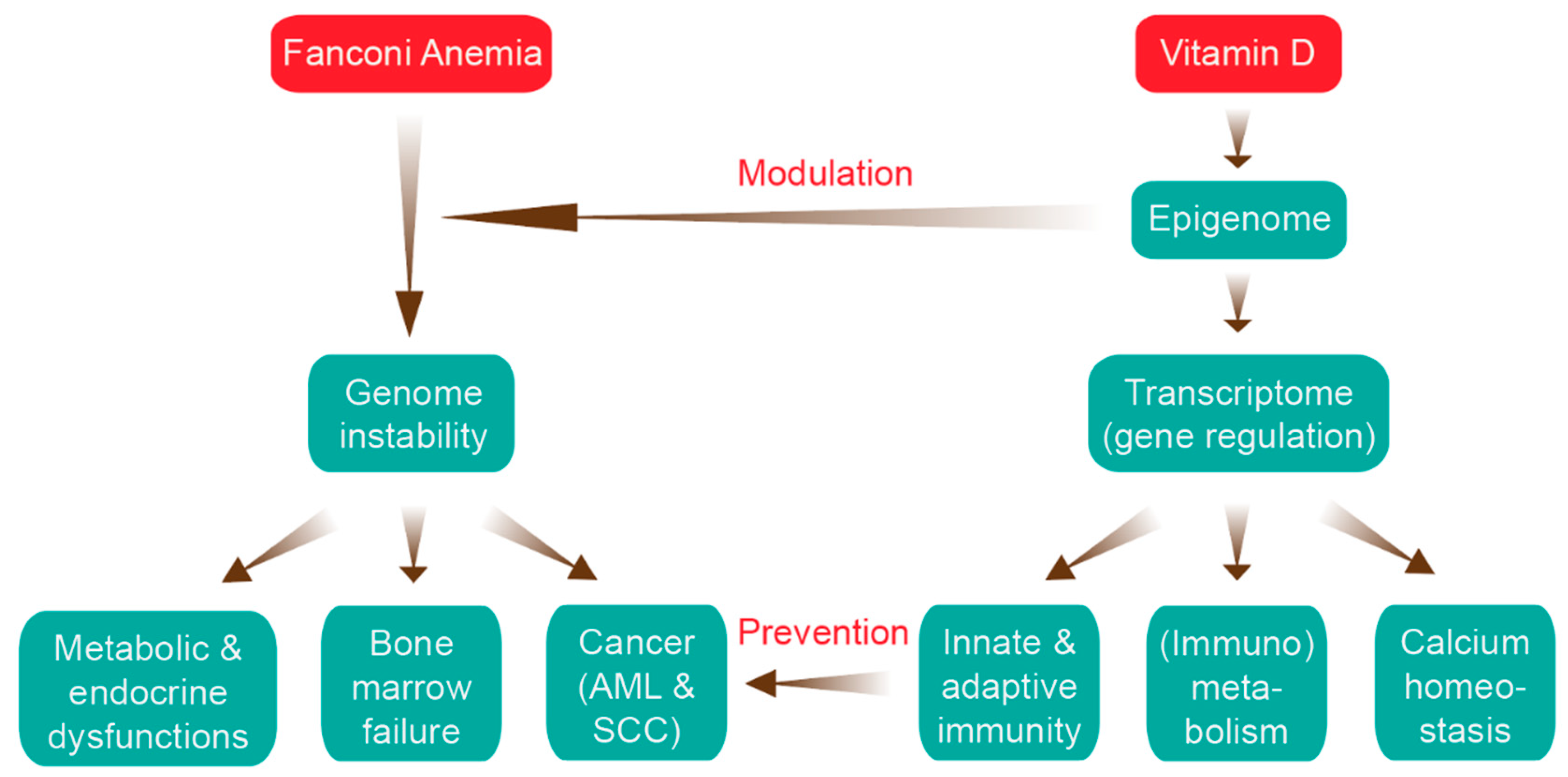
Impact of Epigenetics on Complications of Fanconi Anemia: The Role of Vitamin D-Modulated Immunity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Features of FA
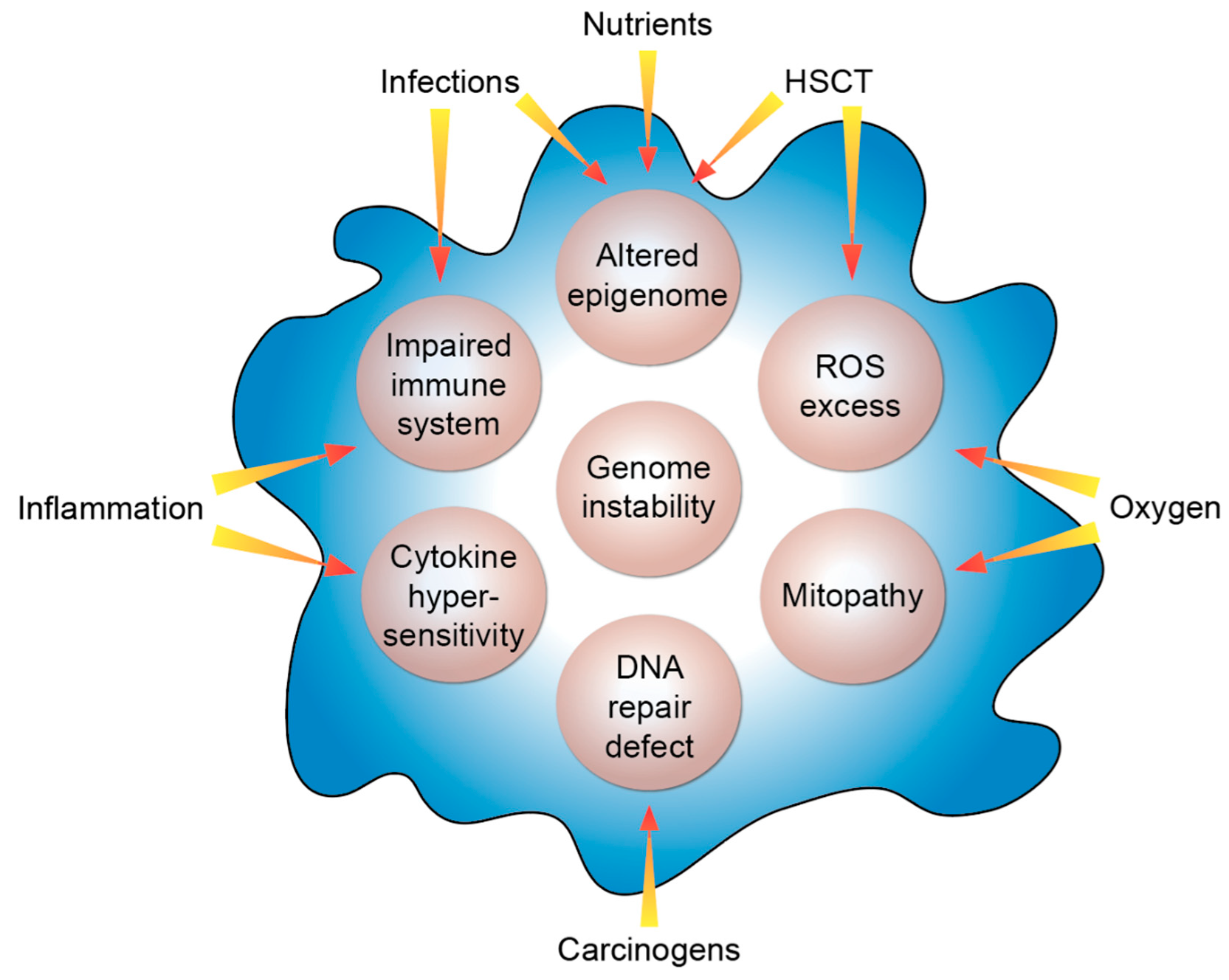
3. Genetic and Molecular Features of FA
4. FA and Cancer
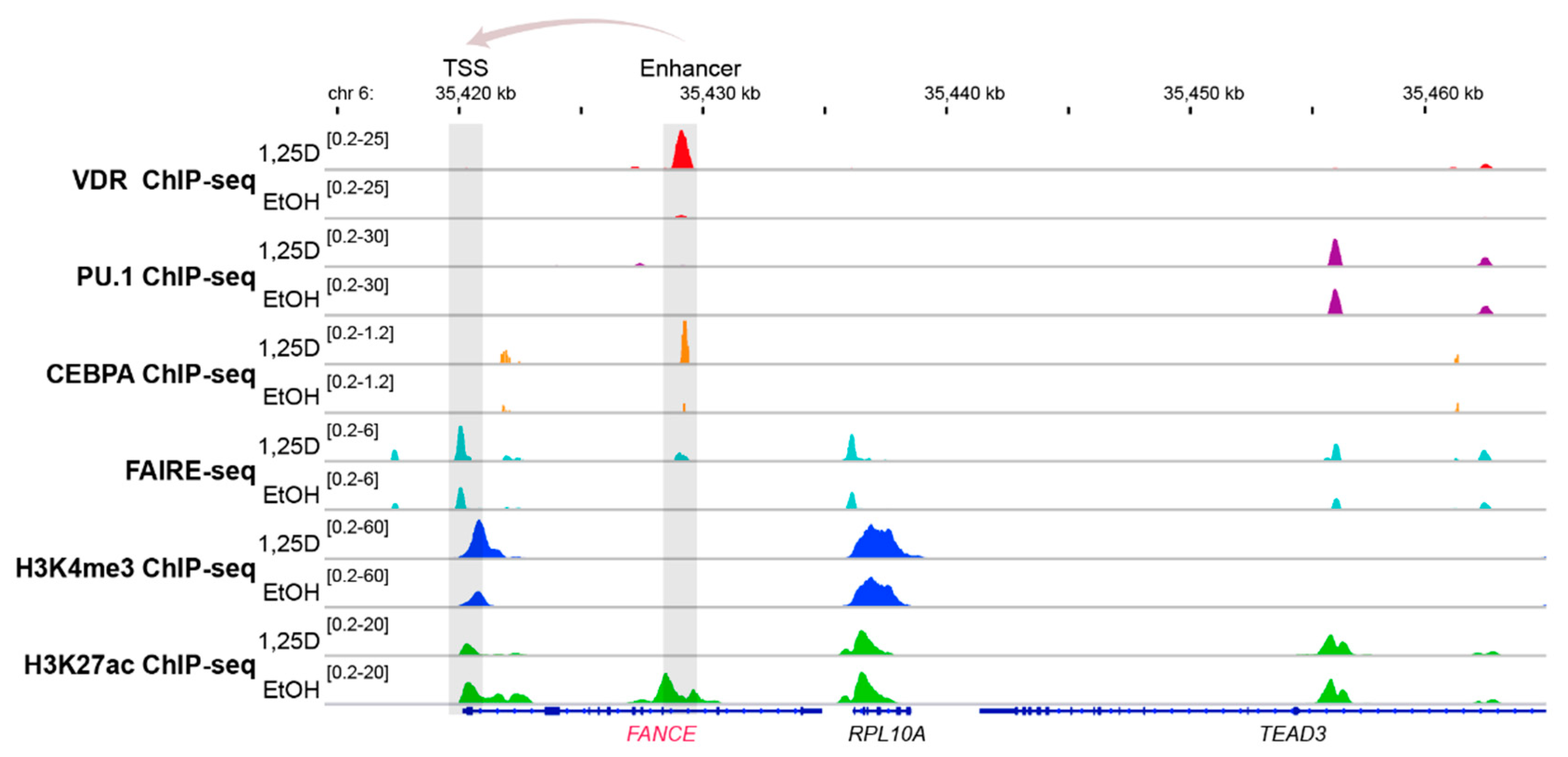
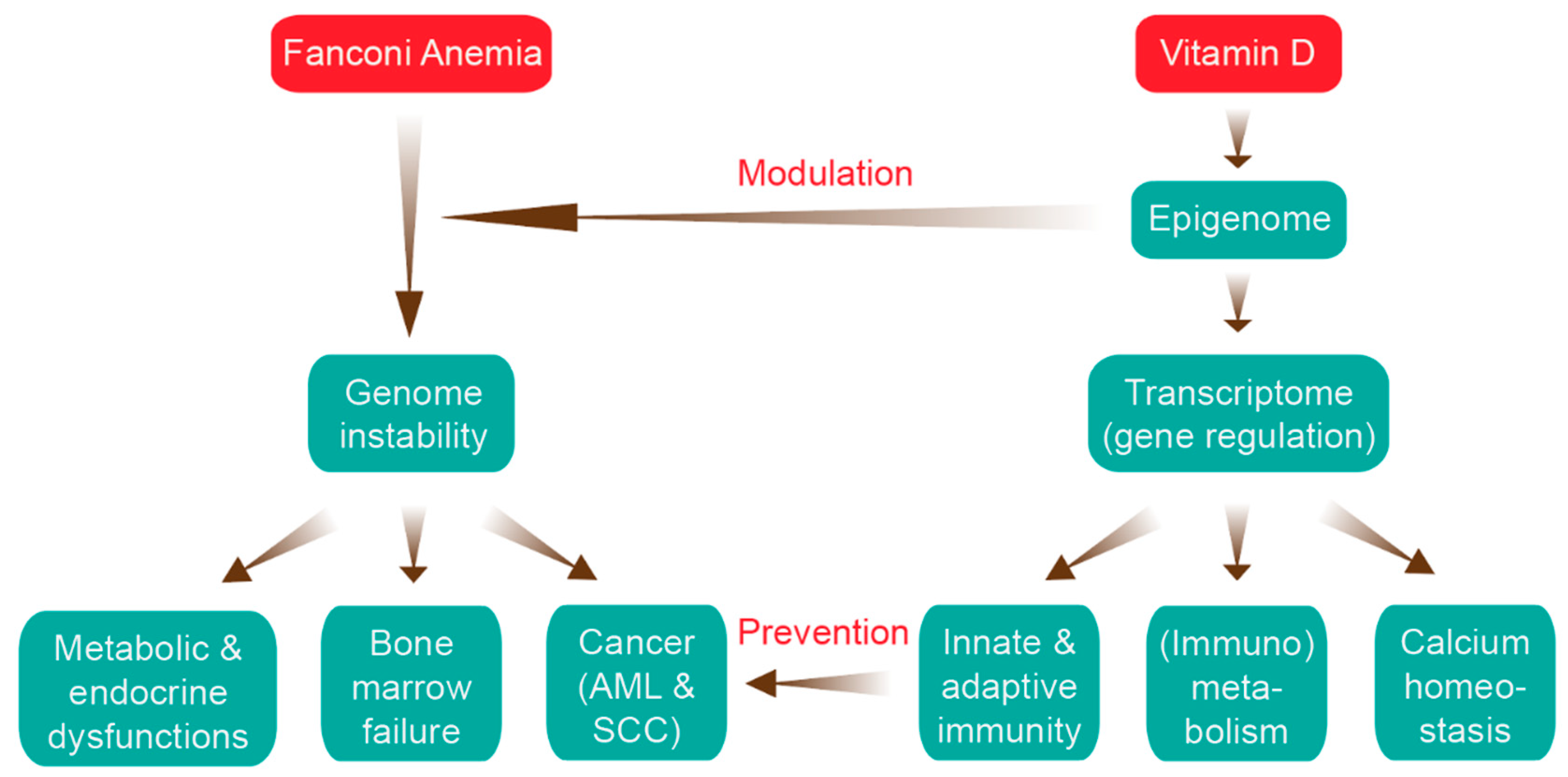
5. The Impact of Epigenetics in FA
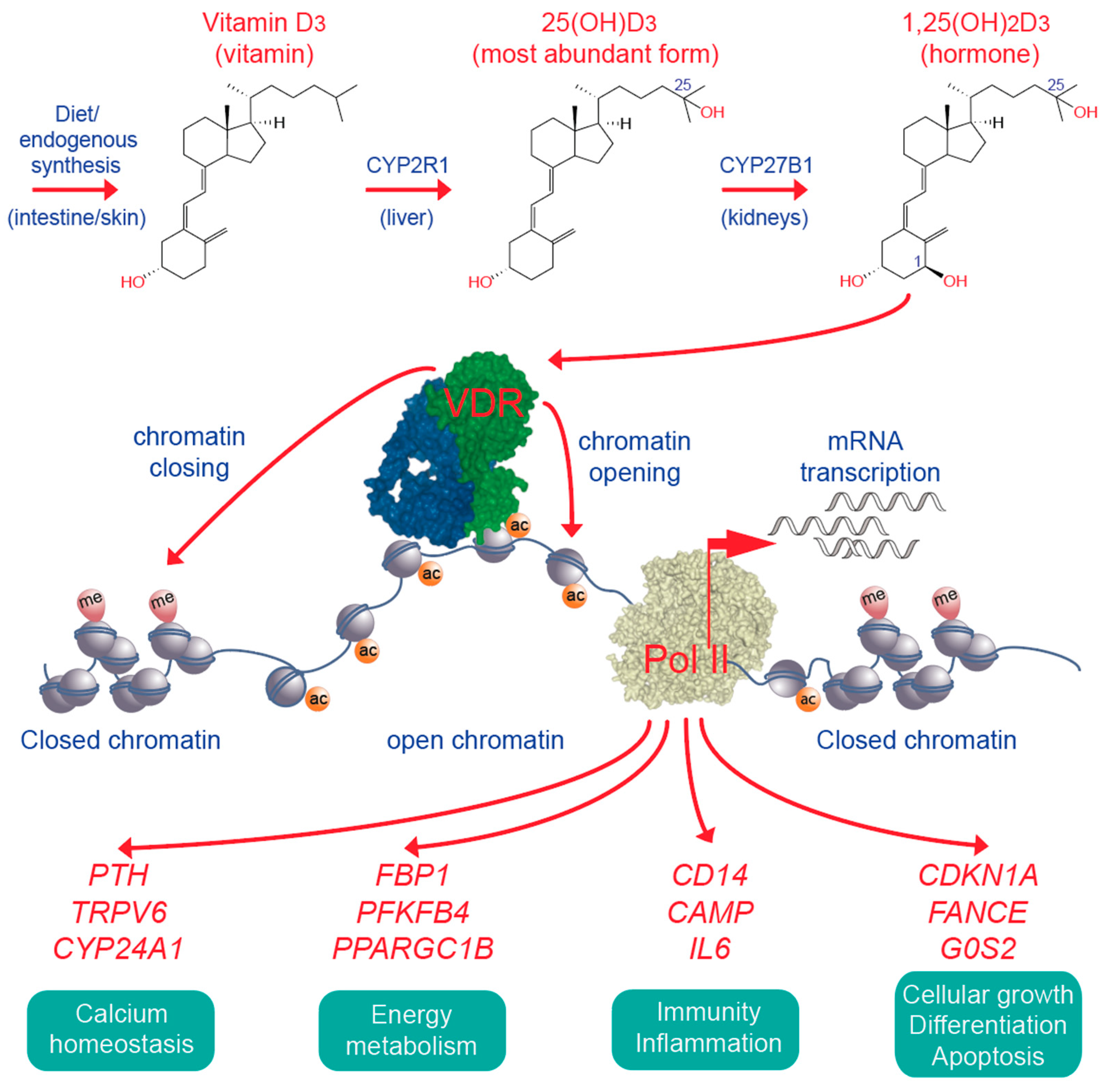
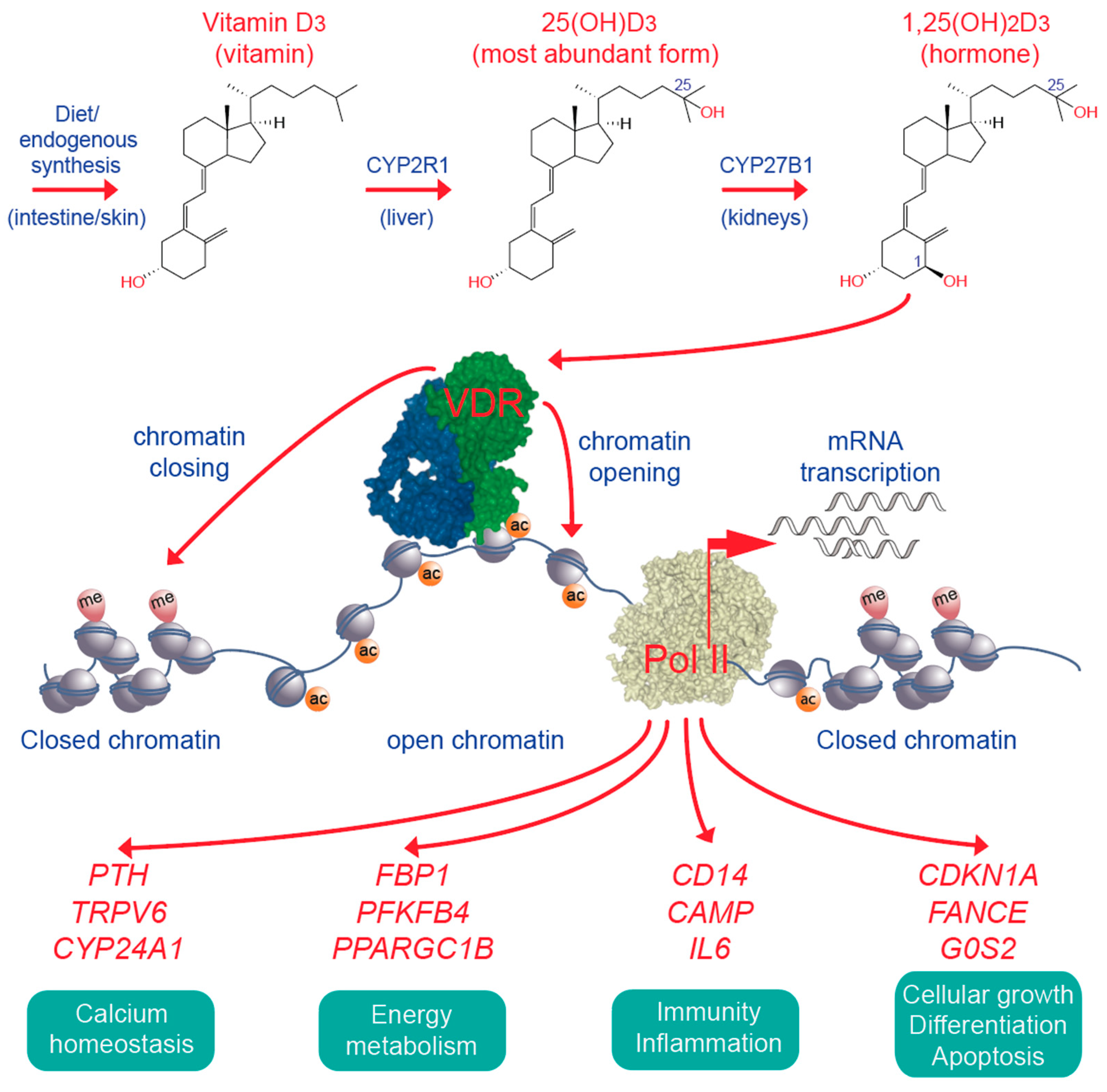
6. Vitamin D, Immunity and Cancer
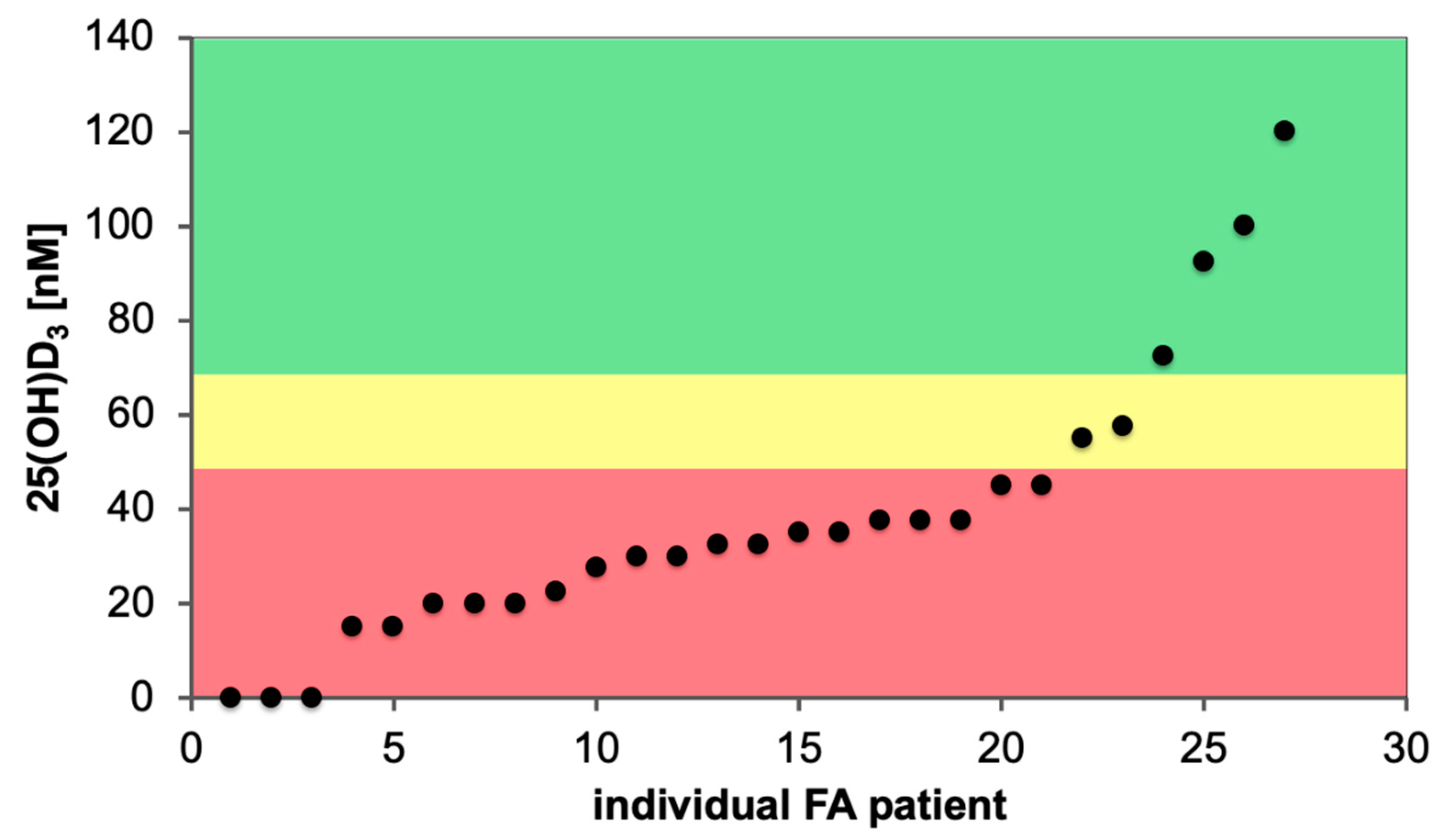
7. Impact of Vitamin D Status and Response Index
8. Limitations and Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alter, B.P. Inherited bone marrow failure syndromes: Considerations pre- and posttransplant. Blood 2017, 130, 2257–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, C. How I manage patients with Fanconi anemia. Br. J. Haematol. 2017, 178, 32–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Wu, C.T.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Rivera, C.M.; Ren, B. Mapping human epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Yadav, T.; Quivy, J.P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Molnár, F. Human Epigenetics: How Science Works; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Carlberg, C.; Ulven, S.M.; Molnár, F. Nutrigenomics: How Science Works; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Liu, L.; Li, Y.; Tollefsbol, T.O. Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 2008, 10, 25–36. [Google Scholar]
- Montgomery, M.; Srinivasan, A. Epigenetic gene regulation by dietary compounds in cancer prevention. Adv. Nutr. 2019, 10, 1012–1028. [Google Scholar] [CrossRef]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 20. [Google Scholar] [CrossRef] [Green Version]
- Brokowski, C.; Adli, M. CRISPR ethics: Moral considerations for applications of a powerful tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef]
- Vears, D.F.; D’Abramo, F. Health, wealth and behavioural change: An exploration of role responsibilities in the wake of epigenetics. J. Community Genet. 2018, 9, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, R.; Velleuer, E. Fanconi anemia: A disease with many faces. Monogr. Hum. Genet. 2007, 15, 9–22. [Google Scholar]
- Poole, S.R.; Smith, A.C.; Hays, T.; McGavran, L.; Auerbach, A.D. Monozygotic twin girls with congenital malformations resembling Fanconi anemia. Am. J. Med. Genet. 1992, 42, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Renaud, E.; Barascu, A.; Rosselli, F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016, 44, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Uncertain verdict as vitamin D goes on trial. Science 2012, 337, 1476–1478. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C. Molecular endocrinology of vitamin D on the epigenome level. Mol. Cell Endocrinol. 2017, 453, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C. Vitamin D genomics: From in vitro to in vivo. Front. Endocrinol. 2018, 9, 250. [Google Scholar] [CrossRef]
- Nurminen, V.; Seuter, S.; Carlberg, C. Primary vitamin D target genes of human monocytes. Front. Physiol. 2019, 10, 194. [Google Scholar] [CrossRef] [Green Version]
