Reactive Oxygen Species in Venous Thrombosis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
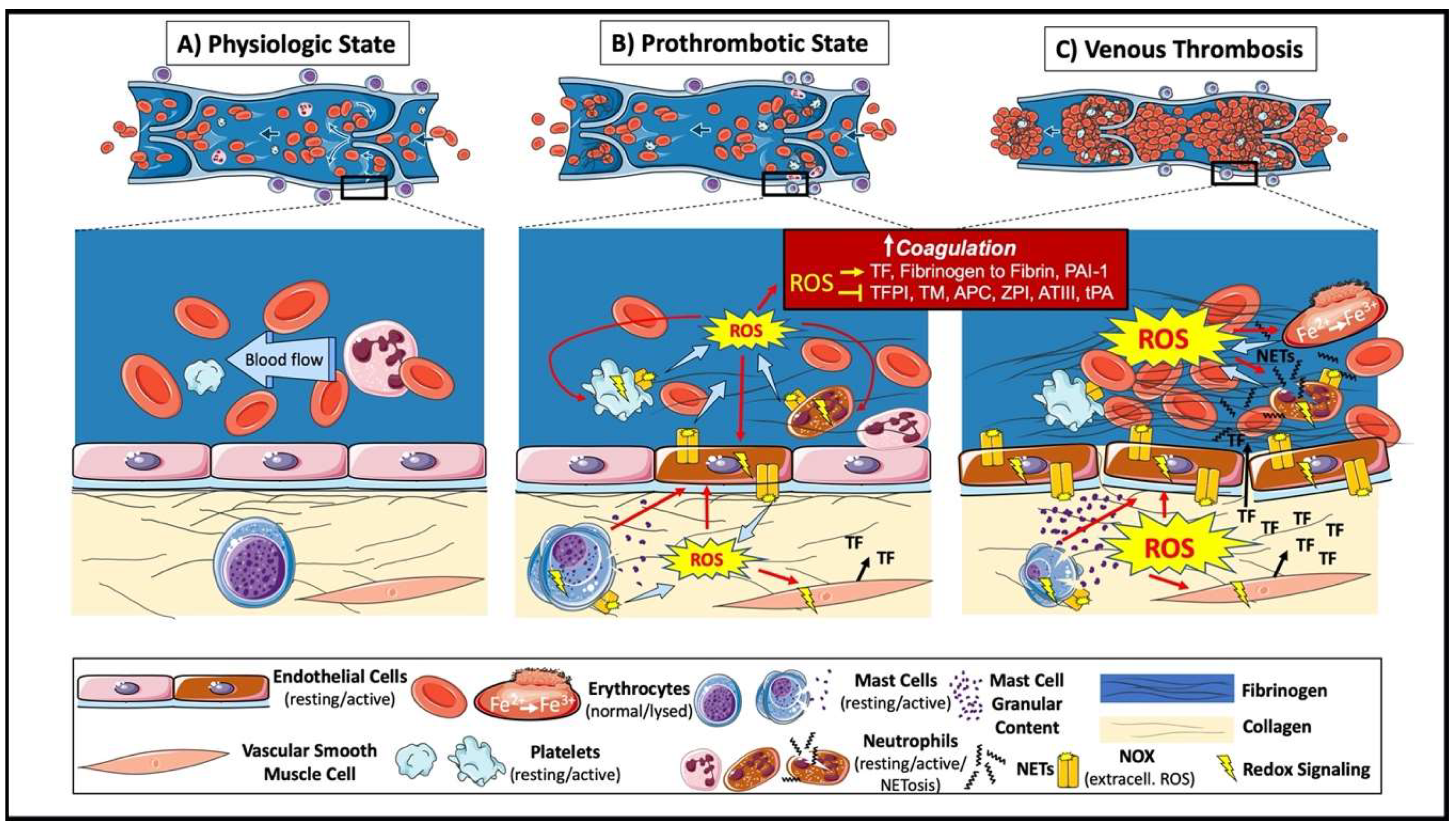
2. Venous Thrombus Formation
3. ROS and Coagulation
3.1. ROS in Signaling Pathways Modulating Procoagulant Responses
3.2. ROS Oxidative Modification of Proteins That Regulate Coagulation
4. ROS and Platelets
5. ROS and Extracellular Traps
6. ROS and Erythrocytes
7. ROS and Mast Cells
8. ROS and the Complement System
9. ROS and Calcium Homeostasis
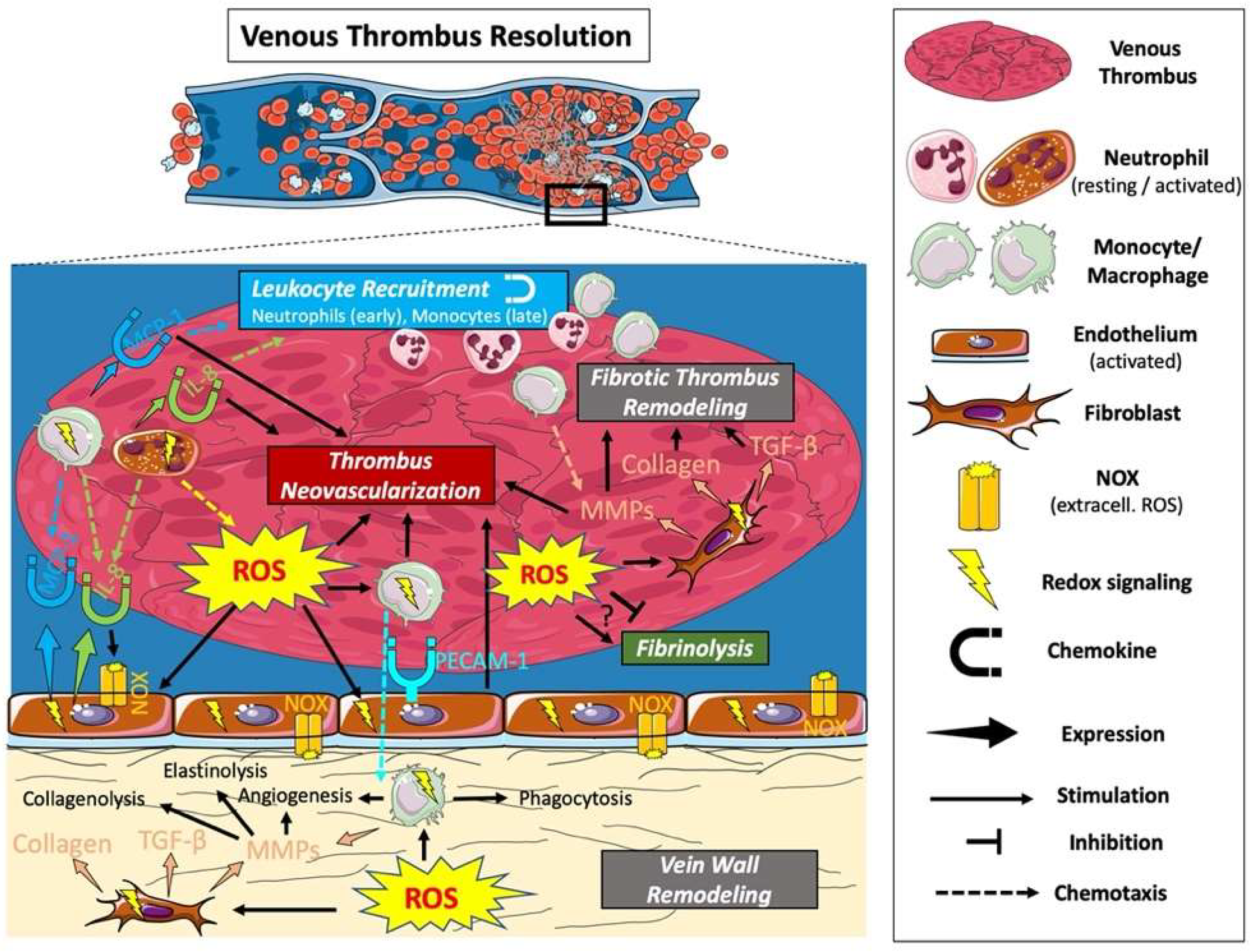
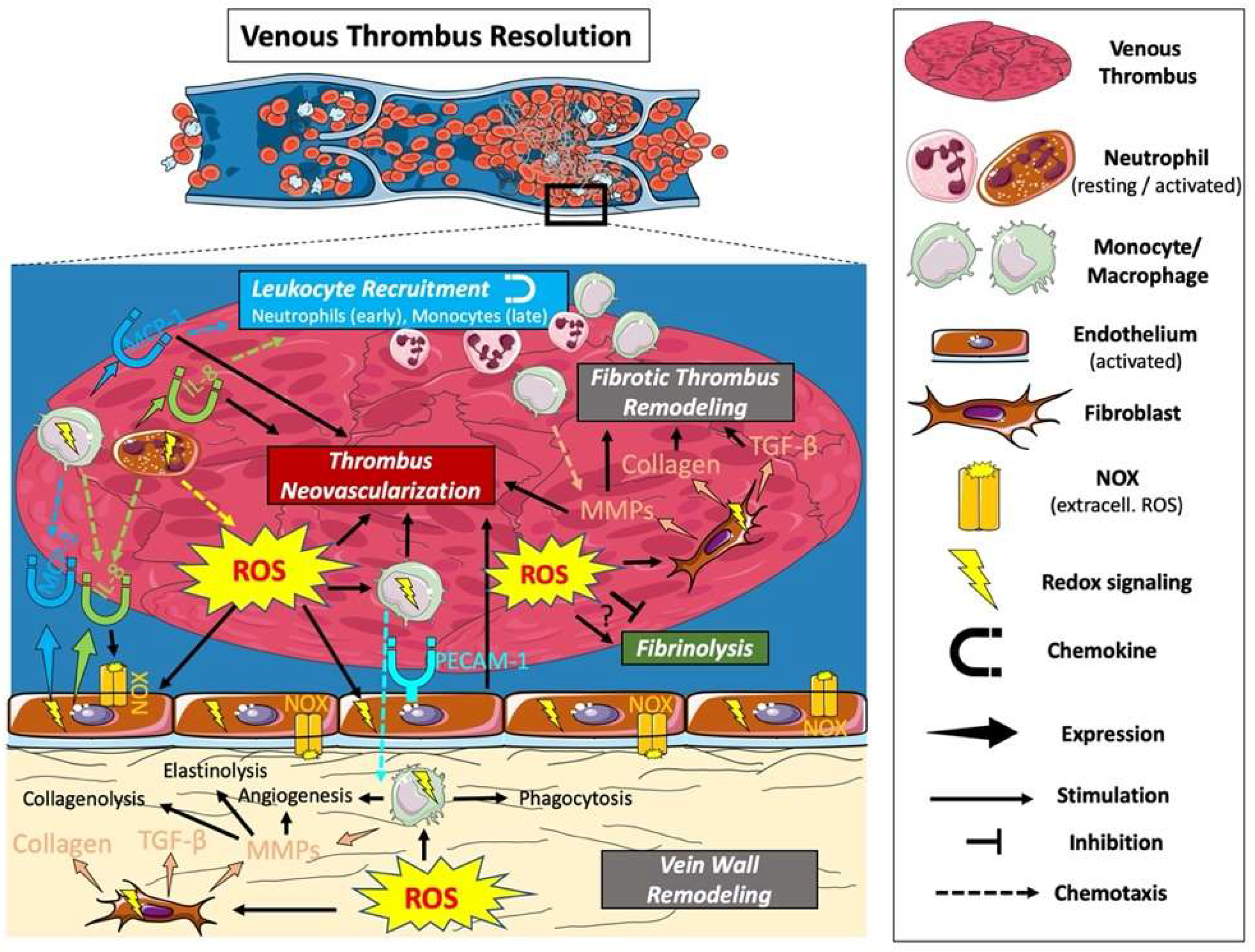
10. Venous Thrombus Resolution
11. ROS and Fibrinolysis
12. ROS and Leukocyte Recruitment
13. ROS and Thrombus Neovascularization
14. ROS and Fibrotic Thrombus Remodeling
15. Diseases with Increased Risk of DVT and Involving ROS
16. Dietary and Pharmacological Antioxidants
16.1. Dietary
16.2. Pharmaceutical
17. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | Activator protein 1 |
| APC | Activated protein C |
| APS | Antiphospholipid Syndrome |
| ATIII | Antithrombin III |
| AQP | Aquaporin channel |
| CAM | Cell adhesion molecule |
| CCR2 | C-C chemokine receptor type 2 |
| CGD | X-linked chronic granulomatous disease |
| COX | Cyclooxygenase |
| CVD | Cardiovascular disease |
| CAM | Cell adhesion molecule |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| DVT | Deep vein thrombosis |
| ECM | Extracellular matrix |
| EVAL | Ethylene-vinyl-alcohol |
| fMLP | N-formyl-met-leu-phe |
| FX | Factor X |
| FXI | Factor XI |
| GPVI | Glycoprotein VI |
| GPX | Glutathione peroxidase |
| Hb | Hemoglobin |
| HCQ | Hydroxychloroquine |
| HIF | Hypoxia-inducible factor |
| HO | Heme oxygenase |
| HUVEC | Human umbilical vein endothelial cell |
| IgE | Immunoglobulin E |
| IL-8 | Interleukin-8 |
| IVC | Inferior vena cava |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MC | Mast cell |
| MCP-1 | Monocyte chemotactic protein 1 |
| MMP | Matrix metalloproteinase |
| MPO | Myeloperoxidase |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MyD88 | Myeloid differentiation primary response 88 |
| NAC | N-acetylcysteine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| NO | Nitric oxide |
| NOX | Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NE | Neutrophil elastase |
| NET | Neutrophil extracellular trap |
| Orai1 | Calcium release-activated calcium channel protein 1 |
| Ox-LDL | Oxidized low-density lipoprotein |
| PA | Pulmonary artery |
| PAD4 | Protein-arginine deiminase type-4 |
| PAI | Plasminogen activator inhibitor |
| PE | Pulmonary embolism |
| PECAM-1 | Platelet endothelial cell adhesion molecule 1 |
| PMA | Phorbol myristate acetate |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| IVC | Inferior vena cava |
| ROS | Reactive oxygen species |
| SOCE | Store-operated calcium entry |
| STIM1 | Stromal interaction molecule 1 |
| TF | Tissue factor |
| TFPI | Tissue factor pathway inhibitor |
| TGF-β | Transforming growth factor beta |
| TIMP | Tissue inhibitor of metalloproteinases |
| TLR | Toll-like receptor |
| TM | Thrombomodulin |
| TNF-α | Tumor necrosis factor alpha |
| tPA | Tissue-type plasminogen activator |
| TRPC6 | Transient receptor potential channel 6 |
| TRX | Thioredoxin |
| uPA | Urokinase-type plasminogen activator |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cells |
| VTE | Venous thromboembolism |
| ZPI | Protein Z-dependent protease inhibitor |
References
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A.; Konstantinides, S.V.; McCumber, M.; et al. Thrombosis: A major contributor to the global disease burden. J. Thromb. Haemost. 2014, 12, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saarinen, J.; Kallio, T.; Lehto, M.; Hiltunen, S.; Sisto, T. The occurrence of the post-thrombotic changes after an acute deep venous thrombosis. A prospective two-year follow-up study. J. Cardiovasc. Surg. 2000, 41, 441–446. [Google Scholar]
- Patterson, B.O.; Hinchliffe, R.; Loftus, I.M.; Thompson, M.M.; Holt, P.J.E. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 669–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
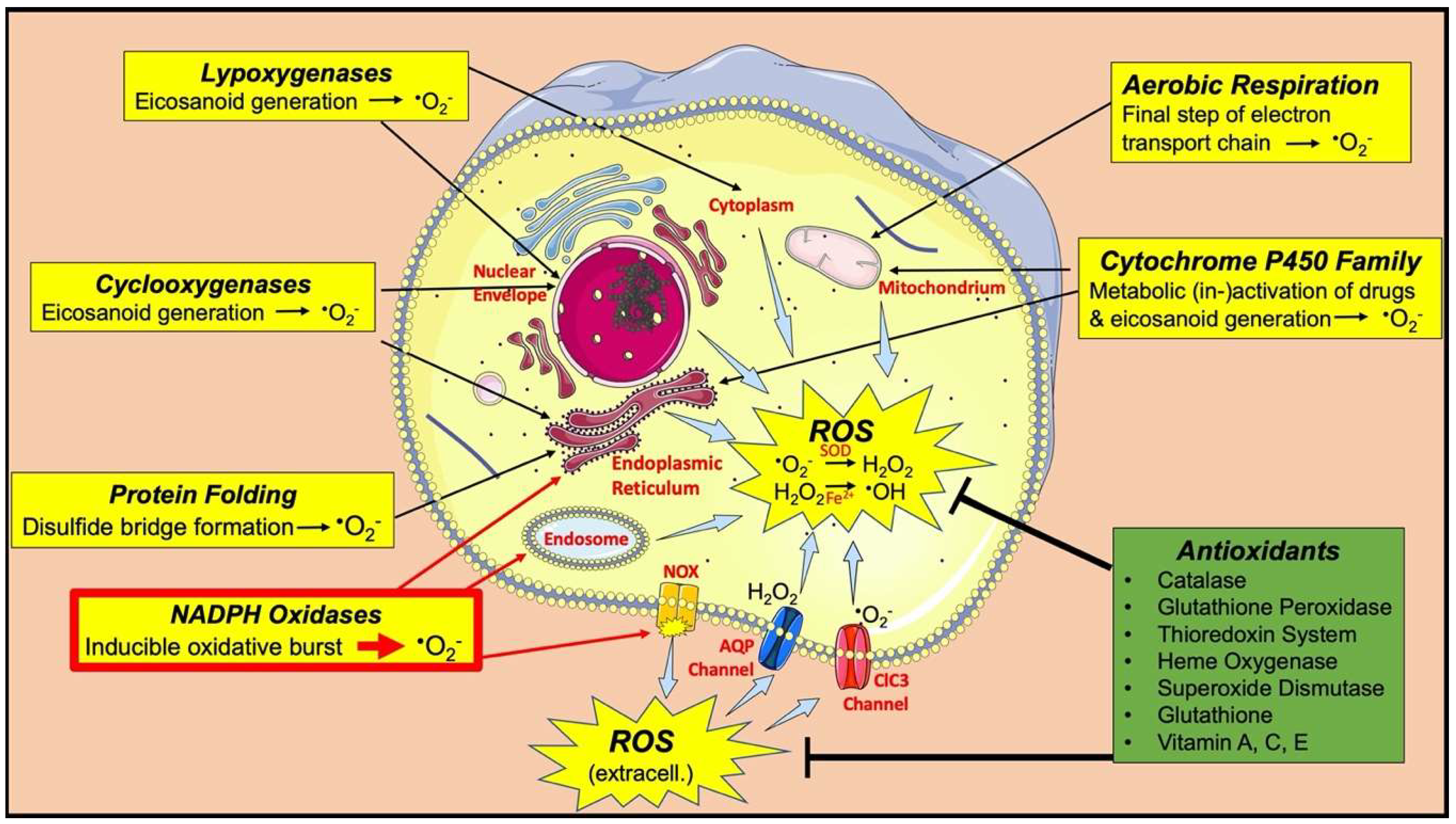
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a healthy proteome during oxidative stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.P.; Weissman, J.S. The FAD-and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Involvement of cytochrome P450 in reactive oxygen species formation and cancer. In Advances in Pharmacology (San Diego, Calif.); Academic Press: Cambridge, MA, USA, 2015; Volume 74. [Google Scholar]
- Silva, G.C.; Abbas, M.; Khemais-Benkhiat, S.; Burban, M.; Ribeiro, T.P.; Toti, F.; Idris-Khodja, N.; Côrtes, S.F.; Schini-Kerth, V.B. Replicative senescence promotes prothrombotic responses in endothelial cells: Role of NADPH oxidase-and cyclooxygenase-derived oxidative stress. Exp. Gerontol. 2017, 93, 7–15. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Enami, S.; Sakamoto, Y.; Colussi, A.J. Fenton chemistry at aqueous interfaces. Proc. Natl. Acad. Sci. USA 2014, 111, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.-T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Leng, Y.; Kufe, D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J. Biol. Chem. 2003, 278, 29667–29675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of heme oxygenase as a modulator of heme-mediated pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Modarai, B.; Guiver Burnand, K.; Humphries, J.; Waltham, M.; Smith, A. The role of neovascularisation in the resolution of venous thrombus. Thromb. Haemost. 2005, 93, 801–809. [Google Scholar] [CrossRef]
