Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages

 ,
,  and
and
Abstract
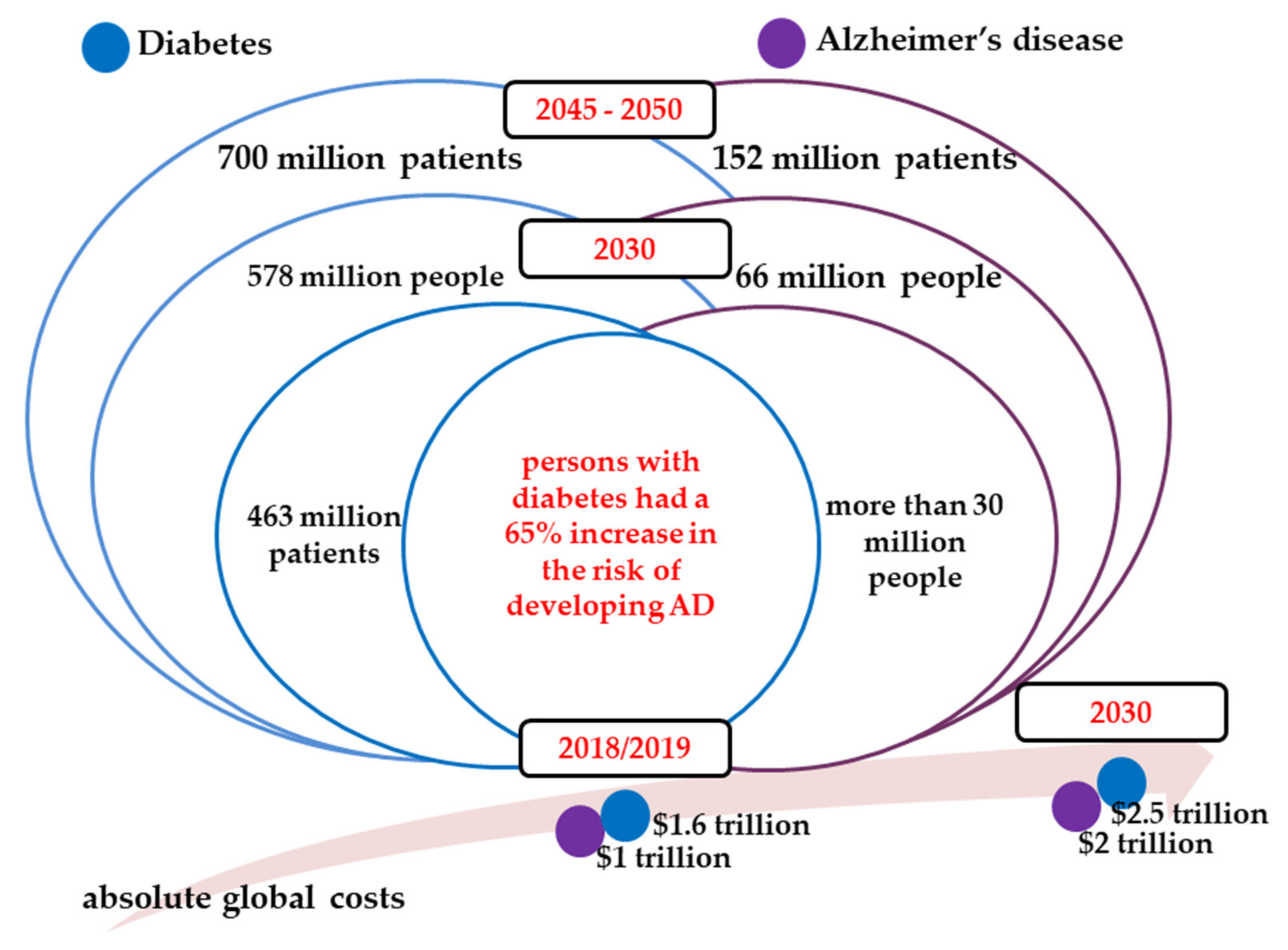
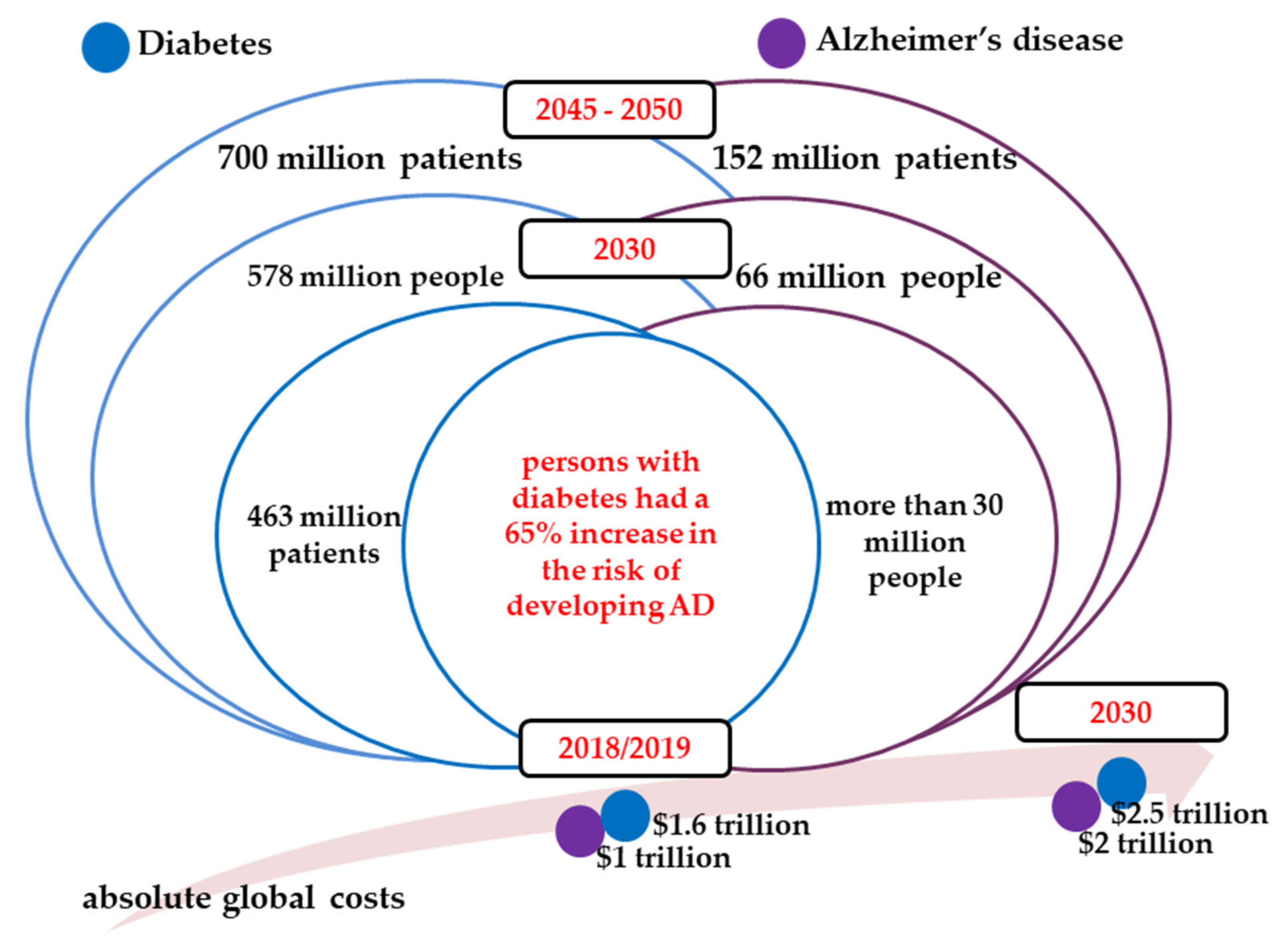
:1. Socio-Economic Burden of Diabetes and Alzheimer’s Disease
2. Amyloid Formation as a Common Pathological Feature in both Diabetes and Alzheimer’s Disease