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Lobitz, S.; Velleuer, E. Guido Fanconi (18921–979): A jack of all trades. Nat. Rev. Cancer 2006, 6, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A.D. A 20-year perspective on the international Fanconi anemia registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.D.; Hodgson, S.V. Fanconi anemia. J. Med. Genet. 2003, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Karalis, A.; Tischkowitz, M.; Millington, G.W. Dermatological manifestations of inherited cancer syndromes in children. Br. J. Dermatol. 2011, 164, 245–256. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N. Thinking of VACTERL-H? Rule out Fanconi anemia according to PHENOS. Am. J. Med. Genet. A 2016, 170, 1520–1524. [Google Scholar] [CrossRef]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, E. Improving survival for Fanconi anemia patients. Blood 2015, 125, 3676. [Google Scholar] [CrossRef] [PubMed]
- Bonfim, C.; Ribeiro, L.; Nichele, S.; Bitencourt, M.; Loth, G.; Koliski, A.; Funke, V.A.M.; Pilonetto, D.V.; Pereira, N.F.; Flowers, M.E.D.; et al. Long-term survival, organ function, and malignancy after hematopoietic stem cell tansplantation for Fanconi anemia. Biol. Blood Marrow. Transplant. 2016, 22, 1257–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMillan, M.L.; DeFor, T.E.; Young, J.A.; Dusenbery, K.E.; Blazar, B.R.; Slungaard, A.; Zierhut, H.; Weisdorf, D.J.; Wagner, J.E. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood 2015, 125, 3798–3804. [Google Scholar] [CrossRef] [Green Version]
- Svahn, J.; Bagnasco, F.; Cappelli, E.; Onofrillo, D.; Caruso, S.; Corsolini, F.; De Rocco, D.; Savoia, A.; Longoni, D.; Pillon, M.; et al. Somatic, hematologic phenotype, long-term outcome, and effect of hematopoietic stem cell transplantation. An analysis of 97 Fanconi anemia patients from the Italian national database on behalf of the Marrow Failure Study Group of the AIEOP (Italian association of pediatric hematology-oncology). Am. J. Hematol. 2016, 91, 666–671. [Google Scholar] [PubMed] [Green Version]
- Bierings, M.; Bonfim, C.; Peffault De Latour, R.; Aljurf, M.; Mehta, P.A.; Knol, C.; Boulad, F.; Tbakhi, A.; Esquirol, A.; McQuaker, G.; et al. Transplant results in adults with Fanconi anemia. Br. J. Haematol. 2018, 180, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, W.; Alter, B.P.; Wang, T.; Spellman, S.R.; Haagenson, M.; Yeager, M.; Lee, S.J.; Chanock, S.J.; Savage, S.A.; et al. Chromosomal aberrations and survival after unrelated donor hematopoietic stem cell transplant in patients with Fanconi anemia. Biol. Blood Marrow. Transplant. 2018, 24, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paustian, L.; Chao, M.M.; Hanenberg, H.; Schindler, D.; Neitzel, H.; Kratz, C.P.; Ebell, W. Androgen therapy in Fanconi anemia: A retrospective analysis of 30 years in Germany. Pediatr. Hematol. Oncol. 2016, 33, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Cle, D.V. Treatment of inherited bone marrow failure syndromes beyond transplantation. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.R.; Kim, M.O.; Korbee, L.; Wilson, K.A.; Ris, M.D.; Eyal, O.; Sherafat-Kazemzadeh, R.; Bollepalli, S.; Harris, R.; Jeng, M.R.; et al. Oxandrolone for the treatment of bone marrow failure in Fanconi anemia. Pediatr. Blood Cancer 2014, 61, 11–19. [Google Scholar] [CrossRef]
- Scheckenbach, K.; Morgan, M.; Filger-Brillinger, J.; Sandmann, M.; Strimling, B.; Scheurlen, W.; Schindler, D.; Gobel, U.; Hanenberg, H. Treatment of the bone marrow failure in Fanconi anemia patients with danazol. Blood Cells Mol. Dis. 2012, 48, 128–131. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Benedetti, E.; Deater, M.; Schubert, K.; Major, A.; Pelz, C.; Impey, S.; Marquez-Loza, L.; Rathbun, R.K.; Kato, S.; et al. Oxymetholone therapy of Fanconi anemia suppresses osteopontin transcription and induces hematopoietic stem cell cycling. Stem. Cell Reports 2015, 4, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, I.; Alter, B.P. Androgens and liver tumors: Fanconi’s anemia and non-Fanconi’s conditions. Am. J. Hematol. 2004, 77, 257–267. [Google Scholar] [CrossRef]
