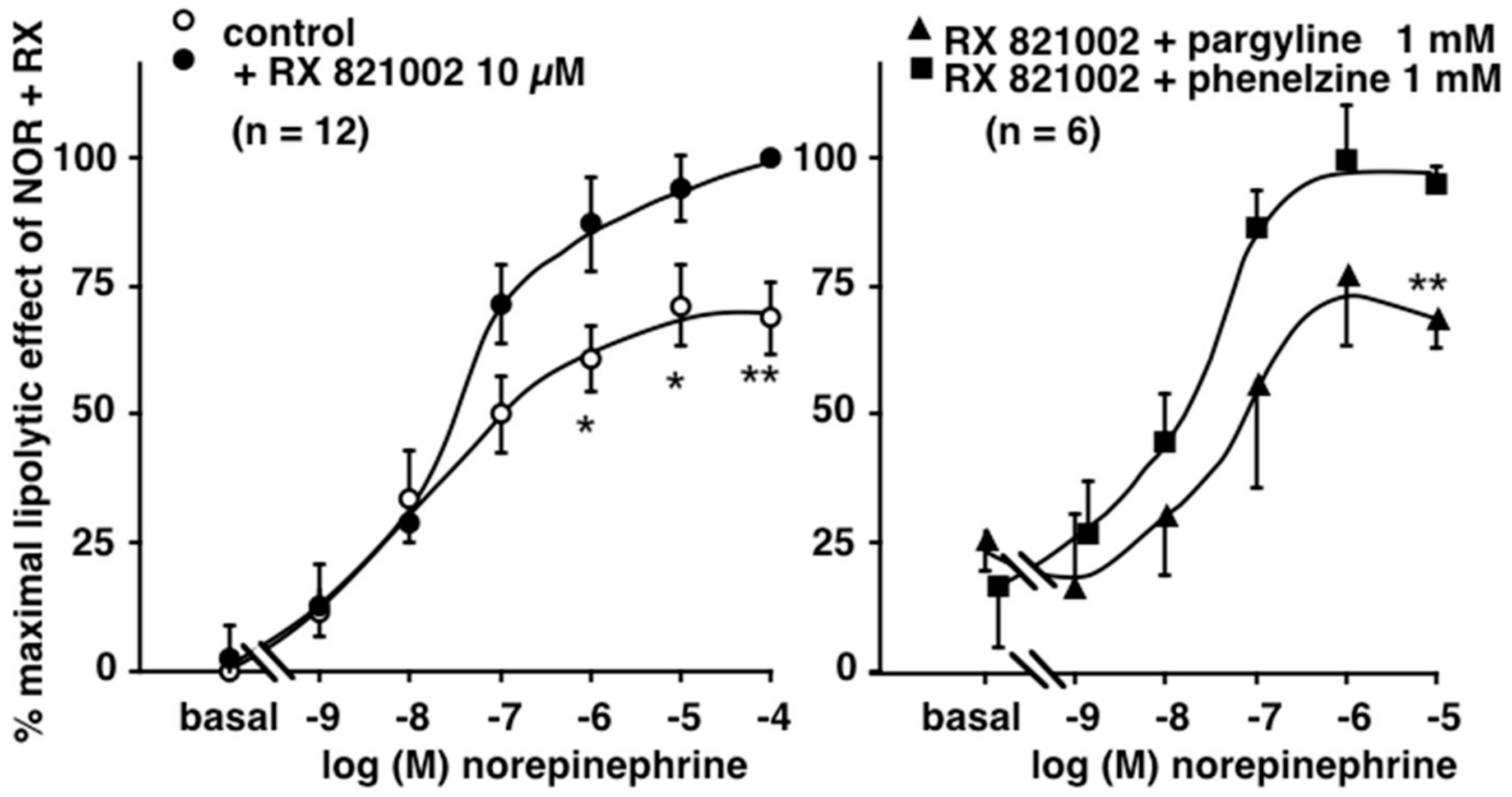
3.3. Influence of Phenelzine and Pargyline on Basal and Stimulated Lipolytic Activity of Human Adipose Cells
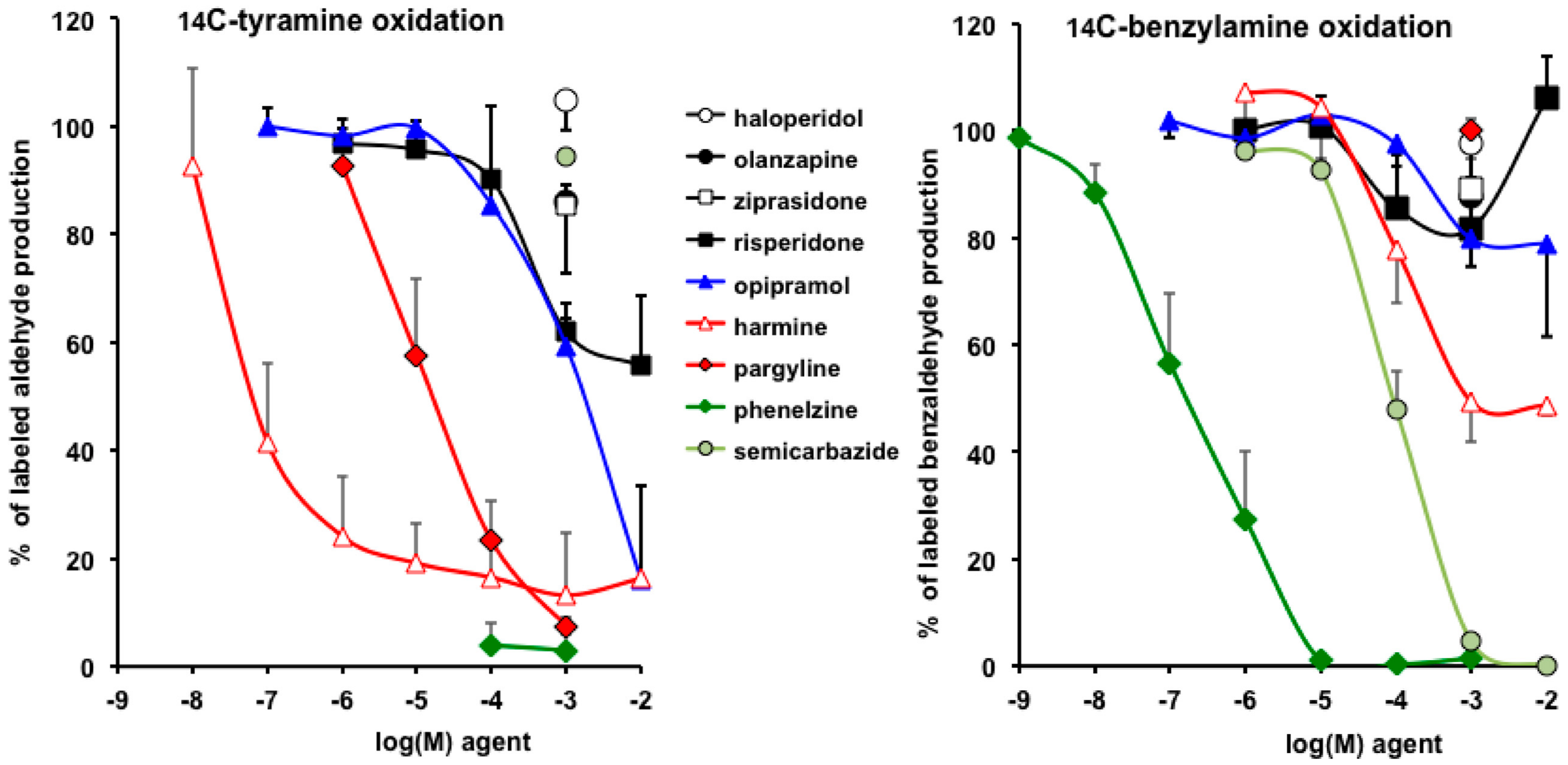
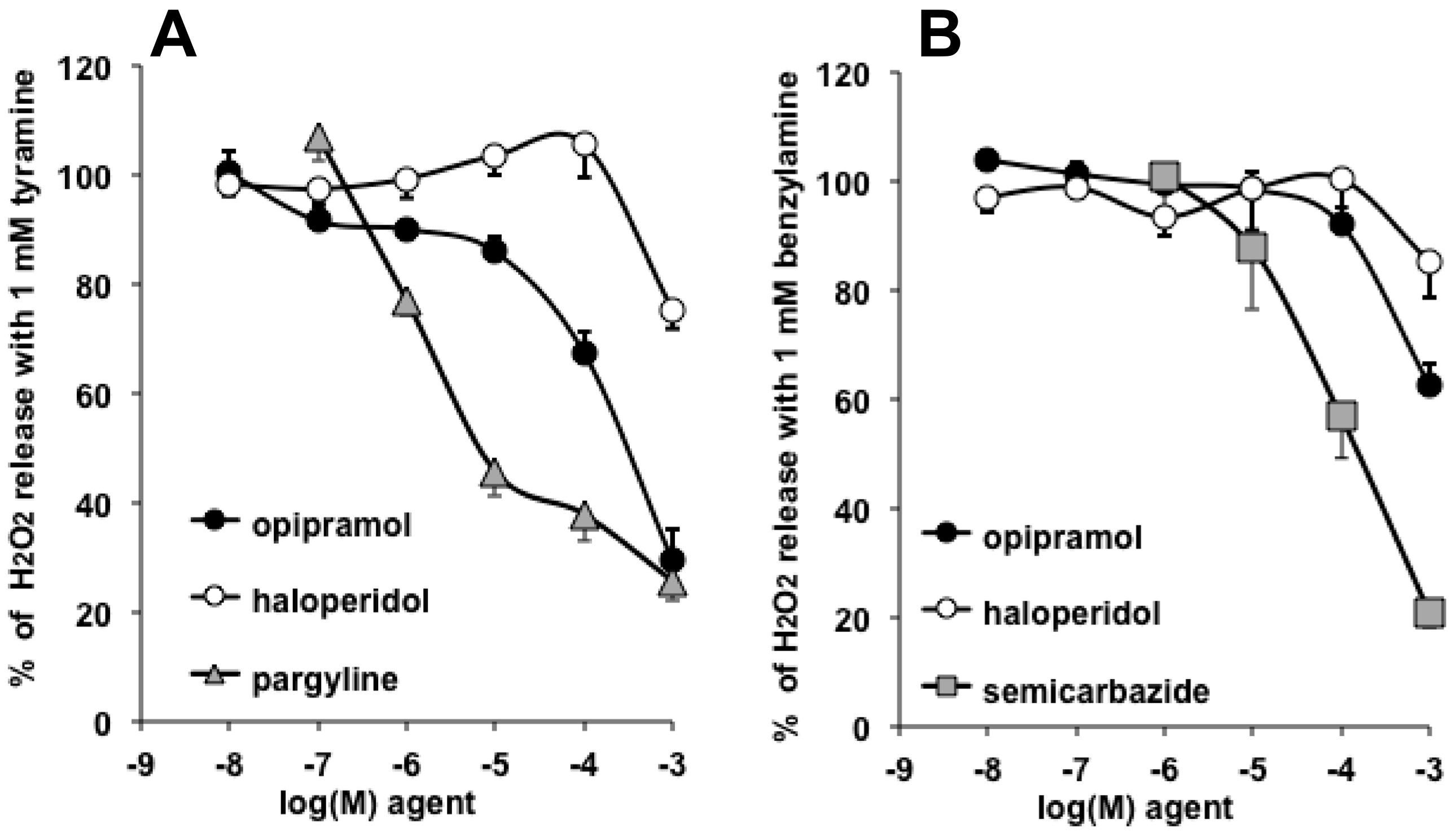
3.4. Interactions of Opipramol and Other Psychotropes with Adipose Tissue Amine Oxidase
3.5. Opipramol Interacted with Hydrogen Peroxide Generation by Amine Oxidases
3.6. Lack of Opipramol Effect on Basal or Insulin-Induced Glucose Uptake in Human Adipocytes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seo, R.J.; MacPherson, H.; Young, A.H. Atypical Antipsychotics and Other Therapeutic Options for Treatment of Resistant Major Depressive Disorder. Pharmaceuticals 2010, 3, 3522–3542. [Google Scholar] [CrossRef] [Green Version]
- Nimura, S.; Yamaguchi, T.; Ueda, K.; Kadokura, K.; Aiuchi, T.; Kato, R.; Obama, T.; Itabe, H. Olanzapine promotes the accumulation of lipid droplets and the expression of multiple perilipins in human adipocytes. Biochem. Biophys. Res. Comm. 2015, 467, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Yacoub Wasef, S.Z.; Buse, M.G. At therapeutic concentrations, olanzapine does not affect basal or insulin-stimulated glucose transport in 3T3-L1 adipocytes. Progr. Neuro-Psychopharmacol. Biol. Psychiatr. 2006, 30, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.; Haack, M.; Schuld, A.; Hinze-Selch, D.; Kühn, M.; Uhr, M.; Pollmächer, T. Body weight and leptin plasma levels during treatment with antipsychotic drugs. Am. J. Psychiatr. 1999, 156, 312–314. [Google Scholar] [PubMed]
- Yamauchi, T.; Tatsumi, K.; Makinodan, M.; Kimoto, S.; Toritsuka, M.; Okuda, H.; Kishimoto, T.; Wanaka, A. Olanzapine increases cell mitotic activity and oligodendrocyte-lineage cells in the hypothalamus. Neurochem. Int. 2010, 57, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Carpéné, C.; Grès, S.; Rascalou, S. The amine oxidase inhibitor phenelzine limits lipogenesis in adipocytes without inhibiting insulin action on glucose uptake. J. Neural Transm. 2013, 120, 997–1003. [Google Scholar] [CrossRef]