- Brooks, E.G.; Trotman, W.; Wadsworth, M.P.; Taatjes, D.J.; Evans, M.F.; Ittleman, F.P.; Callas, P.W.; Esmon, C.T.; Bovill, E.G. Valves of the deep venous system: An overlooked risk factor. Blood 2009, 114, 1276–1279. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.J.; Ritchie, W.G.; Lynch, P.R. Venous endothelial damage produced by massive sticking and emigration of leukocytes. Am. J. Pathol. 1974, 74, 507–532. [Google Scholar]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Day, S.M.; Reeve, J.L.; Pedersen, B.; Farris, D.M.; Myers, D.D.; Im, M.; Wakefield, T.W.; Mackman, N.; Fay, W.P. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood 2005, 105, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacoviello, L.; Kolpakov, V.; Salvatore, L.; Amore, C.; Pintucci, G.; De Gaetano, G.; Donati, M.B. Human endothelial cell damage by neutrophil-derived cathepsin G. Role of cytoskeleton rearrangement and matrix-bound plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Humphries, J.; Modarai, B.; Mattock, K.; Waltham, M.; Evans, C.E.; Ahmad, A.; Patel, A.S.; Premaratne, S.; Lyons, O.T.A.; et al. Leukocytes and the natural history of deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 506–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomaryov, T.; Payne, H.; Fabritz, L.; Wagner, D.D.; Brill, A. Mast cells granular contents are crucial for deep vein thrombosis in mice. Circ. Res. 2017, 121, 941–950. [Google Scholar] [CrossRef]
- Budnik, I.; Brill, A. Immune factors in deep vein thrombosis initiation. Trends Immunol. 2018, 39, 610–623. [Google Scholar] [CrossRef]
- Golino, P.; Ragni, M.; Cirillo, P.; Avvedimento, V.E.; Feliciello, A.; Esposito, N.; Scognamiglio, A.; Trimarco, B.; Iaccarino, G.; Condorelli, M.; et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat. Med. 1996, 2, 35–40. [Google Scholar] [CrossRef]
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancié, H. Polymorphonuclear leukocytes modulate tissue factor production by mononuclear cells: Role of reactive oxygen species. J. Immunol. 2000, 164, 3822–3828. [Google Scholar] [CrossRef] [Green Version]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Görlach, A. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, J.; Kristal, B.; Chezar, J.; Shaul, S.M.; Sela, S. Exogenous superoxide mediates pro-oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures. Free Radic. Biol. Med. 2005, 39, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Banfi, C.; Brioschi, M.; Barbieri, S.S.; Eligini, S.; Barcella, S.; Tremoli, E.; Colli, s.; Mussoni, L. Mitochondrial reactive oxygen species: A common pathway for PAR1-and PAR2-mediated tissue factor induction in human endothelial cells. J. Thromb. Haemost. 2009, 7, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, T.; Pogrebniak, A.; BelAiba, R.S.; Bonello, S.; Wotzlaw, C.; Acker, H.; Hess, J.; Görlach, A. The expression of the NADPH oxidase subunit p22phox is regulated by a redox-sensitive pathway in endothelial cells. Free Radic. Biol. Med. 2005, 38, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Hiraishi, S.; Itabe, H.; Hamuro, T.; Kamikubo, Y.; Takano, T.; Matsuda, J.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid. Redox Signal. 2004, 6, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Nalian, A.; Iakhiaev, A.V. Possible mechanisms contributing to oxidative inactivation of activated protein C: Molecular dynamics study. Thromb. Haemost. 2008, 100, 18–25. [Google Scholar] [CrossRef]
- Glaser, C.B.; Morser, J.; Clarke, J.H.; Blasko, E.; McLean, K.; Kuhn, I.; Chang, R.J.; Lin, J.H.; Vilander, L.; Andrews, W.H.; et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J. Clin. Investig. 1992, 90, 2565–2573. [Google Scholar] [CrossRef] [Green Version]
- Dayal, S.; Gu, S.X.; Hutchins, R.D.; Wilson, K.M.; Wang, Y.; Fu, X.; Lentz, S.R. Deficiency of superoxide dismutase impairs protein C activation and enhances susceptibility to experimental thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1798–1804. [Google Scholar] [CrossRef] [Green Version]
- Upchurch, G.R.; Ramdev, N.; Walsh, M.T.; Loscalzo, J. Prothrombotic consequences of the oxidation of fibrinogen and their inhibition by aspirin. J. Thromb. Thrombolysis 1998, 5, 9–14. [Google Scholar] [CrossRef]
- De Cristofaro, R.; Landolfi, R. Oxidation of human alpha-thrombin by the myeloperoxidase-H2O2-chloride system: Structural and functional effects. Thromb. Haemost. 2000, 83, 253–261. [Google Scholar]
- Van Patten, S.M.; Hanson, E.; Bernasconi, R.; Zhang, K.; Manavalan, P.; Cole, E.S.; McPherson, J.M.; Edmunds, T. Oxidation of methionine residues in antithrombin. Effects on biological activity and heparin binding. J. Biol. Chem. 1999, 274, 10268–10276. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.; Barrowcliffe, T.W. Inhibition of antithrombin III by lipid peroxides. Thromb. Res. 1985, 37, 241–250. [Google Scholar] [CrossRef]
- Huang, X.; Liu, B.; Wei, Y.; Beyea, R.; Yan, H.; Olson, S.T. Lipid oxidation inactivates the anticoagulant function of protein Z-dependent protease inhibitor (ZPI). J. Biol. Chem. 2017, 292, 14625–14635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uderhardt, S.; Ackermann, J.A.; Fillep, T.; Hammond, V.J.; Willeit, J.; Santer, P.; Mayr, M.; Biburger, M.; Miller, M.; Zellner, K.R.; et al. Enzymatic lipid oxidation by eosinophils propagates coagulation, hemostasis, and thrombotic disease. J. Exp. Med. 2017, 214, 2121–2138. [Google Scholar] [CrossRef] [PubMed]
- Von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Montoro-García, S.; Schindewolf, M.; Stanford, S.; Larsen, O.; Thiele, T. The role of platelets in venous thromboembolism. Semin. Thromb. Hemost. 2016, 42, 242–251. [Google Scholar] [CrossRef]