2.1. Relations between Diabetes and Alzheimer’s Disease
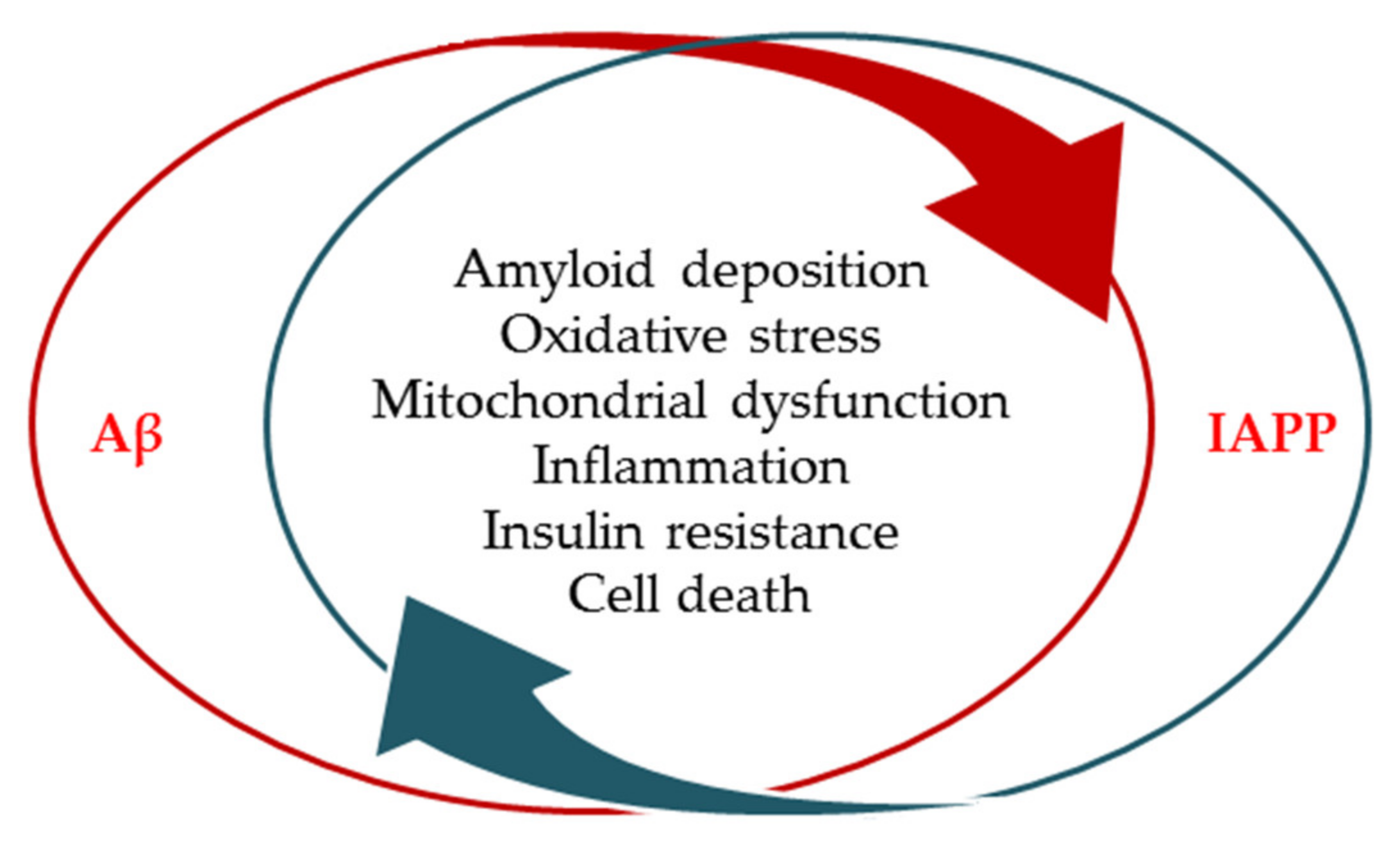
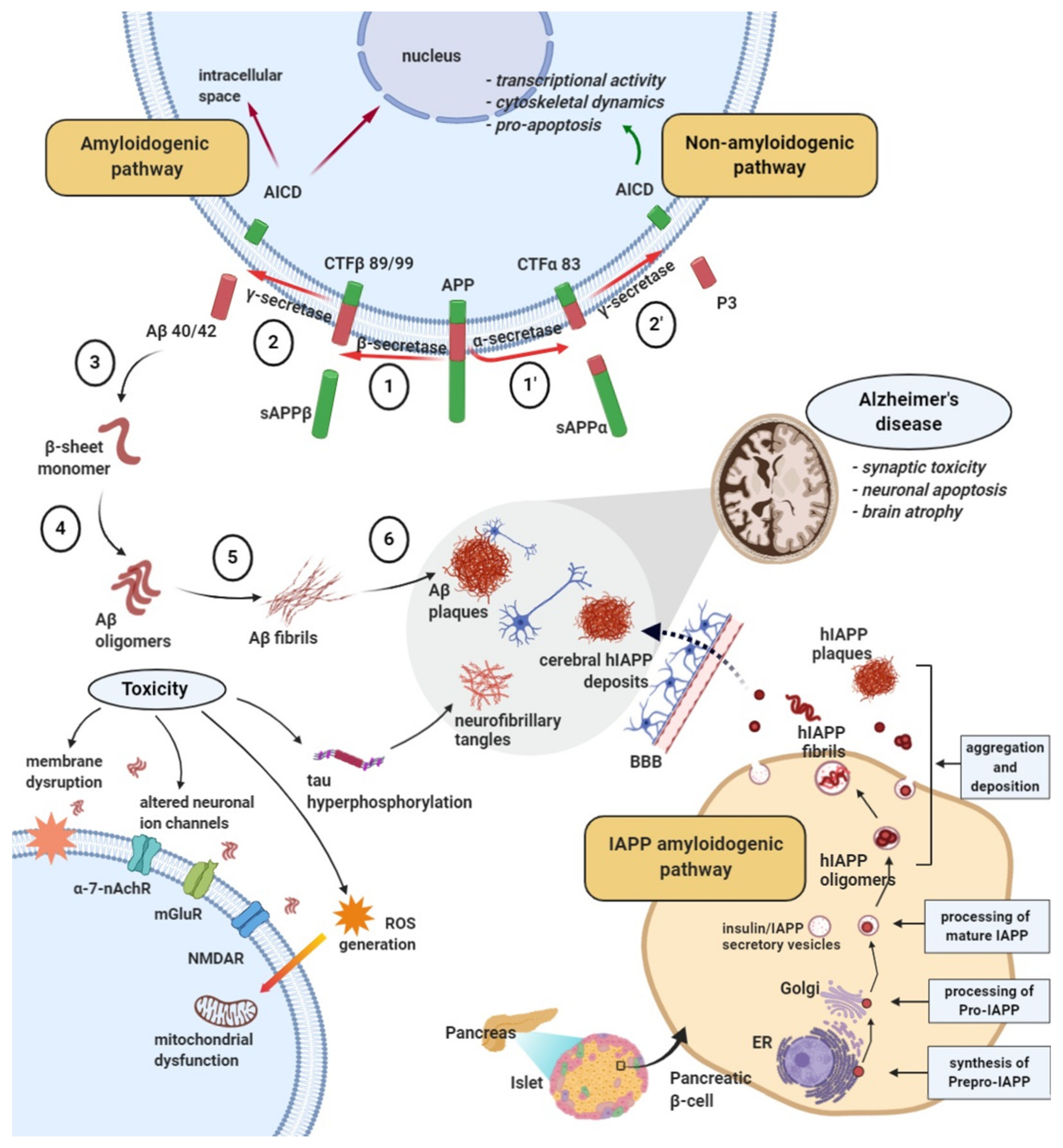
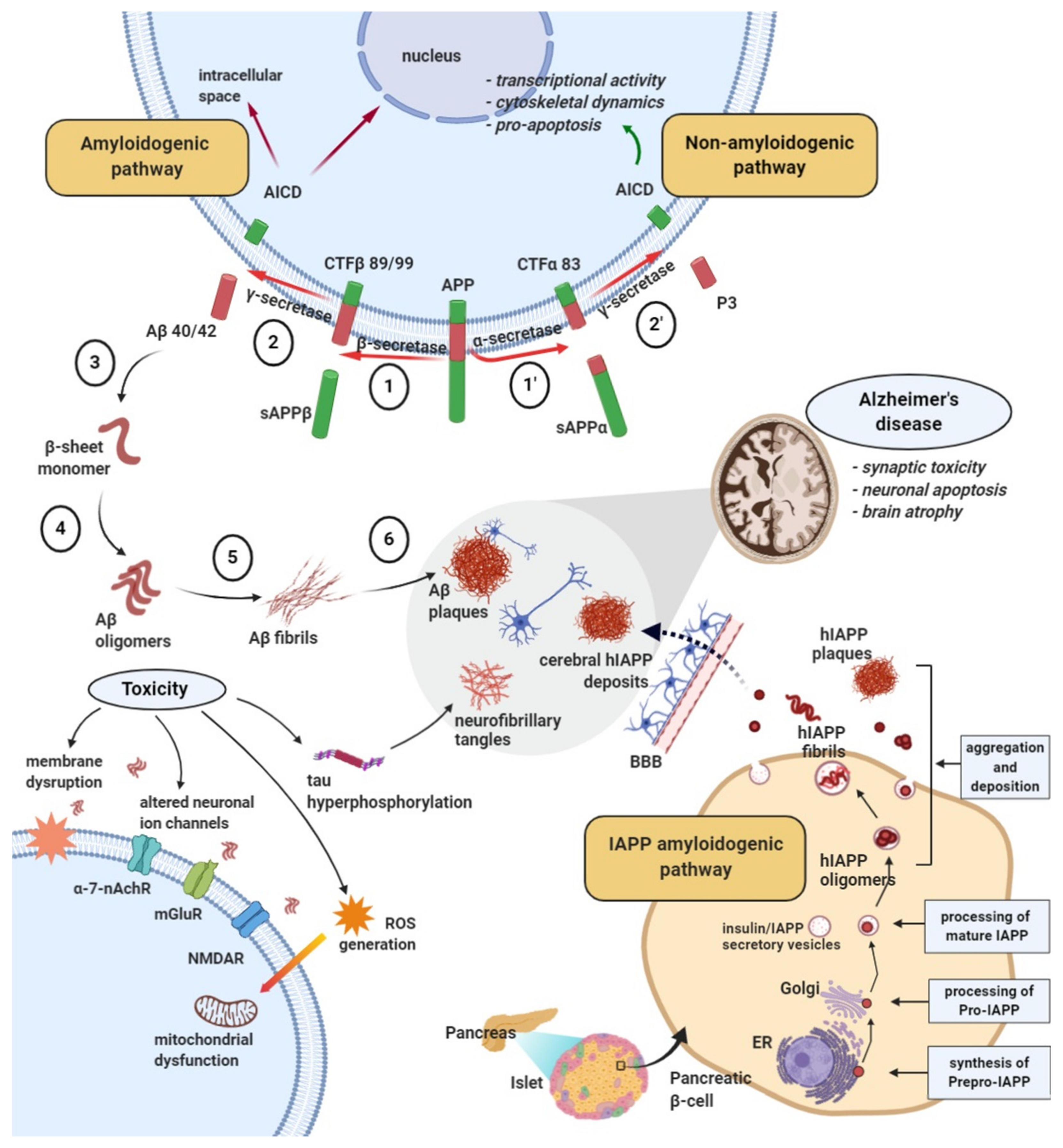
2.2. Amyloid Formation and Deposition Involving both Neurodegenerative Changes and Neurovascular Damage
- ▪
- Misplaced mutations of the APP, presenilin−1 (PSEN−1), and or presenilin−2 (PSEN−2) genes that may result in increased production of Aβ42 peptides throughout life in the dominant forms of AD or,
- ▪
- by impairing the Aβ peptide purification mechanisms that would favor the gradual increase of the Aβ42 peptide level in the brain in the case of non-dominant forms of AD.