- Chiche, F.; Le Guillou, M.; Chetrite, G.; Lasnier, F.; Dugail, I.; Carpéné, C.; Moldes, M.; Fève, B. Antidepressant phenelzine alters differentiation of cultured human and mouse preadipocytes. Mol. Pharmacol. 2009, 75, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Carpéné, C.; Gomez-Zorita, S.; Chaplin, A.; Mercader, J. Metabolic Effects of Oral Phenelzine Treatment on High-Sucrose-Drinking Mice. Int. J. Mol. Sci. 2018, 19, 2904. [Google Scholar] [CrossRef] [Green Version]
- Mercader, J.; Sabater, A.; Le Gonidec, S.; Decaunes, P.; Chaplin, A.; Gomez-Zorita, S.; Milagro, F.; Carpéné, C. Oral Phenelzine Treatment Mitigates Metabolic Disturbances in Mice Fed a High-Fat Diet. J. Pharmacol. Exp. Ther. 2019, in press. [Google Scholar] [CrossRef]
- Minet-Ringuet, J.; Even, P.C.; Valet, P.; Carpéné, C.; Visentin, V.; Prevot, D.; Daviaud, D.; Quignard-Boulange, A.; Tome, D.; de Beaurepaire, R. Alterations of lipid metabolism and gene expression in rat adipocytes during chronic olanzapine treatment. Mol. Psychiatr. 2007, 12, 562–571. [Google Scholar] [CrossRef]
- Vestri, H.S.; Maianu, L.; Moellering, D.R.; Garvey, W.T. Atypical antipsychotic drugs directly impair insulin action in adipocytes: Effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology 2007, 32, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-H.; Chen, T.-M.; Yu, S.-T.; Chen, Y.-H. Olanzapine induces SREBP-1-related adipogenesis in 3T3-L1 cells. Pharmacol. Res. 2007, 56, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Dzitoyeva, S.; Chen, H.; Manev, H. 5-lipoxygenase-activating protein as a modulator of olanzapine-induced lipid accumulation in adipocyte. J. Lipids 2013, 2013, 864593. [Google Scholar] [CrossRef] [PubMed]
- Baba, L.I.; Kolcsar, M.; Kun, I.Z.; Ulakcsai, Z.; Bagamery, F.; Szoko, E.; Tabi, T.; Gall, Z. Effects of Cariprazine, Aripiprazole, and Olanzapine on Mouse Fibroblast Culture: Changes in Adiponectin Contents in Supernatants, Triglyceride Accumulation, and Peroxisome Proliferator-Activated Receptor-gamma Expression. Medicina 2019, 55, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minet-Ringuet, J.; Even, P.C.; Goubern, M.; Tome, D.; Beaurepaire, R.D. Long term treatment with olanzapine mixed with the food in male rats induces body fat deposition with no increase in body weight and no thermogenic alteration. Appetite 2006, 46, 254–262. [Google Scholar] [CrossRef]