- Ay, C.; Jungbauer, L.V.; Sailer, T.; Tengler, T.; Koder, S.; Kaider, A.; Panzer, S.; Quehenberger, P.; Pabinger, I.; Mannhalter, C. High concentrations of soluble P-selectin are associated with risk of venous thromboembolism and the P-selectin Thr715 variant. Clin. Chem. 2007, 53, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Ay, C.; Dunkler, D.; Marosi, C.; Chiriac, A.-L.; Vormittag, R.; Simanek, R.; Quehenberger, P.; Zielinski, C.; Pabinger, I. Prediction of venous thromboembolism in cancer patients. Blood 2010, 116, 5377–5382. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Li, J.; Tseng, A.; Andrews, R.K.; Cho, J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood 2015, 126, 1952–1964. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Colas, R.; Sassolas, A.; Guichardant, M.; Cugnet-Anceau, C.; Moret, M.; Moulin, P.; Lagarde, M.; Calzada, C. LDL from obese patients with the metabolic syndrome show increased lipid peroxidation and activate platelets. Diabetologia 2011, 54, 2931–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevale, R.; Loffredo, L.; Sanguigni, V.; Plebani, A.; Rossi, P.; Pignata, C.; Martire, B.; Finocchi, A.; Pietrogrande, M.C.; Azzari, C.; et al. Different degrees of NADPH oxidase 2 regulation and in vivo platelet activation: Lesson from chronic granulomatous disease. J. Am. Heart Assoc. 2014, 3, e000920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Varghese, S.; Vitseva, O.; Tanriverdi, K.; Freedman, J.E. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2428–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Sanguigni, V.; Lenti, L.; Finocchi, A.; Mendolicchio, L.; Soresina, A.R.; Plebani, A.; et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Davi, G.; Alessandrini, P.; Mezzetti, A.; Minotti, G.; Bucciarelli, T.; Costantini, F.; Cipollone, F.; Bon, G.B.; Ciabattoni, G.; Patrono, C. In vivo formation of 8-epi-prostaglandin F2α is increased in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3230–3235. [Google Scholar] [CrossRef]
- Davì, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Davì, G.; Di Minno, G.; Coppola, A.; Andria, G.; Cerbone, A.M.; Madonna, P.; Tufano, A.; Falco, A.; Marchesani, P.; Ciabattoni, G.; et al. Oxidative stress and platelet activation in homozygous homocystinuria. Circulation 2001, 104, 1124–1128. [Google Scholar] [CrossRef] [Green Version]
- Davì, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women. JAMA 2002, 288, 2008. [Google Scholar] [CrossRef] [Green Version]
- Ray, J.G. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch. Intern. Med. 1998, 158, 2101. [Google Scholar] [CrossRef] [Green Version]
- Gregson, J.; Kaptoge, S.; Bolton, T.; Pennells, L.; Willeit, P.; Burgess, S.; Bell, S.; Sweeting, M.; Rimm, E.B.; Kabrhel, C.; et al. Cardiovascular risk factors associated with venous thromboembolism. JAMA Cardiol. 2019, 4, 163. [Google Scholar] [CrossRef] [Green Version]
- Tajima, M.; Sakagami, H. Tetrahydrobiopterin impairs the action of endothelial nitric oxide via superoxide derived from platelets. Br. J. Pharm. 2000, 131, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yee Aw, T.; Stokes, K.Y. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018, 14, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Olas, B.; Wachowicz, B. Resveratrol and vitamin C as antioxidants in blood platelets. Thromb. Res. 2002, 106, 143–148. [Google Scholar] [CrossRef]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Vitamin E inhibits collagen-induced platelet activation by blunting hydrogen peroxide. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2542–2547. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.C.; Mahoney, C.E.; (Coleman) Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.-Y.; Tang, S.-S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Secor, D.; Li, F.; Ellis, C.G.; Sharpe, M.D.; Gross, P.L.; Wilson, J.X.; Tyml, K. Impaired microvascular perfusion in sepsis requires activated coagulation and P-selectin-mediated platelet adhesion in capillaries. Intensive Care Med. 2010, 36, 1928–1934. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, M.; Luo, D.; Yue, M.; Wang, S.; Chen, X.; Zhou, Y.; Wang, Y.; Cai, Y.; Hu, X.; et al. Class III PI3K positively regulates platelet activation and thrombosis via PI(3)P-directed function of NADPH oxidase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The emerging role of NETs in venous thrombosis and immunothrombosis. Front. Immunol. 2016, 7, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmann, O.; Medina, E. The expanding world of extracellular traps: Not only neutrophils but much more. Front. Immunol. 2013, 3, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.-D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Beckmann, L.; Dicke, C.; Spath, B.; Lehr, C.; Sievers, B.; Klinke, A.; Baldus, S.; Rudolph, V.; Langer, F. Myeloperoxidase is a negative regulator of phospholipid-dependent coagulation. Thromb. Haemost. 2017, 117, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- d’Onofrio, G.; Mancini, R.; Vallone, R.; Alfano, G.; Candido, A.; Palla, M.; Mango, G. Acquired neutrophil myeloperoxidase deficiency: An indicator of subclinical activation of blood coagulation? Blood Cells 1983, 9, 455–466. [Google Scholar]
- Woollard, K.J.; Sturgeon, S.; Chin-Dusting, J.P.F.; Salem, H.H.; Jackson, S.P. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J. Biol. Chem. 2009, 284, 13110–13118. [Google Scholar] [CrossRef] [Green Version]
- Bahl, N.; Winarsih, I.; Tucker-Kellogg, L.; Ding, J.L. Extracellular haemoglobin upregulates and binds to tissue factor on macrophages: Implications for coagulation and oxidative stress. Thromb. Haemost. 2014, 111, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.J.; Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Katusic, Z.S.; Nath, K.A. Induction of heme oxygenase-1 is a beneficial response in a murine model of venous thrombosis. Am. J. Pathol. 2008, 173, 1882–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, S.; Weltermann, A.; Fritsche, R.; Marsik, C.; Wagner, O.; Kyrle, P.A.; Eichinger, S. Genetic variation in heme oxygenase 1 (HMOX1) and the risk of recurrent venous thromboembolism. J. Vasc. Surg. 2008, 47, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; De Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [Green Version]