- Velleuer, E.; Dietrich, R.; Pomjanski, N.; de Santana Almeida Araujo, I.K.; Silva de Araujo, B.E.; Sroka, I.; Biesterfeld, S.; Bocking, A.; Schramm, M. Diagnostic accuracy of brush biopsy-based cytology for the early detection of oral cancer and precursors in Fanconi anemia. Cancer Cytopathol. 2020. [Google Scholar] [CrossRef]
- Kutler, D.I.; Patel, K.R.; Auerbach, A.D.; Kennedy, J.; Lach, F.P.; Sanborn, E.; Cohen, M.A.; Kuhel, W.I.; Smogorzewska, A. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope 2016, 126, 870–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Kutler, D.I. Why otolaryngologists need to be aware of Fanconi anemia. Otolaryngol. Clin. North. Am. 2013, 46, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, E.; Degan, P.; Dufour, C.; Ravera, S. Aerobic metabolism dysfunction as one of the links between Fanconi anemia—Deficient pathway and the aggressive cell invasion in head and neck cancer cells. Oral. Oncol. 2018, 87, 210–211. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Wreesmann, V.B.; Goberdhan, A.; Ben-Porat, L.; Satagopan, J.; Ngai, I.; Huvos, A.G.; Giampietro, P.; Levran, O.; Pujara, K.; et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J. Natl Cancer Inst. 2003, 95, 1718–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zeeburg, H.J.; Snijders, P.J.; Wu, T.; Gluckman, E.; Soulier, J.; Surralles, J.; Castella, M.; van der Wal, J.E.; Wennerberg, J.; Califano, J.; et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2008, 100, 1649–1653. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Quint, W.G.; de Koning, M.N.; Schiffman, M. Squamous cell carcinomas in patients with Fanconi anemia and dyskeratosis congenita: A search for human papillomavirus. Int. J. Cancer 2013, 133, 1513–1515. [Google Scholar] [CrossRef] [Green Version]
- Toptan, T.; Brusadelli, M.G.; Turpin, B.; Witte, D.P.; Surralles, J.; Velleuer, E.; Schramm, M.; Dietrich, R.; Brakenhoff, R.H.; Moore, P.S.; et al. Limited detection of human polyomaviruses in Fanconi anemia related squamous cell carcinoma. PLoS ONE 2018, 13, e0209235. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Bellani, M.; Liu, Y.; Seidman, M.M. Fanconi anemia: A DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res. Rev. 2017, 33, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Velleuer, E.; Dietrich, R. Fanconi anemia: Young patients at high risk for squamous cell carcinoma. Mol. Cell Pediatr. 2014, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Parodi, A.; Kalli, F.; Svahn, J.; Stroppiana, G.; De Rocco, D.; Terranova, P.; Dufour, C.; Fenoglio, D.; Cappelli, E. Impaired immune response to Candida albicans in cells from Fanconi anemia patients. Cytokine 2015, 73, 203–207. [Google Scholar] [CrossRef]