- Richards, A.A.; Hickman, I.J.; Wang, A.Y.H.; Jones, A.L.; Newell, F.; Mowry, B.J.; Whitehead, J.P.; Prins, J.B.; Macdonald, G.A. Olanzapine treatment is associated with reduced high molecular weight adiponectin in serum: A potential mechanism for olanzapine-induced insulin resistance in patients with schizophrenia. J. Clin. Psychopharmacol. 2006, 26, 232–237. [Google Scholar] [CrossRef]
- Sertié, A.L.; Suzuki, A.M.; Sertié, R.A.L.; Andreotti, S.; Lima, F.B.; Passos-Bueno, M.R.; Gattaz, W.F. Effects of antipsychotics with different weight gain liabilities on human in vitro models of adipose tissue differentiation and metabolism. Progr. Neuro-Psychopharmacol. Biol. Psychiatr. 2011, 35, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Sárvári, A.K.; Veréb, Z.; Uray, I.P.; Fésüs, L.; Balajthy, Z. Atypical antipsychotics induce both proinflammatory and adipogenic gene expression in human adipocytes in vitro. Biochem. Biophys. Res. Comm. 2014, 450, 1383–1389. [Google Scholar] [CrossRef]
- Zhang, Q.; He, M.; Deng, C.; Wang, H.; Huang, X.-F. Effects of olanzapine on the elevation of macrophage infiltration and pro-inflammatory cytokine expression in female rats. J. Psychopharmacol. 2014, 28, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Courty, E.; Gobalakichenane, P.; Garcia, M.; Muscat, A.; Kazakian, C.; Ledent, T.; Moldes, M.; Blondeau, B.; Mitanchez, D.; Buyse, M.; et al. Antenatal antipsychotic exposure induces multigenerational and gender-specific programming of adiposity and glucose tolerance in adult mouse offspring. Diabetes Metab. 2018, 44, 281–291. [Google Scholar] [CrossRef]
- Aravagiri, M.; Teper, Y.; Marder, S.R. Pharmacokinetics and tissue distribution of olanzapine in rats. Biopharm. Drug Dispos. 1999, 20, 369–377. [Google Scholar] [CrossRef]
- Song, M.S.; Matveychuk, D.; MacKenzie, E.M.; Duchcherer, M.; Mousseau, D.D.; Baker, G.B. An update on amine oxidase inhibitors: Multifaceted drugs. Prog. Neuropsychopharmacol. Biol. Psychiatr. 2013, 44, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.; Siebert, B.; Holoubek, G.; Gentsch, C. Neuropharmacology of the anxiolytic drug opipramol, a sigma site ligand. Pharmacopsychiatry 2004, 37, S189–S197. [Google Scholar] [CrossRef] [PubMed]