- Bankl, H.C.; Grobschmidt, K.; Pikula, B.; Bankl, H.; Lechner, K.; Valent, P. Mast cells are augmented in deep vein thrombosis and express a profibrinolytic phenotype. Hum. Pathol. 1999, 30, 188–194. [Google Scholar] [CrossRef]
- Evans, C.E.; Humphries, J.; Mattock, K.; Waltham, M.; Wadoodi, A.; Saha, P.; Modarai, B.; Maxwell, P.H.; Maxwell, P.J.; Smith, A. Hypoxia and upregulation of hypoxia-inducible factor 1{alpha} stimulate venous thrombus recanalization. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.C.; Whelan, C.J.; Purcell, W.M. Reactive oxygen species generation and histamine release by activated mast cells: Modulation by nitric oxide synthase inhibition. Br. J. Pharm. 1999, 128, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast cell: A multi-functional master cell. Front. Immunol. 2015, 6, 620. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Jiang, W.Y.; Cui, Z.J. An essential role of NAD(P)H oxidase 2 in UVA-induced calcium oscillations in mast cells. Photochem. Photobiol. Sci. 2015, 14, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Chelombitko, M.A.; Fedorov, A.V.; Ilyinskaya, O.P.; Zinovkin, R.A.; Chernyak, B.V. Role of reactive oxygen species in mast cell degranulation. Biochemistry 2016, 81, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, I.; Nielsen, S.F.; Nordestgaard, B.G. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the copenhagen general population study. Clin. Chem. 2016, 62, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement in activation arteriosclerial and venous thrombosis is mediated by plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, C.D.; Hsu, A.T.; Ellson, C.D.; Y.Miyazawa, B.; Kong, Y.-W.; Greenwood, J.D.; Dhara, S.; Neal, M.D.; Sperry, J.L.; Park, M.S.; et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin. Exp. Immunol. 2018, 194, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Anderson, M.; Wilson, C.; Hagmann, H.; Benzing, T.; Dryer, S.E. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: Essential role of podocin in formation of this complex. Am. J. Physiol. Cell Physiol. 2013, 305, C960–C971. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Liu, B.C.; Lu, X.Y.; Yang, L.L.; Zhai, Y.J.; Eaton, A.F.; Thai, T.L.; Eaton, D.C.; Ma, H.P.; Shen, B.Z. Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Rigutto, S.; Hoste, C.; Grasberger, H.; Milenkovic, M.; Communi, D.; Dumont, J.E.; Corvilain, B.; Miot, F.; De Deken, X. Activation of dual oxidases Duox1 and Duox2: Differential regulation mediated by cAMP-dependent protein kinase and protein kinase C-dependent phosphorylation. J. Biol. Chem. 2009, 284, 6725–6734. [Google Scholar] [CrossRef] [Green Version]
- Jagnandan, D.; Church, J.E.; Banfi, B.; Stuehr, D.J.; Marrero, M.B.; Fulton, D.J.R. Novel mechanism of activation of NADPH oxidase 5: Calcium sensitization via phosphorylation. J. Biol. Chem. 2007, 282, 6494–6507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Jamali, A.; Valente, A.J.; Clark, R.A. Regulation of phagocyte NADPH oxidase by hydrogen peroxide through a Ca2+/c-Abl signaling pathway. Free Radic. Biol. Med. 2010, 48, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Northeast, A.D.; Soo, K.S.; Bobrow, L.G.; Gaffney, P.J.; Burnand, K.G. The tissue plasminogen activator and urokinase response in vivo during natural resolution of venous thrombus. J. Vasc. Surg. 1995, 22, 573–579. [Google Scholar] [CrossRef] [Green Version]
- Soo, K.S.; Northeast, A.D.R.; Happerfield, L.C.; Burnand, K.G.; Bobrow, L.G. Tissue plasminogen activator production by monocytes in venous thrombolysis. J. Pathol. 1996, 178, 190–194. [Google Scholar] [CrossRef]
- Singh, I.; Burnand, K.G.; Collins, M.; Luttun, A.; Collen, D.; Boelhouwer, B.; Smith, A. Failure of thrombus to resolve in urokinase-type plasminogen activator gene–knockout mice. Circulation 2003, 107, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Gossage, J.A.; Humphries, J.; Modarai, B.; Burnand, K.G.; Smith, A. Adenoviral urokinase-type plasminogen activator (uPA) gene transfer enhances venous thrombus resolution. J. Vasc. Surg. 2006, 44, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Siefert, S.A.; Chabasse, C.; Mukhopadhyay, S.; Hoofnagle, M.H.; Strickland, D.K.; Sarkar, R.; Antalis, T.M. Enhanced venous thrombus resolution in plasminogen activator inhibitor type-2 deficient mice. J. Thromb. Haemost. 2014, 12, 1706–1716. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Woodcock-Mitchell, J.; Mitchell, J.; Sakamoto, T.; Marutsuka, K.; Sobel, B.E.; Fujii, S. Induction of plasminogen activator inhibitor type 1 and type 1 collagen expression in rat cardiac microvascular endothelial cells by interleukin-1 and its dependence on oxygen-centered free radicals. Circulation 1998, 97, 2175–2182. [Google Scholar] [CrossRef]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimova, E.Y.; Samoylenko, A.; Kietzmann, T. Oxidative Stress and Hypoxia: Implications for Plasminogen Activator Inhibitor-1 Expression. Antioxid. Redox Signal. 2004, 6, 777–791. [Google Scholar] [CrossRef]
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thromb. Res. 2009, 124, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Vulin, A.I.; Stanley, F.M. Oxidative stress activates the plasminogen activator inhibitor type 1 (PAI-1) promoter through an AP-1 response element and cooperates with insulin for additive effects on PAI-1 transcription. J. Biol. Chem. 2004, 279, 25172–25178. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kawano, H.; Takazoe, K.; Soejima, H.; Sakamoto, T.; Hokamaki, J.; Yoshimura, M.; Nakamura, H.; Yodoi, J.; Ogawa, H. Vitamin E improves fibrinolytic activity in patients with coronary spastic angina. Thromb. Res. 2004, 113, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.A.; Loskutoff, D.J. Inactivation of plasminogen activator inhibitor by oxidants. Biochemistry 1986, 25, 6351–6355. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Berchner-Pfannschmidt, U.; Wotzlaw, C.; Cool, R.H.; Fandrey, J.; Acker, H.; Jungermann, K.; Kietzmann, T. Reactive oxygen species modulate HIF-1 mediated PAI-1 expression: Involvement of the GTPase Rac1. Thromb. Haemost. 2003, 89, 926–935. [Google Scholar]