2.3. Evidence from the Shared Pathological Traits
3. The Influence of Amyloid-β Aggregates on Diabetes Pathology and Islet Amyloid Polypeptide on Alzheimer’s Disease in Animal Models
4. Relevance of Molecular Interaction between Islet Amyloid Polypeptide and Amyloid-β Peptide for Novel Therapeutics
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Magliano, D.J.; Islam, R.M.; Barr, E.L.M.; Gregg, E.W.; Pavkov, M.E.; Harding, J.L.; Tabesh, M.; Koye, D.N.; Shaw, J.E. Trends in incidence of total or type 2 diabetes: Systematic review. BMJ 2019, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, E.W.; Li, Y.; Wang, J.; Rios-Burrows, N.; Ali, M.K.; Rolka, D.; Williams, D.E.; Geiss, L. Changes in diabetes-related complications in the United States, 1990–2010. N. Engl. J. Med. 2014, 370, 1514–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the international diabetes federation diabetes atlas, 9th edition. Diabetes Res. Clin. Pr. 2019, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommer, C.; Sagalova, V.; Heesemann, E.; Manne-Goehler, J.; Atun, R.; Bärnighausen, T.; Davies, J.; Vollmer, S. Global economic burden of diabetes in adults: Projections from 2015 to 2030. Diabetes Care 2018, 41, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. World Alzheimer Report 2018-The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Li, L.; Hölscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.; Guimaraes, I.; Silva, F.; Ribeiro, F. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Ristow, M. Neurodegenetive disorders associated with diabetes mellitus. J. Mol. Med. 2004, 82, 510–529. [Google Scholar] [CrossRef]
- Haan, M.N. Therapy insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pr. Neurol. 2006, 2, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-β peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Roberts, R.O.; Knopman, D.S.; Przybelski, S.A.; Mielke, M.M.; Kantarci, K.; Preboske, G.M.; Senjem, M.L.; Pankratz, V.S.; Geda, Y.E.; Boeve, B.F.; et al. Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology 2014, 82, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef]
- Xu, W.L.; Von Strauss, E.; Qiu, C.X.; Winblad, B.; Fratiglioni, L. Uncontrolled diabetes increases the risk of Alzheimer’s disease: A population-based cohort study. Diabetologia 2009, 52, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.-C.; Woung, L.-C.; Tsai, M.-T.; Liu, C.-C.; Su, Y.-H.; Li, C.-Y. Risk of Alzheimer’s disease in relation to diabetes: A population-based cohort study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef]
- Rowley, W.R.; Bezold, C.; Arikan, Y.; Byrne, E.; Krohe, S. Diabetes 2030: Insights from yesterday, today, and future trends. Popul. Health Manag. 2017, 20, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; De La Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Hauser, C.A.E.; Maurer-Stroh, S.; Martins, I.C. Amyloid-based nanosensors and nanodevices. Chem. Soc. Rev. 2014, 43, 5326–5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimundo, A.F.; Ferreira, S.; Martins, I.C.; Menezes, R. Islet amyloid polypeptide: A partner in crime with Aβ in the pathology of Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Wineman-Fisher, V.; Bloch, D.N.; Miller, Y. Challenges in studying the structures of metal-amyloid oligomers related to type 2 diabetes, Parkinson’s disease, and Alzheimer’s disease. Coord. Chem. Rev. 2016, 327, 20–26. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Y.; Zhang, S.; Song, W. High glucose promotes Aβ production by inhibiting APP degradation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Song, W. Islet amyloid polypeptide: Another key molecule in Alzheimer’s pathogenesis? Prog. Neurobiol. 2017, 153, 100–120. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [Green Version]
- Nishi, M.; Sanke, T.; Nagamatsu, S.; Bell, G.I.; Steiner, D.F. Islet amyloid polypeptide. A new β cell secretory product related to islet amyloid deposits. J. Biol. Chem. 1990, 265, 4173–4176. [Google Scholar]
- Asthana, S.; Mallick, B.; Alexandrescu, A.T.; Jha, S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1765–1782. [Google Scholar] [CrossRef]
- Cao, P.; Marek, P.; Noor, H.; Patsalo, V.; Tu, L.H.; Wang, H.; Abedini, A.; Raleigh, D.P. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013, 587, 1106–1118. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, E.; Ahmad, A.; Singh, S.; Arshad, M.; Khan, A.H.; Khan, R.H. A mechanistic approach for islet amyloid polypeptide aggregation to develop anti-amyloidogenic agents for type-2 diabetes. Biochimie 2011, 93, 793–805. [Google Scholar] [CrossRef]
- Bailey, J.; Potter, K.J.; Verchere, C.B.; Edelstein-Keshet, L.; Coombs, D. Reverse engineering an amyloid aggregation pathway with dimensional analysis and scaling. Phys. Biol. 2011, 8, 66009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 7283–7288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ştefănescu, R.; Stanciu, G.D.; Luca, A.; Caba, I.C.; Tamba, B.I.; Mihai, C.T. Contributions of mass spectrometry to the identification of low molecular weight molecules able to reduce the toxicity of amyloid-β peptide to cell cultures and transgenic mouse models of Alzheimer’s disease. Molecules 2019, 24, 1167. [Google Scholar] [CrossRef] [Green Version]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Morsy, A.; Trippier, P.C. Amyloid-binding alcohol dehydrogenase (ABAD) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2019, 62, 4252–4264. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement. 2020. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, W.; Tong, Y.; He, G.; Song, W. Control of APP processing and Aβ generation level by BACE1 enzymatic activity and transcription. FASEB J. 2006, 20, 285–292. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the amyloid hypothesis in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 68, 493–510. [Google Scholar] [CrossRef]
- Hillen, H. The beta amyloid dysfunction (BAD) hypothesis for Alzheimer’s disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef] [Green Version]
- Manschot, S.M.; Brands, A.M.A.; Van Der Grond, J.; Kessels, R.P.C.; Algra, A.; Kappelle, L.J.; Biessels, G.J. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 2006, 55, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef]
- Maloy, A.L.; Longnecker, D.S.; Greenberg, R.E. The relation of islet amyloid to the clinical type of diabetes. Hum. Pathol. 1981, 12, 917–922. [Google Scholar] [CrossRef]
- Zhao, H.L.; Lai, F.M.M.; Tong, P.C.Y.; Zhong, D.R.; Yang, D.; Tomlinson, B.; Chan, J.C.N. Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes 2003, 52, 2759–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanciu, G.D.; Musteaţă, M.; Armaşu, M.; Solcan, G. Evaluation of central vestibular syndrome in dogs using brainstem auditory evoked responses recorded with surface electrodes. Arq. Bras. Med. Vet. e Zootec. 2016, 68. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisternas, P.; Inestrosa, N.C. Brain glucose metabolism: Role of Wnt signaling in the metabolic impairment in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2017, 80, 316–328. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Aliev, G.; Priyadarshini, M.; Reddy, V.; Grieg, N.H.; Kaminsky, Y.; Cacabelos, R.; Ashraf, G.; Jabir, N.R.; Kamal, M.A.; Nikolenko, V.N.; et al. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014, 21, 2208–2217. [Google Scholar] [CrossRef]