- Pizzinat, N.; Marti, L.; Remaury, A.; Leger, F.; Langin, D.; Lafontan, M.; Carpéné, C.; Parini, A. High expression of monoamine oxidases in human white adipose tissue: Evidence for their involvement in noradrenaline clearance. Biochem. Pharmacol. 1999, 58, 1735–1742. [Google Scholar] [CrossRef]
- Carpéné, C.; Mercader, J.; Le Gonidec, S.; Schaak, S.; Mialet-Perez, J.; Zakaroff-Girard, A.; Galitzky, J. Body fat reduction without cardiovascular changes in mice after oral treatment by the MAO inhibitor phenelzine. Br. J. Pharmacol. 2018, 175, 2428–2440. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.T.; Koncsos, G.; Varga, Z.V.; Baranyai, T.; Tuza, S.; Kassai, F.; Ernyey, A.J.; Gyertyan, I.; Kiraly, K.; Olah, A.; et al. Selegiline reduces adiposity induced by high-fat, high-sucrose diet in male rats. Br. J. Pharmacol. 2018, 175, 3713–3726. [Google Scholar] [CrossRef] [Green Version]
- Morin, N.; Lizcano, J.M.; Fontana, E.; Marti, L.; Smih, F.; Rouet, P.; Prévot, D.; Zorzano, A.; Unzeta, M.; Carpéné, C. Semicarbazide-sensitive amine oxidase substrates stimulate glucose transport and inhibit lipolysis in human adipocytes. J. Pharmacol. Exp. Ther. 2001, 297, 563–572. [Google Scholar]
- Carpéné, C.; Boulet, N.; Chaplin, A.; Mercader, J. Past, Present and Future Anti-Obesity Effects of Flavin-Containing and/or Copper-Containing Amine Oxidase Inhibitors. Medicines 2019, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Zorita, S.; Treguer, K.; Mercader, J.; Carpéné, C. Resveratrol directly affects in vitro lipolysis and glucose transport in human fat cells. J. Physiol. Biochem. 2013, 69, 585–593. [Google Scholar] [CrossRef]
- Visentin, V.; Prevot, D.; Marti, L.; Carpéné, C. Inhibition of rat fat cell lipolysis by monoamine oxidase and semicarbazide-sensitive amine oxidase substrates. Eur. J. Pharmacol. 2003, 466, 235–243. [Google Scholar] [CrossRef]
- Carpéné, C.; Galitzky, J.; Belles, C.; Zakaroff-Girard, A. Mechanisms of the antilipolytic response of human adipocytes to tyramine, a trace amine present in food. J. Physiol. Biochem. 2018, 74, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Carpéné, C.; Hasnaoui, M.; Balogh, B.; Matyus, P.; Fernandez-Quintela, A.; Rodriguez, V.; Mercader, J.; Portillo, M.P. Dietary phenolic compounds interfere with the fate of hydrogen peroxide in human adipose tissue but do not directly inhibit primary amine oxidase activity. Oxid. Med. Cell. Longev. 