- Uchida, Y.; Ohba, K.; Yoshioka, T.; Irie, K.; Muraki, T.; Maru, Y. Cellular carbonyl stress enhances the expression of plasminogen activator inhibitor-1 in rat white adipocytes via reactive oxygen species-dependent pathway. J. Biol. Chem. 2004, 279, 4075–4083. [Google Scholar] [CrossRef] [Green Version]
- Wagenaar, G.T.; Ter Horst, S.A.; Van Gastelen, M.A.; Leijser, L.M.; Mauad, T.; Van Der Velden, P.A.; De Heer, E.; Hiemstra, P.S.; Poorthuis, B.J.H.; Walther, F.J. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic. Biol. Med. 2004, 36, 782–801. [Google Scholar] [CrossRef]
- Franke, K.; Curth, K.; Lenart, J.; Knochenhauer, D.; Kietzmann, T. Enhanced plasminogen activator inhibitor-1 expression in transgenic mice with hepatocyte-specific overexpression of superoxide dismutase or glutathione peroxidase. Antioxid. Redox Signal. 2004, 6, 721–728. [Google Scholar] [CrossRef]
- Varma, M.R.; Varga, A.J.; Knipp, B.S.; Sukheepod, P.; Upchurch, G.R.; Kunkel, S.L.; Wakefield, T.W.; Henke, P.K. Neutropenia impairs venous thrombosis resolution in the rat. J. Vasc. Surg. 2003, 38, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.; Humphries, J.; Burnand, K.; Sawyer, B.; Bursill, C.; Channon, K.; Greaves, D.; Rollins, B.; Charo, I.F.; Smith, A. Monocyte recruitment in venous thrombus resolution. J. Vasc. Surg. 2006, 43, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klyubin, I.V.; Kirpichnikova, K.M.; Gamaley, I.A. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur. J. Cell Biol. 1996, 70, 347–351. [Google Scholar] [PubMed]
- Tur, E.; Bolton, L.; Constantine, B.E. Topical hydrogen peroxide treatment of ischemic ulcers in the guinea pig: Blood recruitment in multiple skin sites. J. Am. Acad. Dermatol. 1995, 33, 217–221. [Google Scholar] [CrossRef]
- Nakamura, H.; Herzenberg, L.A.; Bai, J.; Araya, S.; Kondo, N.; Nishinaka, Y.; Herzenberg, L.A.; Yodoi, J. Circulating thioredoxin suppresses lipopolysaccharide-induced neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2001, 98, 15143–15148. [Google Scholar] [CrossRef] [Green Version]
- Fraticelli, A.; Serrano, C.V.; Bochner, B.S.; Capogrossi, M.C.; Zweier, J.L. Hydrogen peroxide and superoxide modulate leukocyte adhesion molecule expression and leukocyte endothelial adhesion. Biochim. Biophys. Acta Mol. Cell Res. 1996, 1310, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Kellermair, J.; Redwan, B.; Alias, S.; Jabkowski, J.; Panzenboeck, A.; Kellermair, L.; Winter, M.P.; Weltermann, A.; Lang, I.M. Platelet endothelial cell adhesion molecule 1 deficiency misguides venous thrombus resolution. Blood 2013, 122, 3376–3384. [Google Scholar] [CrossRef] [Green Version]
- Rattan, V.; Sultana, C.; Shen, Y.; Kalra, V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Am. J. Physiol. Metab. 1997, 273, E453. [Google Scholar] [CrossRef]
- Saragih, H.; Zilian, E.; Jaimes, Y.; Paine, A.; Figueiredo, C.; Eiz-Vesper, B.; Blasczyk, R.; Larmann, J.; Theilmeier, G.; Burg-Roderfeld, M.; et al. PECAM-1-dependent heme oxygenase-1 regulation via an Nrf2-mediated pathway in endothelial cells. Thromb. Haemost. 2014, 111, 1077–1088. [Google Scholar]
- Van Aken, B.E.; Reitsma, P.H.; Rosendaal, F.R. Interleukin 8 and venous thrombosis: Evidence for a role of inflammation in thrombosis. Br. J. Haematol. 2002, 116, 173–177. [Google Scholar] [CrossRef]
- Henke, P.K.; Wakefield, T.W.; Kadell, A.M.; Linn, M.J.; Varma, M.R.; Sarkar, M.; Hawley, A.; Fowlkes, J.B.; Strieter, R.M. Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J. Surg. Res. 2001, 99, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Lekstrom-Himes, J.A.; Kuhns, D.B.; Alvord, W.G.; Gallin, J.I. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J. Immunol. 2005, 174, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmijewski, J.W.; Zhao, X.; Xu, Z.; Abraham, E. Exposure to hydrogen peroxide diminishes NF-κB activation, IκB-α degradation, and proteasome activity in neutrophils. Am. J. Physiol. Physiol. 2007, 293, C255–C266. [Google Scholar] [CrossRef] [PubMed]
- DeForge, L.E.; Preston, A.M.; Takeuchi, E.; Kenney, J.; Boxer, L.A.; Remick, D.G. Regulation of interleukin 8 gene expression by oxidant stress. J. Biol. Chem. 1993, 268, 25568–25576. [Google Scholar]
- Monteseirín, J.; Chacón, P.; Vega, A.; El Bekay, R.; Alvarez, M.; Alba, G.; Conde, M.; Jiménez, J.; Asturias, J.A.; Martínez, A.; et al. Human neutrophils synthesize IL-8 in an IgE-mediated activation. J. Leukoc. Biol. 2004, 76, 692–700. [Google Scholar] [CrossRef]
- Hidalgo, M.A.; Carretta, M.D.; Teuber, S.E.; Zárate, C.; Cárcamo, L.; Concha, I.I.; Burgos, R.A. fMLP-induced IL-8 release is dependent on NADPH oxidase in human neutrophils. J. Immunol. Res. 2015, 2015, 120348. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, T.; Yamashita, K.; Arai, T.; Yamamoto, K.; Mizugishi, K.; Uchiyama, T. The role of endothelial interleukin-8/NADPH oxidase 1 axis in sepsis. Immunology 2010, 131, 331–339. [Google Scholar] [CrossRef]
- Yadav, A.; Saini, V.; Arora, S. MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 2010, 411, 1570–1579. [Google Scholar] [CrossRef]
- Humphries, J.; McGuinness, C.L.; Smith, A.; Waltham, M.; Poston, R.; Burnand, K.G. Monocyte chemotactic protein-1 (MCP-1) accelerates the organization and resolution of venous thrombi. J. Vasc. Surg. 1999, 30, 894–899. [Google Scholar] [CrossRef]