- Boccardi, V.; Murasecco, I.; Mecocci, P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res. Rev. 2019, 54, 100936. [Google Scholar] [CrossRef] [PubMed]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Ashby, E.L.; Miners, J.S.; Kehoe, P.G.; Love, S. Effects of hypertension and anti-hypertensive treatment on amyloid-β (Aβ) plaque load and Aβ-synthesizing and Aβ-degrading enzymes in frontal cortex. J. Alzheimer’s Dis. 2016, 50, 1191–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saedi, E.; Gheini, M.R.; Faiz, F.; Arami, M.A. Diabetes mellitus and cognitive impairments. World J. Diabetes 2016, 7, 412. [Google Scholar] [CrossRef]
- Kim, H.-G. Cognitive dysfunctions in individuals with diabetes mellitus. Yeungnam Univ. J. Med. 2019, 36, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Le Roith, D.; Zick, Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001, 24, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Sankar, S.B.; Infante-Garcia, C.; Weinstock, L.D.; Ramos-Rodriguez, J.J.; Hierro-Bujalance, C.; Fernandez-Ponce, C.; Wood, L.B.; Garcia-Alloza, M. Amyloid beta and diabetic pathology cooperatively stimulate cytokine expression in an Alzheimer’s mouse model. J. Neuroinflammation 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Cukierman-Yaffe, T.; Gerstein, H.C.; Williamson, J.D.; Lazar, R.M.; Lovato, L.; Miller, M.E.; Coker, L.H.; Murray, A.; Sullivan, M.D.; Marcovina, S.M.; et al. Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular rIsk factors the action to control cardiovascular risk in diabetes-memory in diabetes (ACCORD-MIND) trial. Diabetes Care 2009, 32, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Bucala, R.; Striker, L. Pathogenic effects of advanced glycosylation: Biochemical, biologic, and clinical implications for diabetes and aging. Lab. Invest. 1994, 70, 138–151. [Google Scholar]
- Serban, D.; Anton, E.; Chirita, R.; Bild, V.; Ciobica, A.; Alexinschi, O.; Arcan, O.; Popescu, R.; Paduraru, L.; Timofte, D. Current aspects of the interactions between dementia, the brain renin-angiotensin system and oxidative stress. Arch. Biol. Sci. 2015, 67, 903–907. [Google Scholar] [CrossRef]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Differential effects of glycation on protein aggregation and amyloid formation. Front. Mol. Biosci. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Role of glycation in amyloid: Effect on the aggregation process and cytotoxicity. In Exploring New Findings on Amyloidosis; InTech: London, UK, 2016. [Google Scholar]
- Kapurniotu, A.; Bernhagen, J.; Greenfield, N.; Al-Abed, Y.; Teichberg, S.; Frank, R.W.; Voelter, W.; Bucala, R. Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur. J. Biochem. 1998, 251, 208–216. [Google Scholar] [CrossRef]
- Chen, K.; Maley, J.; Yu, P.H. Potential implications of endogenous aldehydes in?-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Carafa, V.; Altucci, L.; Vitiello, M.; Balestrieri, M.L.; Ricci, G.; Irace, G.; Sirangelo, I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The Rotterdam study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Akomolafe, A.; Beiser, A.; Meigs, J.B.; Au, R.; Green, R.C.; Farrer, L.A.; Wolf, P.A.; Seshadri, S. Diabetes mellitus and risk of developing Alzheimer disease: Results from the Framingham study. Arch. Neurol. 2006, 63, 1551–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Matsubara, T.; Sobue, K.; Tanida, M.; Kasahara, R.; Naruse, K.; Taniura, H.; Sato, T.; Suzuki, K. Brain insulin resistance accelerates Aβ fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 2012, 121, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Bourdel-Marchasson, I.; Lapre, E.; Laksir, H.; Puget, E. Insulin resistance, diabetes and cognitive function: Consequences for preventative strategies. Diabetes Metab. 2010, 36, 173–181. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Haj-ali, V.; Mohaddes, G.; Babri, S.H. Intracerebroventricular insulin improves spatial learning and memory in male Wistar rats. Behav. Neurosci. 2009, 123, 1309–1314. [Google Scholar] [CrossRef]
- Kim, B.; Backus, C.; Oh, S.S.; Hayes, J.M.; Feldman, E.L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.D.; Delerue, F.; Gladbach, A.; Götz, J.; Ittner, L.M. Experimental diabetes mellitus exacerbates Tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2009, 4, e7917. [Google Scholar] [CrossRef] [Green Version]
- Mcclean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Escribano, L.; Simón, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; De Maturana, R.L.; García-Osta, A.; Ricobaraza, A.; Pérez-Mediavilla, A.; Del Río, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, M. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889. [Google Scholar] [CrossRef] [PubMed]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Experimental induction of β-amyloid plaques and cerebral angiopathy in primates. Ann. NY Acad. Sci. 1993, 695, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Callahan, M.J.; Bian, F.; Durham, R.A.; Roher, A.E.; Lipinski, W.J. Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides 2002, 23, 1241–1247. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [Green Version]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of β-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; De Armond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Pype, S.; Moechars, D.; Dillen, L.; Mercken, M. Characterization of amyloid β peptides from brain extracts of transgenic mice overexpressing the London mutant of human amyloid precursor protein. J. Neurochem. 2003, 84, 602–609. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Fritschi, S.K.; Hamaguchi, T.; Obermüller, U.; Füger, P.; Skodras, A.; Schäfer, C.; Odenthal, J.; Heikenwalder, M.; Staufenbiel, M.; et al. Multiple factors contribute to the peripheral induction of cerebral β-amyloidosis. J. Neurosci. 2014, 34, 10264–10273. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Lamb, B.T.; Call, L.M.; Slunt, H.H.; Bardel, K.A.; Lawler, A.M.; Eckman, C.B.; Younkin, S.G.; Holtz, G.; Wagner, S.L.; Price, D.L.; et al. Altered metabolism of familial Alzheimer’s disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum. Mol. Genet. 1997, 6, 1535–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Palomares, M.; Ramos-Rodríguez, J.J.; López-Acosta, J.F.; Pacheco-Herrero, M.; Lechuga-Sancho, A.M.; Perdomo, G.; García-Alloza, M.; Cózar-Castellano, I. Increased Aβ production prompts the onset of glucose intolerance and insulin resistance. Am. J. Physiol. Metab. 2012, 302, E1373–E1380. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renov. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glastras, S.J.; Chen, H.; Teh, R.; McGrath, R.T.; Chen, J.; Pollock, C.A.; Wong, M.G.; Saad, S. Mouse models of diabetes, obesity and related kidney disease. PLoS ONE 2016, 11, e0162131. [Google Scholar] [CrossRef]