2016, 2016, 2427618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Les, F.; Deleruyelle, S.; Cassagnes, L.E.; Boutin, J.A.; Balogh, B.; Arbones-Mainar, J.M.; Biron, S.; Marceau, P.; Richard, D.; Nepveu, F.; et al. Piceatannol and resveratrol share inhibitory effects on hydrogen peroxide release, monoamine oxidase and lipogenic activities in adipose tissue, but differ in their antilipolytic properties. Chem. Biol. Interact. 2016, 258, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Carpéné, C.; Mauriège, P.; Boulet, N.; Biron, S.; Grolleau, J.L.; Garcia-Barrado, M.J.; Iglesias-Osma, M.C. Methylamine Activates Glucose Uptake in Human Adipocytes Without Overpassing Action of Insulin or Stimulating its Secretion in Pancreatic Islets. Medicines 2019, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Visentin, V.; Morin, N.; Fontana, E.; Prevot, D.; Boucher, J.; Castan, I.; Valet, P.; Grujic, D.; Carpéné, C. Dual action of octopamine on glucose transport into adipocytes: Inhibition via beta3-adrenoceptor activation and stimulation via oxidation by amine oxidases. J. Pharmacol. Exp. Ther. 2001, 299, 96–104. [Google Scholar] [PubMed]
- Lafontan, M.; Moro, C.; Berlan, M.; Crampes, F.; Sengenes, C.; Galitzky, J. Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol. Metabol. 2008, 19, 130–137. [Google Scholar] [CrossRef]
- Sarsenbayeva, A.; Marques-Santos, C.M.; Thombare, K.; Di Nunzio, G.; Almby, K.E.; Lundqvist, M.; Eriksson, J.W.; Pereira, M.J. Effects of second-generation antipsychotics on human subcutaneous adipose tissue metabolism. Psychoneuroendocrinology 2019, 110, 104445. [Google Scholar] [CrossRef]
- Rozanski, V.; Laux, G.; Schwarz, J. The Dopamine Receptor Antagonism of Opipramol: Relevance to Parkinsonism? Clin. Neuropharmacol. 2019, 42, 77–79. [Google Scholar] [CrossRef]
- Betschart, H.R.; Jondorf, W.R.; Bickel, M.H. Differences in adipose tissue distribution of basic lipophilic drugs between intraperitoneal and other routes of administration. Xenobiotica 1988, 18, 113–121. [Google Scholar] [CrossRef]