- Barnum, J.L.; Petryk, A.; Zhang, L.; DeFor, T.E.; Baker, K.S.; Steinberger, J.; Nathan, B.; Wagner, J.E.; MacMillan, M.L. Endocrinopathies, bone health, and insulin resistance in patients with Fanconi anemia after hematopoietic cell transplantation. Biol. Blood Marrow. Transplant. 2016, 22, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Petryk, A.; Kanakatti Shankar, R.; Giri, N.; Hollenberg, A.N.; Rutter, M.M.; Nathan, B.; Lodish, M.; Alter, B.P.; Stratakis, C.A.; Rose, S.R. Endocrine disorders in Fanconi anemia: Recommendations for screening and treatment. J. Clin. Endocrinol. Metab. 2015, 100, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravera, S.; Degan, P.; Sabatini, F.; Columbaro, M.; Dufour, C.; Cappelli, E. Altered lipid metabolism could drive the bone marrow failure in Fanconi anemia. Br. J. Haematol. 2019, 184, 693–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagundzin, D.; Hu, W.F.; Law, H.C.H.; Krieger, K.L.; Qiao, F.; Clement, E.J.; Drincic, A.T.; Nedic, O.; Naldrett, M.J.; Alvarez, S.; et al. Delineating the role of FANCA in glucose-stimulated insulin secretion in beta cells through its protein interactome. PLoS ONE 2019, 14, e0220568. [Google Scholar] [CrossRef]
- Rose, S.R.; Myers, K.C.; Rutter, M.M.; Mueller, R.; Khoury, J.C.; Mehta, P.A.; Harris, R.E.; Davies, S.M. Endocrine phenotype of children and adults with Fanconi anemia. Pediatr. Blood Cancer 2012, 59, 690–696. [Google Scholar] [CrossRef]
- Tsui, V.; Crismani, W. The Fanconi anemia pathway and fertility. Trends Genet. 2019, 35, 199–214. [Google Scholar] [CrossRef]
- Wang, A.T.; Smogorzewska, A. SnapShot: Fanconi anemia and associated proteins. Cell 2015, 160, 354. [Google Scholar] [CrossRef] [Green Version]
- Ameziane, N.; May, P.; Haitjema, A.; van de Vrugt, H.J.; van Rossum-Fikkert, S.E.; Ristic, D.; Williams, G.J.; Balk, J.; Rockx, D.; Li, H.; et al. A novel Fanconi anemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015, 6, 8829. [Google Scholar] [CrossRef]
- Meetei, A.R.; Levitus, M.; Xue, Y.; Medhurst, A.L.; Zwaan, M.; Ling, C.; Rooimans, M.A.; Bier, P.; Hoatlin, M.; Pals, G.; et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004, 36, 1219–1224. [Google Scholar] [CrossRef]
- Joo, W.; Xu, G.; Persky, N.S.; Smogorzewska, A.; Rudge, D.G.; Buzovetsky, O.; Elledge, S.J.; Pavletich, N.P. Structure of the FANCI-FANCD2 complex: Insights into the Fanconi anemia DNA repair pathway. Science 2011, 333, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.J.; Minguillon, J.; Loveless, S.; Lake, K.; Carrasco, E.; Stjepanovic, N.; Balmana, J.; Catala, A.; Mehta, P.A.; Surralles, J. Chromosome fragility in the buccal epithelium in patients with Fanconi anemia. Cancer Lett. 2020, 472, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Hodskinson, M.R.; Bolner, A.; Sato, K.; Kamimae-Lanning, A.N.; Rooijers, K.; Witte, M.; Mahesh, M.; Silhan, J.; Petek, M.; Williams, D.M.; et al. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 2020, 579, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernandez, A.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, A.; Rajendran, A.; Mohan, S.; Raj, R.; Vundinti, B.R. Mitochondrial DNA variations and mitochondrial dysfunction in Fanconi anemia. PLoS ONE 2020, 15, e0227603. [Google Scholar] [CrossRef]
- Chatla, S.; Du, W.; Wilson, A.F.; Meetei, A.R.; Pang, Q. Fancd2-deficient hematopoietic stem and progenitor cells depend on augmented mitochondrial translation for survival and proliferation. Stem Cell Res. 2019, 40, 101550. [Google Scholar] [CrossRef]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef]