- Lee, Y.W.; Lee, W.H.; Kim, P.H. Role of NADPH oxidase in interleukin-4-induced monocyte chemoattractant protein-1 expression in vascular endothelium. Inflamm. Res. 2010, 59, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Rojas, M.; Lilly, B.; Tsai, N.-T.; Lemtalsi, T.; Liou, G.I.; Caldwell, R.W.; Caldwell, R.B. NAD(P)H oxidase-dependent regulation of CCL2 production during retinal inflammation. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Dehnad, A.; Fish, S.; Sato, A.; Jiang, J.; Tian, J.; Schröder, K.; Brandes, R.; Török, N.J. NOX4 regulates CCR2 and CCL2 mRNA stability in alcoholic liver disease. Sci. Rep. 2017, 7, 46144. [Google Scholar] [CrossRef] [PubMed]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modarai, B.; Humphries, J.; Gossage, J.A.; Waltham, M.; Burnand, K.G.; Kanaganayagam, G.S.; Afuwape, A.; Paleolog, E.; Smith, A.; Wadoodi, A. Adenovirus-mediated VEGF gene therapy enhances venous thrombus recanalization and resolution. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1753–1759. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.E.; Grover, S.P.; Humphries, J.; Saha, P.; Patel, A.P.; Patel, A.S.; Lyons, O.T.; Waltham, M.; Modarai, B.; Smith, A. Antiangiogenic therapy inhibits venous thrombus resolution. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Altmann, J.; Sharma, S.; Lang, I.M. Advances in our understanding of mechanisms of venous thrombus resolution. Expert Rev. Hematol. 2016, 9, 69–78. [Google Scholar] [CrossRef]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef]
- Kim, Y.-W.; Byzova, T. V Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [Green Version]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Podrez, E.A.; Salomon, R.G.; Byzova, T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef]
- Gengrinovitch, S.; Berman, B.; David, G.; Witte, L.; Neufeld, G.; Ron, D. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J. Biol. Chem. 1999, 274, 10816–10822. [Google Scholar] [CrossRef] [Green Version]
- BelAiba, R.S.; Djordjevic, T.; Petry, A.; Diemer, K.; Bonello, S.; Banfi, B.; Hess, J.; Pogrebniak, A.; Bickel, C.; Görlach, A. NOX5 variants are functionally active in endothelial cells. Free Radic. Biol. Med. 2007, 42, 446–459. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Platt, D.H.; Matragoon, S.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am. J. Pathol. 2005, 167, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Fassina, G.; Morini, M.; Aluigi, M.G.; Masiello, L.; Fontanini, G.; D’Agostini, F.; De Flora, S.; Noonan, D.M.; Albini, A. N-acetylcysteine inhibits endothelial cell invasion and angiogenesis. Lab. Investig. 1999, 79, 1151–1159. [Google Scholar]
- Wheeler, M.D.; Smutney, O.M.; Samulski, R.J. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of B16 melanomas in mice. Mol. Cancer Res. 2003, 1, 871–881. [Google Scholar]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.-C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary Arteriosclery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-β1–induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-Y.; Wu, L.-C.; Dai, T.; Chen, S.-Y.; Wang, A.-Y.; Lin, K.; Lin, D.-M.; Yang, J.-Q.; Cheng, B.; Zhang, L.; et al. NADPH oxidase-2 is a key regulator of human dermal fibroblasts: A potential therapeutic strategy for the treatment of skin fibrosis. Exp. Dermatol. 2014, 23, 639–644. [Google Scholar] [CrossRef]
- Lévigne, D.; Modarressi, A.; Krause, K.-H.; Pittet-Cuénod, B. NADPH oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic. Biol. Med. 2016, 96, 374–384. [Google Scholar] [CrossRef]
- Brandt, M.; Giokoglu, E.; Garlapati, V.; Bochenek, M.L.; Molitor, M.; Hobohm, L.; Schönfelder, T.; Münzel, T.; Kossmann, S.; Karbach, S.H.; et al. Pulmonary arteriosclerial hypertension and endothelial dysfunction is linked to NADPH oxidase-derived superoxide formation in venous thrombosis and pulmonary embolism in mice. Oxid. Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Jobi, K.; Rauch, B.H.; Dangwal, S.; Freidel, K.; Doller, A.; Eberhardt, W.; Fischer, J.W.; Schrör, K.; Rosenkranz, A.C. Redox regulation of human protease-activated receptor-2 by activated factor X. Free Radic. Biol. Med. 2011, 51, 1758–1764. [Google Scholar] [CrossRef]
- Henke, P.; Varma, M.; Deatrick, K.; Dewyer, N.; Lynch, E.; Moore, A.; Dubay, D.; Sukheepod, P.; Pearce, C.; Upchurch, G.; et al. Neutrophils modulate post-thrombotic vein wall remodeling but not thrombus neovascularization. Thromb. Haemost. 2006, 95, 272–281. [Google Scholar]
- Henke, P.K.; Varma, M.R.; Moaveni, D.K.; Dewyer, N.A.; Moore, A.J.; Lynch, E.M.; Longo, C.; Deatrick, C.B.; Kunkel, S.L.; Upchurch, G.R.; et al. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb. Haemost. 2007, 98, 1045–1055. [Google Scholar]
- Levi, E.; Fridman, R.; Miao, H.Q.; Ma, Y.S.; Yayon, A.; Vlodavsky, I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc. Natl. Acad. Sci. USA 1996, 93, 7069–7074. [Google Scholar] [CrossRef] [Green Version]
- Ugwu, F.; Van Hoef, B.; Bini, A.; Collen, D.; Lijnen, H.R. Proteolytic cleavage of urokinase-type plasminogen activator by stromelysin-1 (MMP-3). Biochemistry 1998, 37, 7231–7236. [Google Scholar] [CrossRef]
- Collen, A.; Hanemaaijer, R.; Lupu, F.; Quax, P.H.A.; Van Lent, N.; Grimbergen, J.; Peters, E.; Koolwijk, P.; Van Hinsbergh, V.W.M. Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix. Blood 2003, 101, 1810–1817. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-D.; Li, X.-Q. Endothelial progenitor cells accelerate the resolution of deep vein thrombosis. Vasc. Pharm. 2016, 83, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Deatrick, K.B.; Eliason, J.L.; Lynch, E.M.; Moore, A.J.; Dewyer, N.A.; Varma, M.R.; Pearce, C.G.; Upchurch, G.R.; Wakefield, T.W.; Henke, P.K. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J. Vasc. Surg. 2005, 42, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Deatrick, K.B.; Obi, A.; Luke, C.E.; Elfline, M.A.; Sood, V.; Upchurch, G.R.; Jaffer, F.; Wakefield, T.W.; Henke, P.K. Matrix metalloproteinase-9 deletion is associated with decreased mid-term vein wall fibrosis in experimental stasis DVT. Thromb. Res. 2013, 132, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Henke, P.K.; Pearce, C.G.; Moaveni, D.M.; Moore, A.J.; Lynch, E.M.; Longo, C.; Varma, M.; Dewyer, N.A.; Deatrick, K.B.; Upchurch, G.R.; et al. Targeted deletion of CCR2 impairs deep vein thombosis resolution in a mouse model. J. Immunol. 