- Carpéné, C.; Garcia-Vicente, S.; Serrano, M.; Marti, L.; Belles, C.; Royo, M.; Galitzky, J.; Zorzano, A.; Testar, X. Insulin-mimetic compound hexaquis (benzylammonium) decavanadate is antilipolytic in human fat cells. World J. Diabetes 2017, 8, 143–153. [Google Scholar] [CrossRef]
- Baker, G.B.; Coutts, R.T.; Greenshaw, A.J. Neurochemical and metabolic aspects of antidepressants: An overview. J. Psychiatry Neurosci. 2000, 25, 481–496. [Google Scholar] [PubMed]
- Carpéné, C.; Bousquet-Melou, A.; Galitzky, J.; Berlan, M.; Lafontan, M. Lipolytic effects of beta 1-, beta 2-, and beta 3-adrenergic agonists in white adipose tissue of mammals. Ann. N. Y. Acad. Sci. 1998, 839, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.H.; Wertz, D.L.; Klinman, J.P. Implication for functions of the ectopic adipocyte copper amine oxidase (AOC3) from purified enzyme and cell-based kinetic studies. PLoS ONE 2012, 7, e29270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Sablin, S.O.; Ramsay, R.R. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.; Rico, D.; Khiari, Z.; Henehan, G.; O’Sullivan, J.; Tipton, K. From caffeine to fish waste: Amine compounds present in food and drugs and their interactions with primary amine oxidase. J. Neural Transm. 2011, 118, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, H.; Sho, S.; Kamijo, K. Difference in actions of harmine on the oxidations of serotonin and tyramine by beef brain mitochondrial MAO. Jpn. J. Pharmacol. 1972, 22, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Miralles, A.; Esteban, S.; Sastre-Coll, A.; Moranta, D.; Asensio, V.J.; Garcia-Sevilla, J.A. High-affinity binding of ß-carbolines to imidazoline I2B receptors and MAO-A in rat tissues: Norharman blocks the effect of morphine withdrawal on DOPA/noradrenaline synthesis in the brain. Eur. J. Pharmacol. 2005, 518, 234–242. [Google Scholar] [CrossRef]
- Holt, A.; Wieland, B.; Baker, G.B. Allosteric modulation of semicarbazide-sensitive amine oxidase activities in vitro by imidazoline receptor ligands. Br. J. Pharmacol. 2004, 143, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, A.; Smith, D.J.; Cendron, L.; Zanotti, G.; Rigo, A.; Di Paolo, M.L. Multiple binding sites for substrates and modulators of semicarbazide-sensitive amine oxidases: Kinetic consequences. Mol. Pharmacol. 2008, 73, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Shertzer, H.G.; Kendig, E.L.; Nasrallah, H.A.; Johansson, E.; Genter, M.B. Protection from olanzapine-induced metabolic toxicity in mice by acetaminophen and tetrahydroindenoindole. Int. J. Obes. 2010, 34, 970–979. [Google Scholar] [CrossRef] [Green Version]