- Marzban, L.; Park, K.; Verchere, C.B. Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 2003, 38, 347–351. [Google Scholar] [CrossRef]
- Tesch, G.H.; Allen, T.J. Rodent models of streptozotocin-induced diabetic nephropathy (methods in renal research). Nephrology 2007, 12, 261–266. [Google Scholar] [CrossRef]
- Kalafatakis, K.; Zarros, A. Intracerebroventricular administration of streptozotocin as an experimental approach to Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 944–946. [Google Scholar] [CrossRef]
- Pruzin, J.J.; Nelson, P.T.; Abner, E.L.; Arvanitakis, Z. Review: Relationship of type 2 diabetes to human brain pathology. Neuropathol. Appl. Neurobiol. 2018, 44, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Chargé, S.B.P.; Esiri, M.M.; Bethune, C.A.; Hansen, B.C.; Clark, A. Apolipoprotein E is associated with islet amyloid and other amyloidoses: Implications for Alzheimer’s disease. J. Pathol. 1996, 179, 443–447. [Google Scholar] [CrossRef]
- Powell, D.S.; Maksoud, H.; Chargé, S.B.P.; Moffitt, J.H.; Desai, M.; Da Silva Fihlo, R.L.; Hattersley, A.T.; Stratton, I.M.; Matthews, D.R.; Levy, J.C.; et al. Apolipoprotein E genotype, islet amyloid deposition and severity of Type 2 diabetes. Diabetes Res. Clin. Pr. 2003, 60, 105–110. [Google Scholar] [CrossRef]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-b and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Gonzalez, I.; Edwards, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabayashi, S.; Shimozawa, N.; Yasutomi, Y.; Yanagisawa, K.; Kimura, N. Diabetes mellitus accelerates Aβ pathology in brain accompanied by enhanced GAβ generation in nonhuman primates. PLoS ONE 2015, 10, e0117362. [Google Scholar] [CrossRef] [Green Version]
- Infante-Garcia, C.; Ramos-Rodriguez, J.J.; Hierro-Bujalance, C.; Ortegon, E.; Pickett, E.; Jackson, R.; Hernandez-Pacho, F.; Spires-Jones, T.; Garcia-Alloza, M. Antidiabetic polypill improves central pathology and cognitive impairment in a mixed model of Alzheimer’s disease and type 2 diabetes. Mol. Neurobiol. 2018, 55, 6130–6144. [Google Scholar] [CrossRef] [Green Version]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain. Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra-Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Fernandez-Martos, C.M.; Atkinson, R.A.K.; Chuah, M.I.; King, A.E.; Vickers, J.C. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 92–106. [Google Scholar] [CrossRef]
- Ma, D.L.; Chen, F.Q.; Xu, W.J.; Yue, W.Z.; Yuan, G.; Yang, Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J. Neurochem. 2015, 135, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Hölscher, C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, S47–S54. [Google Scholar]
- Wang, J.; Gallagher, D.; Devito, L.M.; Cancino, G.I.; Tsui, D.; He, L.; Keller, G.M.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 2012, 11, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Pathak, N.M.; Pathak, V.; Gault, V.A.; McClean, S.; Irwin, N.; Flatt, P.R. Novel dual incretin agonist peptide with antidiabetic and neuroprotective potential. Biochem. Pharm. 2018, 155, 264–274. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 2014, 86, 241–258. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-term metformin usage and cognitive function among older adults with diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: A population-based case-control study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.H.; Bahari, B.; Salari, A.A. ATP-sensitive potassium-channel inhibitor glibenclamide attenuates HPA axis hyperactivity, depression-and anxiety-related symptoms in a rat model of Alzheimer’s disease. Brain Res. Bull. 2018, 137, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Rosenblat, J.D.; Brietzke, E.; Park, C.; Lee, Y.; Musial, N.; Pan, Z.; Mansur, R.B.; McIntyre, R.S. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2018, 20, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Armaşu, M.; Musteaţă, M.; Stanciu, G.D.; Mocanu, D.; Solcan, G. Brainstem auditory evoked responses in healthy Argentine Mastiffdogs recorded with surface electrodes. Arq. Bras. Med. Vet. e Zootec. 2015, 67. [Google Scholar] [CrossRef] [Green Version]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.B.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau-Hind, M.E.; Hosford, D.A.; Roses, A.D. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharm. J. 2006, 6, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.M.; Saunders, A.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.H.; van Dyck, C.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimers disease: Two phase 3 studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef]
- Cai, H.Y.; Wang, Z.J.; Hölscher, C.; Yuan, L.; Zhang, J.; Sun, P.; Li, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide attenuates the detrimental effects of amyloid β protein on spatial working memory and hippocampal neurons in rats. Behav. Brain Res. 2017, 318, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Stanciu, G.-D.; Packer, R.M.A.; Pakozdy, A.; Solcan, G.; Volk, H.A. Clinical reasoning in feline epilepsy: Which combination of clinical information is useful? Vet. J. 2017, 225, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Chen, S.; Peng, P.; Gu, Z.; Yu, J.; Zhao, G.; Deng, Y. Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3β. Biochem. Biophys. Res. Commun. 2019, 511, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.Q.; Chen, Z.; Wang, Y.P.; Liu, X.Y.; Liu, X.H.; Ke, L.F.; Zheng, Z.; Lin, X.W.; Zhou, Y.; Wu, L.J.; et al. Subcutaneous liraglutide ameliorates methylglyoxal-induced alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3β. Am. J. Transl. Res. 2017, 9, 247–260. [Google Scholar] [PubMed]
- Han, W.N.; Hölscher, C.; Yuan, L.; Yang, W.; Wang, X.H.; Wu, M.N.; Qi, J.S. Liraglutide protects against amyloid-β protein-induced impairment of spatial learning and memory in rats. Neurobiol. Aging 2013, 34, 576–588. [Google Scholar] [CrossRef]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide protects against brain amyloid-β1–42 accumulation in female mice with early Alzheimer’s disease-like pathology by partially rescuing oxidative/nitrosative stress and inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.M.D.; Madhunapantula, V.S.R.; Nataraj, S.K.M.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Muthureddy Nataraj, S.K.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharm. 2013, 65, 1773–1784. [Google Scholar] [CrossRef]
- Ma, Q.H.; Jiang, L.F.; Mao, J.L.; Xu, W.X.; Huang, M. Vildagliptin prevents cognitive deficits and neuronal apoptosis in a rat model of Alzheimer’s disease. Mol. Med. Rep. 2018, 17, 4113–4119. [Google Scholar] [CrossRef]
- Zhang, D.D.; Shi, N.; Fang, H.; Ma, L.; Wu, W.P.; Zhang, Y.Z.; Tian, J.L.; Tian, L.B.; Kang, K.; Chen, S. Vildagliptin, a DPP4 inhibitor, alleviates diabetes-associated cognitive deficits by decreasing the levels of apoptosis-related proteins in the rat hippocampus. Exp. Med. 2018, 15, 5100–5106. [Google Scholar] [CrossRef]