2006, 177, 3388–3397. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Belkhiri, A.; Richards, C.; Whaley, M.; McQueen, S.A.; Orr, F.W. Increased expression of activated matrix metalloproteinase-2 by human endothelial cells after sublethal H2O2 exposure. Lab. Investig. 1997, 77, 533–539. [Google Scholar] [PubMed]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.-O.; Park, S.-J.; Yoon, S.Y.; Yun, C.-H.; Chung, A.-S. Sustained production of H2O2 activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-κB pathway. J. Biol. Chem. 2002, 277, 30271–30282. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.H.; Moon, Y.J.; Seo, J.-E.; Lee, Y.; Kim, K.H.; Chung, J.H. Reactive oxygen species produced by NADPH oxidase, xanthine oxidase, and mitochondrial electron transport system mediate heat shock-induced MMP-1 and MMP-9 expression. Free Radic. Biol. Med. 2008, 44, 635–645. [Google Scholar] [CrossRef]
- Poitevin, S.; Garnotel, R.; Antonicelli, F.; Gillery, P.; Nguyen, P. Type I collagen induces tissue factor expression and matrix metalloproteinase 9 production in human primary monocytes through a redox-sensitive pathway. J. Thromb. Haemost. 2008, 6, 1586–1594. [Google Scholar] [CrossRef]
- Galis, Z.S.; Asanuma, K.; Godin, D.; Meng, X. N-acetyl-cysteine decreases the matrix-degrading capacity of macrophage-derived foam cells: New target for antioxidant therapy? Circulation 1998, 97, 2445–2453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.J.; Zhao, W.; Venkataraman, S.; Robbins, M.E.C.; Buettner, G.R.; Kregel, K.C.; Oberley, L.W. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J. Biol. Chem. 2002, 277, 20919–20926. [Google Scholar] [CrossRef] [Green Version]
- Svineng, G.; Ravuri, C.; Rikardsen, O.; Huseby, N.-E.; Winberg, J.-O. The role of reactive oxygen species in integrin and matrix metalloproteinase expression and function. Connect. Tissue Res. 2008, 49, 197–202. [Google Scholar] [CrossRef]
- Lackner, K.J.; Manukyan, D.; Müller-Calleja, N. Endosomal redox signaling in the antiphospholipid syndrome. Curr. Rheumatol. Rep. 2017, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, D.; Müller-Calleja, N.; Jäckel, S.; Luchmann, K.; Mönnikes, R.; Kiouptsi, K.; Reinhardt, C.; Jurk, K.; Walter, U.; Lackner, K.J. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J. Thromb. Haemost. 2016, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakopoulos, B.; Gao, L.; Qi, M.; Wong, J.W.; Yu, D.M.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M.; Atsumi, T.; Koike, T.; Hogg, P.; et al. Factor XI is a substrate for oxidoreductases: Enhanced activation of reduced FXI and its role in antiphospholipid syndrome thrombosis. J. Autoimmun. 2012, 39, 121–129. [Google Scholar] [CrossRef]
- Müller-Calleja, N.; Manukyan, D.; Canisius, A.; Strand, D.; Lackner, K.J. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann. Rheum. Dis. 2017, 76, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Emmi, G.; Silvestri, E.; Bruschi, G.; Ciucciarelli, L.; Squatrito, D.; Vaglio, A.; Taddei, N.; Abbate, R.; Emmi, L.; et al. Neutrophil activation promotes fibrinogen oxidation and thrombus formation in Behçet disease. Circulation 2016, 133, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Martinelli, I.; Zanobetti, A.; Grillo, P.; Hou, L.-F.; Bertazzi, P.A.; Mannucci, P.M.; Schwartz, J. Exposure to particulate air pollution and risk of deep vein thrombosis. Arch. Intern. Med. 2008, 168, 920–927. [Google Scholar] [CrossRef]
- Snow, S.J.; Cheng, W.; Wolberg, A.S.; Carraway, M.S. Air pollution upregulates endothelial cell procoagulant activity via ultrafine particle-induced oxidant signaling and tissue factor expression. Toxicol. Sci. 2014, 140, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Ageno, W.; Prandoni, P.; Romualdi, E.; Ghirarduzzi, A.; Dentali, F.; Pesavento, R.; Crowther, M.; Venco, A. The metabolic syndrome and the risk of venous thrombosis: A case–control study. J. Thromb. Haemost. 2006, 4, 1914–1918. [Google Scholar] [CrossRef]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.W.; Sadeh, N. Traumatic stress, oxidative stress and post-traumatic stress disorder: Neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatry 2014, 19, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.; Cheng, Y.-W.; Yang, F.; Sun, P.-W.; Zhu, C.-J.; Zhu, L.; Zhang, G.-X. Chronic stress facilitates the development of deep venous thrombosis. Oxid. Med. Cell. Longev. 2015, 2015, 384535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yisireyili, M.; Takeshita, K.; Hayashi, M.; Wu, H.; Uchida, Y.; Yamamoto, K.; Kikuchi, R.; Hao, C.-N.; Nakayama, T.; Cheng, X.W.; et al. Dipeptidyl peptidase-IV inhibitor alogliptin improves stress-induced insulin resistance and prothrombotic state in a murine model. Psychoneuroendocrinology 2016, 73, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, X.; Fan, X.; Xiao, M.; Yang, D.; Liang, B.; Dai, M.; Shan, L.; Lu, J.; Lin, Z.; et al. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood 2016, 128, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Milman, U.; Blum, S.; Shapira, C.; Aronson, D.; Miller-Lotan, R.; Anbinder, Y.; Alshiek, J.; Bennett, L.; Kostenko, M.; Landau, M.; et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crutchley, D.J.; Que, B.G. Copper-induced tissue factor expression in human monocytic THP-1 cells and its inhibition by antioxidants. Circulation 1995, 92, 238–243. [Google Scholar] [CrossRef]
- Ferro, D.; Basili, S.; Praticó, D.; Iuliano, L.; FitzGerald, G.A.; Violi, F. Vitamin E reduces monocyte tissue factor expression in cirrhotic patients. Blood 1999, 93, 2945–2950. [Google Scholar] [CrossRef]