- Ravera, S.; Dufour, C.; Degan, P.; Cappelli, E. Fanconi anemia: From DNA repair to metabolism. Eur. J. Hum. Genet. 2018, 26, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Garbati, M.R.; Hays, L.E.; Rathbun, R.K.; Jillette, N.; Chin, K.; Al-Dhalimy, M.; Agarwal, A.; Newell, A.E.; Olson, S.B.; Bagby, G.C., Jr. Cytokine overproduction and crosslinker hypersensitivity are unlinked in Fanconi anemia macrophages. J. Leukoc. Biol. 2016, 99, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnan, I.; Gunel-Ozcan, A.; Aerts-Kaya, F.; Ameziane, N.; Kuskonmaz, B.; Dorsman, J.; Gumruk, F.; Uckan, D. Bone marrow mesenchymal stem cells carrying FANCD2 mutation differ from the other Fanconi anemia complementation groups in terms of TGF-beta1 production. Stem. Cell Rev. Rep. 2018, 14, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.S.; Taniguchi, T. Recent insights into the molecular basis of Fanconi anemia: Genes, modifiers, and drivers. Int. J. Hematol. 2017, 106, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Del Valle, J.; Rofes, P.; Moreno-Cabrera, J.M.; Lopez-Doriga, A.; Belhadj, S.; Vargas-Parra, G.; Teule, A.; Cuesta, R.; Munoz, X.; Campos, O.; et al. Exploring the role of mutations in Fanconi anemia genes in hereditary cancer patients. Cancers 2020, 12, 829. [Google Scholar] [CrossRef] [Green Version]
- Pouliot, G.P.; Degar, J.; Hinze, L.; Kochupurakkal, B.; Vo, C.D.; Burns, M.A.; Moreau, L.; Ganesa, C.; Roderick, J.; Peirs, S.; et al. Fanconi-BRCA pathway mutations in childhood T-cell acute lymphoblastic leukemia. PLoS ONE 2019, 14, e0221288. [Google Scholar] [CrossRef]
- Masserot, C.; Peffault de Latour, R.; Rocha, V.; Leblanc, T.; Rigolet, A.; Pascal, F.; Janin, A.; Soulier, J.; Gluckman, E.; Socie, G. Head and neck squamous cell carcinoma in 13 patients with Fanconi anemia after hematopoietic stem cell transplantation. Cancer 2008, 113, 3315–3322. [Google Scholar] [CrossRef]
- Rosenberg, P.S.; Socie, G.; Alter, B.P.; Gluckman, E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood 2005, 105, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Neitzel, H.; Tonnies, H. Chromosomal aberrations associated with clonal evolution and leukemic transformation in fanconi anemia: Clinical and biological implications. Anemia 2012, 2012, 349837. [Google Scholar] [CrossRef]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pages, M.P.; Vasquez, N.; Dubois d’Enghien, C.; Larghero, J.; Peffault de Latour, R.; et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, e161–e170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnies, H.; Huber, S.; Kuhl, J.S.; Gerlach, A.; Ebell, W.; Neitzel, H. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: Gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood 2003, 101, 3872–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.J.; Unwin, R.D.; Bindels, E.; Pierce, A.; Teng, H.Y.; Muter, J.; Greystoke, B.; Somerville, T.D.; Griffiths, J.; Lovell, S.; et al. Phosphorylation of the leukemic oncoprotein EVI1 on serine 196 modulates DNA binding, transcriptional repression and transforming ability. PLoS ONE 2013, 8, e66510. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Riboli, E. The EPIC study: An update. Recent Results Cancer Res. 2009, 181, 63–70. [Google Scholar]
- Gatherer, D. A stroll across the epigenetic landscape: Bringing Waddington’s ideas into molecular biology. Early Pregnancy 1996, 2, 241–243. [Google Scholar]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Neme, A.; Seuter, S.; Carlberg, C. Selective regulation of biological processes by vitamin D based on the spatio-temporal cistrome of its receptor. Biochim. Biophys. Acta 2017, 1860, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Seuter, S.; Neme, A.; Carlberg, C. Epigenomic PU.1-VDR crosstalk modulates vitamin D signaling. Biochim. Biophys. Acta 2017, 1860, 405–415. [Google Scholar] [CrossRef]
- Nurminen, V.; Neme, A.; Seuter, S.; Carlberg, C. Modulation of vitamin D signaling by the pioneer factor CEBPA. Biochim. Biophys. Acta 2019, 1862, 96–106. [Google Scholar] [CrossRef]