- Bush, N.D.; Townsend, L.K.; Wright, D.C. AICAR Prevents Acute Olanzapine-Induced Disturbances in Glucose Homeostasis. J. Pharmacol. Exp. Ther. 2018, 365, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engl, J.; Laimer, M.; Niederwanger, A.; Kranebitter, M.; Starzinger, M.; Pedrini, M.T.; Fleischhacker, W.W.; Patsch, J.R.; Ebenbichler, C.F. Olanzapine impairs glycogen synthesis and insulin signaling in L6 skeletal muscle cells. Mol. Psychiatry 2005, 10, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpéné, C.; Balogh, B.; Mercader, J.; Brea, J.; Gomez-Ruiz, A.; Dunkel, P.; Matyus, P.; Loza, M.I. Direct inhibitory effect of phenelzine on human primary amine oxidase, an enzyme formerly known as SSAO/VAP-1. In Proceedings of the 10th Congress of the European Association for Clinical Phamacology and Therapeutics (EACPT), Budapest, Hungary, 26–29 June 2011; Vas, A., Ed.; Editografica: Bologna, Italy, 2011; pp. 77–83. [Google Scholar]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, G.; Matveychuk, D.; MacKenzie, E.M.; Holt, A.; Wang, Y.; Kar, S. Attenuation of the effects of oxidative stress by the MAO-inhibiting antidepressant and carbonyl scavenger phenelzine. Chem. Biol. Interact. 2019, 304, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Wampers, M.; Hanssens, L.; van Winkel, R.; Heald, A.; Collette, J.; Peuskens, J.; Reginster, J.Y.; Scheen, A.; De Hert, M. Differential effects of olanzapine and risperidone on plasma adiponectin levels over time: Results from a 3-month prospective open-label study. Eur. Neuropsychopharmacol. 2012, 22, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Peng, Y.; Zhang, C.; Li, Z.; Su, Y.; Qi, Y.; Xing, M.; Li, J.; Kim, G.E.; Su, K.N.; et al. Macrophage migration inhibitory factor mediates metabolic dysfunction induced by atypical antipsychotic therapy. J. Clin. Invest. 2018, 128, 4997–5007. [Google Scholar] [CrossRef]