- Dong, Q.; Teng, S.W.; Wang, Y.; Qin, F.; Li, Y.; Ai, L.L.; Yu, H. Sitagliptin protects the cognition function of the Alzheimer’s disease mice through activating glucagon-like peptide-1 and BDNF-TrkB signalings. Neurosci. Lett. 2019, 696, 184–190. [Google Scholar] [CrossRef]
- Bild, V.; Ababei, D.C.; Neamtu, M.; Vasincu, A.; Bild, W.; Stanciu, G.D.; Tamba, B.I.; Solcan, G.; Beschea Chiriac, S. Isobolar analysis of the binary fixed-ratio combination of acetylsalicilic acid-acetaminophen. Farmacia 2017, 65, 563–566. [Google Scholar]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci. 2015, 21, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Arafa, N.M.S.; Ali, E.H.A.; Hassan, M.K. Canagliflozin prevents scopolamine-induced memory impairment in rats: Comparison with galantamine hydrobromide action. Chem. Biol. Interact. 2017, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Shingo, A.S.; Kanabayashi, T.; Kito, S.; Murase, T. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav. Brain Res. 2013, 241, 105–111. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Frey, W.H.; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Solcan, G. Acute idiopathic polyradiculoneuritis concurrent with acquired myasthenia gravis in a West Highland white terrier dog. BMC Vet. Res. 2016, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: Potential therapeutics of Alzheimer’s disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Qian, Z.; Pei, D. Macrocycles as protein-protein interaction inhibitors. Biochem. J. 2017, 474, 1109–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.-M.; Velkova, A.; Tatarek-Nossol, M.; Rammes, G.; Sibaev, A.; Andreetto, E.; Kracklauer, M.; Bakou, M.; Malideli, E.; Göke, B.; et al. Selectively N-methylated soluble IAPP mimics as potent IAPP receptor agonists and nanomolar inhibitors of cytotoxic self-assembly of both IAPP and Aβ40. Angew. Chem. Int. Ed. 2013, 52, 10378–10383. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanopoulou, A.; Heidrich, L.; Chen, H.R.; Frost, C.; Hrle, D.; Malideli, E.; Hille, K.; Grammatikopoulos, A.; Bernhagen, J.; Zacharias, M.; et al. Designed macrocyclic peptides as nanomolar amyloid inhibitors based on minimal recognition elements. Angew. Chem. Int. Ed. 2018, 57, 14503–14508. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Diabetes | Insulin |
| Biguanides: Metformin | |
| Sulphonylureas: Glibenclamide, Glibornuride, Glipizide, Gliquidone, Glisoxepide, Glyclopyramide, Glimepiride | |
| Alpha-glucosidase inhibitors: Acarbose, Miglitol, Voglibose | |
| Incretins Dipeptidyl peptidase−4 inhibitors: Sitagliptin, Saxagliptin, Linagliptin, Alogliptin Glucagon-like peptide−1 receptor agonists: Exenatide, Liraglutide, Albiglutide, Dulaglutide | |
| Thiazolidinediones: Pioglitazone, Rosiglitazone | |
| SGLT2 Inhibitors: Empagliflozin, Canagliflozin, Dapagliflozin, Ipragliflozin | |
| Meglitinides: Repaglinide, Nateglinide | |
| Amylin analog: Pramlintide | |
| Alzheimer’s Disease | Cholinesterase inhibitors: Tacrine, Donepezil, Rivastigmine, Galantamine |
| N-methyl-D-aspartate receptor: Memantine |
| Antidiabetic Medication | Experimental Model | Findings | References |
|---|---|---|---|
| Biguanides | |||
| Metformin | mouse neuroblastoma cell lines under sustained hyperinsulinemic conditions treated with different concentrations of metformin (0.4–3.2 mM) | resensitization of insulin signaling; prevention of the molecular and pathological alterations detected in AD neurons | [130] |
| transgenic APPswe/PSd1E9 mouse model of AD; intraperitoneal delivery of 200 mg/kg metformin for 14 days | amelioration of spatial memory deficits, neural cellular proliferation; in the cortex and hippocampus, reduction of local inflammation, decrease of Aβ plaque deposition | [119] | |
| PDAPP (J9) mouse model of AD; 350 mg/kg/day metformin delivered in drinking water for several months | attenuation of memory impairment in female subjects and intensification of it in males | [131] | |
| longitudinal aging study in adults with diabetes | long-term metformin therapy (over 6 years) could diminish the risk of developing AD | [132] | |
| case-control study, older adults with an incident diagnosis of AD; 1–9, 10–29, 30–59, or ≥60 metformin prescriptions | long-term treatment (60 or more prescriptions) has been correlated with a slight augmented risk of developing AD | [133] | |
| Sulphonylureas | |||
| Glibenclamide | Aβ25–35-induced rat AD model; 6 mg/kg/day of glibenclamide for 20 days by gavage | reduction of Aβ25–35-treated behavioral anomalies | [134] |
| Thiazolidinediones | |||
| Pioglitazone | meta-analysis of randomized clinical trials; 15 to 30 mg of pioglitazone, as adjunct therapy for AD | doses of 15 to 30 mg pioglitazone but not 45 mg improve cognitive capacity | [135] |
| transgenic APPswe/PSEN1dE9 AD mouse model; combined therapy with 0.03 mg/kg/day of leptin intranasal delivery + intraperitoneal administration of 10 mg/kg/day pioglitazone for 2 weeks | decrease of spatial memory impairments and brain Aβ levels | [122] | |
| APPV717I transgenic mice, a model for AD; acute 7 days gavage therapy with 40 mg/kg/day of pioglitazone | reduction of soluble Aβ1–42 peptide levels by 27% and glial inflammation | [125] | |
| controlled trial in cases with mild Alzheimer’s disease and an accompanying diagnosis of diabetes; daily doses of 15–30 mg pioglitazone for 6 months | cognitive and functional improvements and stabilization of the disease in diabetics with AD | [136] | |
| controlled pilot trial in individuals with AD without diabetes; daily 45 mg of pioglitazone | 18 months of pioglitazone therapy were well tolerated by patients, but no important efficacy data were detected | [137] | |
| Rosiglitazone | meta-analysis of randomized clinical trials; 2 to 8 mg of rosiglitazone, as adjunct therapy for mild to moderate AD patients | pro-cognitive effects | [135] |
| pilot study that randomized individuals with AD or amnestic mild cognitive damage | better delayed recall and selective attention | [138] | |
| large study in population with mild to moderate AD; 2, 4, or 8 mg of rosiglitazone for 6 months | in week 24 an improvement (−2.9 points) of cognition in apolipoprotein Eε4-negative people treated with 8 mg of rosiglitazone was registered | [139] | |
| phase III trials of rosiglitazone in AD; 2 mg or 8 mg rosiglitazone for 48 weeks, as adjunctive agent to ongoing acetylcholine esterase inhibitors | rosiglitazone did not lead to an improvement in cognition or overall function | [140] | |
| Glucagon-like peptide−1 receptor agonists | |||
| Lixisenatide | transgenic APPswe/PSd1E9 mouse model of AD; intraperitoneal injection with 1 or 10 nmol/kg of compound for 10 weeks | several biomarkers have been improved such as learning, inflammation, or plate loading | [129] |
| cell culture, 100μM of lixisenatide were applied 24 h before Aβ25–35 application rat model of AD; 5 nmol/μL of lixisenatide before intrahippocampal application of Aβ25–35 (5 nmol/μL) | reversal Aβ25–35-triggered cytotoxicity, normalization of intracellular calcium levels prevention of memory loss caused by amyloid intracerebroventricular injection | [141] | |