- Nurminen, V.; Neme, A.; Seuter, S.; Carlberg, C. The impact of the vitamin D-modulated epigenome on VDR target gene regulation. Biochim. Biophys. Acta 2018, 1861, 697–705. [Google Scholar] [CrossRef]
- Seuter, S.; Neme, A.; Carlberg, C. Epigenome-wide effects of vitamin D and their impact on the transcriptome of human monocytes involve CTCF. Nucleic Acids Res. 2016, 44, 4090–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- ENCODE-Project-Consortium; Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Swuec, P.; Renault, L.; Borg, A.; Shah, F.; Murphy, V.J.; van Twest, S.; Snijders, A.P.; Deans, A.J.; Costa, A. The FA core complex contains a homo-dimeric catalytic module for the symmetric mono-ubiquitination of FANCI-FANCD2. Cell Rep. 2017, 18, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Alegria-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F.; Frommer, J.E.; McNeill, S.C.; Richtand, N.M.; Henley, J.W.; Potts, J.T., Jr. Photometabolism of 7-dehydrocholesterol to previtamin D3 in skin. Biochem. Biophys. Res. Commun. 1977, 76, 107–114. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A critical and essential micronutrient for human health. Front. Physiol. 2014, 5, 248. [Google Scholar] [CrossRef]
- Zerwekh, J.E. Blood biomarkers of vitamin D status. Am. J. Clin. Nutr. 2008, 87, 1087S–1091S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Medicine. Dietary Reference Intakes for Calcium and Vitamin D; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Heaney, R.P.; Holick, M.F. Why the IOM recommendations for vitamin D are deficient. J. Bone Miner. Res. 2011, 26, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Dawson-Hughes, B.; Heaney, R.P.; Holick, M.F.; Lips, P.; Meunier, P.J.; Vieth, R. Estimates of optimal vitamin D status. Osteoporos. Int. 2005, 16, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)2vitamin D3: Genomic and non-genomic mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef]
- Evans, R.M. The nuclear receptor superfamily: A Rosetta stone for physiology. Mol. Endocrinol. 2005, 19, 1429–1438. [Google Scholar] [CrossRef]
- Haussler, M.R.; Haussler, C.A.; Jurutka, P.W.; Thompson, P.D.; Hsieh, J.C.; Remus, L.S.; Selznick, S.H.; Whitfield, G.K. The vitamin D hormone and its nuclear receptor: Molecular actions and disease states. J. Endocrinol 1997, 154, S57–S73. [Google Scholar]
- Whitfield, G.K.; Dang, H.T.; Schluter, S.F.; Bernstein, R.M.; Bunag, T.; Manzon, L.A.; Hsieh, G.; Dominguez, C.E.; Youson, J.H.; Haussler, M.R.; et al. Cloning of a functional vitamin D receptor from the lamprey (Petromyzon marinus), an ancient vertebrate lacking a calcified skeleton and teeth. Endocrinology 2003, 144, 2704–2716. [Google Scholar] [CrossRef] [Green Version]
- Hanel, A.; Carlberg, C. Vitamin D and evolution: Pharmacologic implications. Biochem. Pharmacol. 2020, 173, 113595. [Google Scholar] [CrossRef]
- Escriva, H.; Bertrand, S.; Laudet, V. The evolution of the nuclear receptor superfamily. Essays Biochem. 2004, 40, 11–26. [Google Scholar]
- Krasowski, M.D.; Ni, A.; Hagey, L.R.; Ekins, S. Evolution of promiscuous nuclear hormone receptors: LXR, FXR, VDR, PXR, and CAR. Mol. Cell Endocrinol. 2011, 334, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Muller, V.; de Boer, R.J.; Bonhoeffer, S.; Szathmary, E. An evolutionary perspective on the systems of adaptive immunity. Biol. Rev. Camb. Philos. Soc. 2018, 93, 505–528. [Google Scholar] [CrossRef] [Green Version]