- Langin, D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol. Res. 2006, 53, 482–491. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hexose Uptake into Fat Cells (Fold Increase of Basal 2-DG Uptake) | ||||
|---|---|---|---|---|
| Experiment 1 | Experiment 2 | |||
| Control | +Opipramol | Control | +Phenelzine | |
| basal | 1.00 ± 0.16 | 1.12 ± 0.14 | 1.00 ± 0.15 | 1.09 ± 0.15 |
| insulin 10 nM | 2.05 ± 0.38 * | 2.12 ± 0.55 * | 3.18 ± 0.42 ** | ND |
| insulin 100 nM | 2.66 ± 0.29 ** | 2.41 ± 0.47 ** | 3.91 ± 0.55 ** | 4.09 ± 0.63 ** |
| benzylamine 1 mM | ND | ND | 1.60 ± 0.14 * | 1.03 ± 0.20 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpéné, C.; Les, F.; Mercader, J.; Gomez-Zorita, S.; Grolleau, J.-L.; Boulet, N.; Fontaine, J.; Iglesias-Osma, M.C.; Garcia-Barrado, M.J. Opipramol Inhibits Lipolysis in Human Adipocytes without Altering Glucose Uptake and Differently from Antipsychotic and Antidepressant Drugs with Adverse Effects on Body Weight Control. Pharmaceuticals 2020, 13, 41. https://doi.org/10.3390/ph13030041
Carpéné C, Les F, Mercader J, Gomez-Zorita S, Grolleau J-L, Boulet N, Fontaine J, Iglesias-Osma MC, Garcia-Barrado MJ. Opipramol Inhibits Lipolysis in Human Adipocytes without Altering Glucose Uptake and Differently from Antipsychotic and Antidepressant Drugs with Adverse Effects on Body Weight Control. Pharmaceuticals. 2020; 13(3):41. https://doi.org/10.3390/ph13030041
Chicago/Turabian StyleCarpéné, Christian, Francisco Les, Josep Mercader, Saioa Gomez-Zorita, Jean-Louis Grolleau, Nathalie Boulet, Jessica Fontaine, Mari Carmen Iglesias-Osma, and Maria José Garcia-Barrado. 2020. "Opipramol Inhibits Lipolysis in Human Adipocytes without Altering Glucose Uptake and Differently from Antipsychotic and Antidepressant Drugs with Adverse Effects on Body Weight Control" Pharmaceuticals 13, no. 3: 41. https://doi.org/10.3390/ph13030041
APA StyleCarpéné, C., Les, F., Mercader, J., Gomez-Zorita, S., Grolleau, J.-L., Boulet, N., Fontaine, J., Iglesias-Osma, M. C., & Garcia-Barrado, M. J. (2020). Opipramol Inhibits Lipolysis in Human Adipocytes without Altering Glucose Uptake and Differently from Antipsychotic and Antidepressant Drugs with Adverse Effects on Body Weight Control. Pharmaceuticals, 13(3), 41. https://doi.org/10.3390/ph13030041