- Castilla, P.; Dávalos, A.; Teruel, J.L.; Cerrato, F.; Fernández-Lucas, M.; Merino, J.L.; Sánchez-Martín, C.C.; Ortuño, J.; Lasunción, M.A. Comparative effects of dietary supplementation with red grape juice and vitamin E on production of superoxide by circulating neutrophil NADPH oxidase in hemodialysis patients. Am. J. Clin. Nutr. 2008, 87, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Cariello, M.; Simone, S.; Loverre, A.; Gigante, M.; Incampo, F.; Pietanza, S.; Colucci, M.; Schena, F.P.; Gesualdo, L.; Grandaliano, G.; et al. Coagulation activation is associated with nicotinamide adenine dinucleotide phosphate oxidase-dependent reactive oxygen species generation in hemodialysis patients. Antioxid. Redox Signal. 2012, 16, 428–439. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Yamaguchi, Y.; Kikuchi, A.; Nagasaki, Y. Creation of a blood-compatible surface: A novel strategy for suppressing blood activation and coagulation using a nitroxide radical-containing polymer with reactive oxygen species scavenging activity. Acta Biomater. 2012, 8, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Wei, Z.; Ji, C.; Zheng, H.; Gu, J.; Ma, L.; Huang, W.; Morris-Natschke, S.L.; Yeh, J.-L.; Zhang, R.; et al. Xanthohumol isolated from Humulus lupulus prevents thrombosis without increased bleeding risk by inhibiting platelet activation and mtDNA release. Free Radic. Biol. Med. 2017, 108, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Pomp, E.R.; Rosendaal, F.R.; Doggen, C.J.M. Alcohol consumption is associated with a decreased risk of venous thrombosis. Thromb. Haemost. 2008, 99, 59–63. [Google Scholar] [PubMed]
- Gaborit, F.; Overvad, K.; Nørgaard, M.; Kristensen, S.; Tjønneland, A.; Severinsen, M. Alcohol intake and risk of venous thromboembolism. Thromb. Haemost. 2013, 110, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollny, T.; Aiello, L.; Di Tommaso, D.; Bellavia, V.; Rotilio, D.; Donati, M.B.; De Gaetano, G.; Iacoviello, L. Modulation of haemostatic function and prevention of experimental thrombosis by red wine in rats: A role for increased nitric oxide production. Br. J. Pharm. 1999, 127, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Gresele, P.; Pignatelli, P.; Guglielmini, G.; Carnevale, R.; Mezzasoma, A.M.; Ghiselli, A.; Momi, S.; Violi, F. Resveratrol, at concentrations attainable with moderate wine consumption, stimulates human platelet nitric oxide production. J. Nutr. 2008, 138, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Di Castelnuovo, A.; Rotondo, S.; Iacoviello, L.; Donati, M.B.; De Gaetano, G. Meta-analysis of wine and beer consumption in relation to vascular risk. Circulation 2002, 105, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Pignatelli, P.; Nocella, C.; Loffredo, L.; Pastori, D.; Vicario, T.; Petruccioli, A.; Bartimoccia, S.; Violi, F. Extra virgin olive oil blunt post-prandial oxidative stress via NOX2 down-regulation. Atherosclerosis 2014, 235, 649–658. [Google Scholar] [CrossRef]
- Puccetti, L.; Pasqui, A.L.; Pastorelli, M.; Bova, G.; Cercignani, M.; Palazzuoli, A.; Angori, P.; Auteri, A.; Bruni, F. Time-dependent effect of statins on platelet function in hypercholesterolaemia. Eur. J. Clin. Investig. 2002, 32, 901–908. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Pastori, D.; Cangemi, R.; Napoleone, L.; Bartimoccia, S.; Nocella, C.; Basili, S.; Violi, F. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation 2012, 126, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Patti, G.; Cannon, C.P.; Murphy, S.A.; Mega, S.; Pasceri, V.; Briguori, C.; Colombo, A.; Yun, K.H.; Jeong, M.H.; Kim, J.-S.; et al. Clinical benefit of statin pretreatment in patients undergoing percutaneous coronary intervention. Circulation 2011, 123, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Kessinger, C.W.; Kim, J.W.; Henke, P.K.; Thompson, B.; McCarthy, J.R.; Hara, T.; Sillesen, M.; Margey, R.J.P.; Libby, P.; Weissleder, R.; et al. Statins improve the resolution of established murine venous thrombosis: Reductions in thrombus burden and vein wall scarring. PLoS ONE 2015, 10, e0116621. [Google Scholar] [CrossRef] [Green Version]
- Basili, S.; Pignatelli, P.; Tanzilli, G.; Mangieri, E.; Carnevale, R.; Nocella, C.; Di Santo, S.; Pastori, D.; Ferroni, P.; Violi, F. Anoxia-reoxygenation enhances platelet thromboxane A2 production via reactive oxygen species–generated NOX2. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1766–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammisotto, V.; Carnevale, R.; Nocella, C.; Stefanini, L.; Bartimoccia, S.; Coluccia, A.; Silvestri, R.; Pignatelli, P.; Pastori, D.; Violi, F. Nox2-mediated platelet activation by glycoprotein (GP) VI: Effect of rivaroxaban alone and in combination with aspirin. Biochem. Pharm. 2019, 163, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Ramasubramoni, A.; Pula, G.; Harper, M.T.; Mundell, S.J.; Coxon, C.H. Thioredoxin inhibitors attenuate platelet function and thrombus formation. PLoS ONE 2016, 11, e0163006. [Google Scholar] [CrossRef] [PubMed]
- Vitseva, O.; Flockhart, D.A.; Jin, Y.; Varghese, S.; Freedman, J.E. The effects of tamoxifen and its metabolites on platelet function and release of reactive oxygen intermediates. J. Pharm. Exp. 2004, 312, 1144–1150. [Google Scholar] [CrossRef]
- SMART—Servier Medical ART. Available online: https://smart.servier.com/ (accessed on 30 October 2019).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. https://doi.org/10.3390/ijms21061918
Gutmann C, Siow R, Gwozdz AM, Saha P, Smith A. Reactive Oxygen Species in Venous Thrombosis. International Journal of Molecular Sciences. 2020; 21(6):1918. https://doi.org/10.3390/ijms21061918
Chicago/Turabian StyleGutmann, Clemens, Richard Siow, Adam M. Gwozdz, Prakash Saha, and Alberto Smith. 2020. "Reactive Oxygen Species in Venous Thrombosis" International Journal of Molecular Sciences 21, no. 6: 1918. https://doi.org/10.3390/ijms21061918
APA StyleGutmann, C., Siow, R., Gwozdz, A. M., Saha, P., & Smith, A. (2020). Reactive Oxygen Species in Venous Thrombosis. International Journal of Molecular Sciences, 21(6), 1918. https://doi.org/10.3390/ijms21061918