- Vanherwegen, A.S.; Gysemans, C.; Mathieu, C. Vitamin D endocrinology on the cross-road between immunity and metabolism. Mol. Cell Endocrinol. 2017, 453, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Chen, M.J.; Stachura, D.L.; Liu, S.Y.; Kwan, W.; Wright, F.; Vo, L.T.; Theodore, L.N.; Esain, V.; Frost, I.M.; et al. Developmental vitamin D availability impacts hematopoietic stem cell production. Cell Rep. 2016, 17, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R.; Suda, T. Vitamin D: Calcium and bone homeostasis during evolution. BoneKEy Rep. 2014, 3, 480. [Google Scholar] [CrossRef]
- Veldurthy, V.; Wei, R.; Oz, L.; Dhawan, P.; Jeon, Y.H.; Christakos, S. Vitamin D, calcium homeostasis and aging. Bone Res. 2016, 4, 16041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F. Resurrection of vitamin D deficiency and rickets. J. Clin. Investig. 2006, 116, 2062–2072. [Google Scholar] [CrossRef] [Green Version]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; McComish, B.J.; Burdon, K.P.; Taylor, B.V.; Körner, H. The association between vitamin D and multiple sclerosis risk: 1,25(OH)2D3 induces super-enhancers bound by VDR. Front. Immunol. 2019, 10, 488. [Google Scholar] [CrossRef]
- Dankers, W.; Colin, E.M.; van Hamburg, J.P.; Lubberts, E. Vitamin D in autoimmunity: Molecular mechanisms and therapeutic potential. Front. Immunol 2016, 7, 697. [Google Scholar] [CrossRef] [Green Version]
- Hart, P.H.; Gorman, S.; Finlay-Jones, J.J. Modulation of the immune system by UV radiation: More than just the effects of vitamin D? Nat. Rev. Immunol. 2011, 11, 584–596. [Google Scholar] [CrossRef]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Saramäki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21waf1/cip1 gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Lee, M.-H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros-Soto, J.; Anthias, C.; Madrigal, A.; Snowden, J.A. Vitamin D: Is it important in haematopoietic stem cell transplantation? A review. Bone Marrow Transplant. 2019, 54, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D receptor controls cell stemness in acute myeloid leukemia and in normal bone marrow. Cell Rep. 2020, 30, 739–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Hu, W.; Lu, L.; Zhao, Y.; Zhou, Y.; Xiao, Z.; Zhang, L.; Zhang, H.; Li, X.; Li, W.; et al. Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm. Sin. B 2019, 9, 203–219. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Karkeni, E.; Morin, S.O.; Bou Tayeh, B.; Goubard, A.; Josselin, E.; Castellano, R.; Fauriat, C.; Guittard, G.; Olive, D.; Nunes, J.A. Vitamin D controls tumor growth and CD8+ T cell infiltration in breast cancer. Front. Immunol. 2019, 10, 1307. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, C.M.; Kazantzidis, A.; Ryan, M.J.; Barber, N.; Sempos, C.T.; Durazo-Arvizu, R.A.; Jorde, R.; Grimnes, G.; Eiriksdottir, G.; Gudnason, V.; et al. Seasonal changes in vitamin D-effective UVB availability in Europe and associations with population serum 25-hydroxyvitamin D. Nutrients 2016, 8, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M.; Endocrine, S. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, G.; He, X.; Gao, J.; Wang, R.; Wang, Y.; Zhao, W. Serum 25-hydroxyvitamin D levels and prognosis in hematological malignancies: A systematic review and meta-analysis. Cell Physiol. Biochem. 2015, 35, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Lauter, B.; Schmidt-Wolf, I.G. Prevalence, supplementation, and impact of vitamin D deficiency in multiple myeloma patients. Cancer Investig. 