| transgenic APP/PS1/tau mouse model of AD; daily intraperitoneal injection of 10 nmol/kg lixisenatide for 60 days | reduction of amyloid plaques, neuroinflammation, and neurofibrillary tangles | [142] | |
| Dulaglutide | intracerebral injection of streptozotocin-induced mouse AD-like condition; 0.6 mg/kg/week of dulaglutide with intraperitoneal delivery for 4 weeks | amelioration of learning and memory deficits | [143] |
| Liraglutide | transgenic APPswe/PSd1E9 mouse model of AD; intraperitoneal injection with 2.5 or 25 nmol/kg of drug for 10 weeks | improvement of learning, reduction of amyloid plaque deposits by 40%–50%, and decrease inflammatory response | [129] |
| methylglyoxal-induced mouse Alzheimer-like condition; daily subcutaneous administration of 25 nmol/kg liraglutide for 2 months | attenuation of hippocampal damage and cognitive deficits in C57BL/6J mice | [144] | |
| cell culture; liraglutide (300 nm) was added to cultures 40 min before Aβ oligomers Aβ oligomers-induced AD mouse model; daily intraperitoneal injections of liraglutide (25 nmol/kg) for 7 days Aβ oligomer-induced non-human primate model of AD; subcutaneous delivery of liraglutide (0.006 mg/kg/day for the first week and 0.012 mg/kg thereafter) for 24 days | reduction of Aβ oligomer-induced synaptotoxicity, protective effects on synapses; prevention and reversal of cognitive abnormalities, and insulin receptor loss produced by intracerebroventricular injection of Aβ oligomers; the agent was less effective, but still provided partial protection against insulin resistance loss; synapses and phosphorylation of tau | [121] | |
| Aβ protein-induced rat model of AD; 2 μL liraglutide trough intrahippocampal administration | liraglutide pre-therapy remarkably protected against Aβ-induced damage of spatial memory and long-term potentiation | [145] | |
| transgenic 3xTg-AD female mice; 0.2 mg/kg/day of liraglutide, intraperitoneal injections | reduction of cortical Aβ1–42 levels, partial attenuation of cerebral estradiol, inflammation, and oxidative/nitrosative stress | [146] | |
| a pilot clinical trial in AD patients lasting 26 weeks; in the first week, the drug was daily delivered subcutaneously at a dose of 0.6 mg; hereafter 1.2 mg daily for another week before finally increasing to 1.8 mg daily | prevention of brain glucose metabolism decline; there were no important cognitive changes compared with placebo group | [147] | |
| Dipeptidyl peptidase−4 inhibitors | |||
| Saxagliptin | intracerebral injection of streptozotocin-induced rat model of AD; 0.25, 0.5, and 1 mg/kg of saxagliptin administered orally for 60 days | reduction of amyloid plaque formation, a marked decrease of Aβ42 level, and phosphorylation of tau protein; total reversal of cognitive impairments | [148] |
| Vildagliptin | intracerebral injection of streptozotocin-induced rat model of AD; daily oral doses of 2.5, 5, and 10 mg/kg vildagliptin for 30 days | attenuation of Aβ, phosphorylation of tau protein, and inflammatory markers | [149] |
| Aβ protein-induced rat model of AD; daily gavage of 5 or 10 mg/kg vildagliptin for 4 weeks | anti-apoptotic effect, attenuation of memory abnormalities, reduction of tau phosphorylation, and increase of neurotrophic protein expression | [150] | |
| streptozotocin-induced rat diabetes model associated cognitive decline; daily gavage of 5 mg/kg vildagliptin for 4 weeks | prevention of memory impairment and diminution of apoptosis in hippocampal neurons | [151] | |
| Sitagliptin | APP/PS1 AD mice model; 20 mg/kg/day of sitagliptin for an 8-weeks period | protective effect of cognitive function, reduction of amyloid plaque deposits | [152] |
| transgenic APPswe/PSd1E9 mouse model of AD; daily gavage of 5, 10, and 20 mg/kg sitagliptin for 12 weeks | much more obvious effects for the 20 mg/kg sitagliptin dose—reduction of nitrosative stress and inflammation markers; an important diminution in the number and area of APP and Aβ deposition | [153] | |
| Linagliptin | 3xTg-AD mouse model of AD; daily oral administration of 5, 10, and 20 mg/kg linagliptin for 8 weeks | improvement of cognitive performance; reduction of Aβ42 levels, but not Aβ40; diminution of tau phosphorylation and neuroinflammation | [154] |
| human neuroblastoma SK-N-MC cell culture; exposure to 10 to 100 μM linagliptin for 24 h | protection of cells against Aβ-induced intracellular reactive oxygen species accumulation and mitochondria dysfunction | [155] | |
| Amylin analog | |||
| Pramlintide | SAMP8 mice, a model of sporadic AD; subcutaneous infusion of 0.24 mg/kg/day pramlintide for 5 weeks | may improve memory, decrease neuroinflammation, and reduce oxidative stress | [156] |
| Sodium-glucose cotransporter 2 (SGLT−2) inhibitors | |||
| Canagliflozin | scopolamine-induced rat model of memory impairment; daily oral gavage of 10 mg/kg for 2 weeks | improvement of memory dysfunction | [157] |
| Insulin analogues | |||
| intracerebral injection of streptozotocin rat model of cognitive decline; 0.5 units = 12 nmol of detemir | alleviating cognitive dysfunction with a significant increase in learning ability; change in insulin degrading enzyme, insulin receptor, and somatostatin | [158] | |
| patients with early AD; intranasal administration of 20 or 40 IU insulin | facilitation of verbal memory recall in memory-impaired ɛ4− patients; no influence on glucose or plasma insulin levels | [159] | |
| patients with early AD; intranasal administration of 20 or 40 IU insulin for 21 days | improvement of attention, functional status, and verbal memory; modulation of Aβ peptide | [87] | |
| placebo-controlled pilot clinical trial in people with AD; intranasal delivery of 20 or 40 IU insulin for 4 months | improvement of cognition and functional ability compared to control group | [160] | |
| clinical trial; 20 or 40 IU of insulin detemir for 21 days, intranasal administration in AD | therapy effect for the memory composite outcome for the 40 IU patients, influenced by the APOE status | [161] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Cobzaru, A.; Paduraru, L.; Bulea, D. Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. J. Clin. Med. 2020, 9, 1713. https://doi.org/10.3390/jcm9061713
Stanciu GD, Bild V, Ababei DC, Rusu RN, Cobzaru A, Paduraru L, Bulea D. Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. Journal of Clinical Medicine. 2020; 9(6):1713. https://doi.org/10.3390/jcm9061713
Chicago/Turabian StyleStanciu, Gabriela Dumitrita, Veronica Bild, Daniela Carmen Ababei, Razvan Nicolae Rusu, Alina Cobzaru, Luminita Paduraru, and Delia Bulea. 2020. "Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages" Journal of Clinical Medicine 9, no. 6: 1713. https://doi.org/10.3390/jcm9061713
APA StyleStanciu, G. D., Bild, V., Ababei, D. C., Rusu, R. N., Cobzaru, A., Paduraru, L., & Bulea, D. (2020). Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. Journal of Clinical Medicine, 9(6), 1713. https://doi.org/10.3390/jcm9061713