2015, 33, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Pezeshki, S.M.S.; Asnafi, A.A.; Khosravi, A.; Shahjahani, M.; Azizidoost, S.; Shahrabi, S. Vitamin D and its receptor polymorphisms: New possible prognostic biomarkers in leukemias. Oncol. Rev. 2018, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Verma, N.; Kumar, A. Prevalence of vitamin D deficiency in childhood acute lymphoblastic leukemia and its association with adverse outcomes during induction phase of treatment. Nutr. Cancer 2020. [Google Scholar] [CrossRef]
- Lips, P.; Cashman, K.D.; Lamberg-Allardt, C.; Bischoff-Ferrari, H.A.; Obermayer-Pietsch, B.; Bianchi, M.L.; Stepan, J.; El-Hajj Fuleihan, G.; Bouillon, R. Current vitamin D status in European and Middle East countries and strategies to prevent vitamin D deficiency: A position statement of the European calcified tissue society. Eur. J. Endocrinol. 2019, 180, P23–P54. [Google Scholar] [CrossRef] [Green Version]
- Laviano, E.; Sanchez Rubio, M.; Gonzalez-Nicolas, M.T.; Palacian, M.P.; Lopez, J.; Gilaberte, Y.; Calmarza, P.; Rezusta, A.; Serrablo, A. Association between preoperative levels of 25-hydroxyvitamin D and hospital-acquired infections after hepatobiliary surgery: A prospective study in a third-level hospital. PLoS ONE 2020, 15, e0230336. [Google Scholar] [CrossRef]
- Carlberg, C.; Haq, A. The concept of the personal vitamin D response index. J. Steroid Biochem. Mol. Biol. 2018, 175, 12–17. [Google Scholar] [CrossRef]
- Neme, A.; Seuter, S.; Malinen, M.; Nurmi, T.; Tuomainen, T.P.; Virtanen, J.K.; Carlberg, C. In vivo transcriptome changes of human white blood cells in response to vitamin D. J. Steroid Biochem. Mol. Biol. 2019, 188, 71–76. [Google Scholar] [CrossRef]
- Mangin, M.; Sinha, R.; Fincher, K. Inflammation and vitamin D: The infection connection. Inflamm. Res. 2014, 63, 803–819. [Google Scholar] [CrossRef] [Green Version]
- Salzer, J.; Hallmans, G.; Nystrom, M.; Stenlund, H.; Wadell, G.; Sundstrom, P. Vitamin D as a protective factor in multiple sclerosis. Neurology 2012, 79, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Seuter, S.; de Mello, V.D.; Schwab, U.; Voutilainen, S.; Pulkki, K.; Nurmi, T.; Virtanen, J.; Tuomainen, T.P.; Uusitupa, M. Primary vitamin D target genes allow a categorization of possible benefits of vitamin D3 supplementation. PLoS ONE 2013, 8, e71042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seuter, S.; Virtanen, J.K.; Nurmi, T.; Pihlajamaki, J.; Mursu, J.; Voutilainen, S.; Tuomainen, T.P.; Neme, A.; Carlberg, C. Molecular evaluation of vitamin D responsiveness of healthy young adults. J. Steroid Biochem. Mol. Biol. 2017, 174, 314–321. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velleuer, E.; Carlberg, C. Impact of Epigenetics on Complications of Fanconi Anemia: The Role of Vitamin D-Modulated Immunity. Nutrients 2020, 12, 1355. https://doi.org/10.3390/nu12051355
Velleuer E, Carlberg C. Impact of Epigenetics on Complications of Fanconi Anemia: The Role of Vitamin D-Modulated Immunity. Nutrients. 2020; 12(5):1355. https://doi.org/10.3390/nu12051355
Chicago/Turabian StyleVelleuer, Eunike, and Carsten Carlberg. 2020. "Impact of Epigenetics on Complications of Fanconi Anemia: The Role of Vitamin D-Modulated Immunity" Nutrients 12, no. 5: 1355. https://doi.org/10.3390/nu12051355
APA StyleVelleuer, E., & Carlberg, C. (2020). Impact of Epigenetics on Complications of Fanconi Anemia: The Role of Vitamin D-Modulated Immunity. Nutrients, 12(5), 1355. https://doi.org/10.3390/nu12051355