Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options


 ,
, 
 ,
, 
Abstract
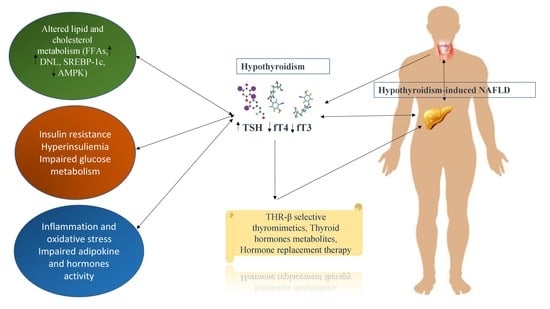
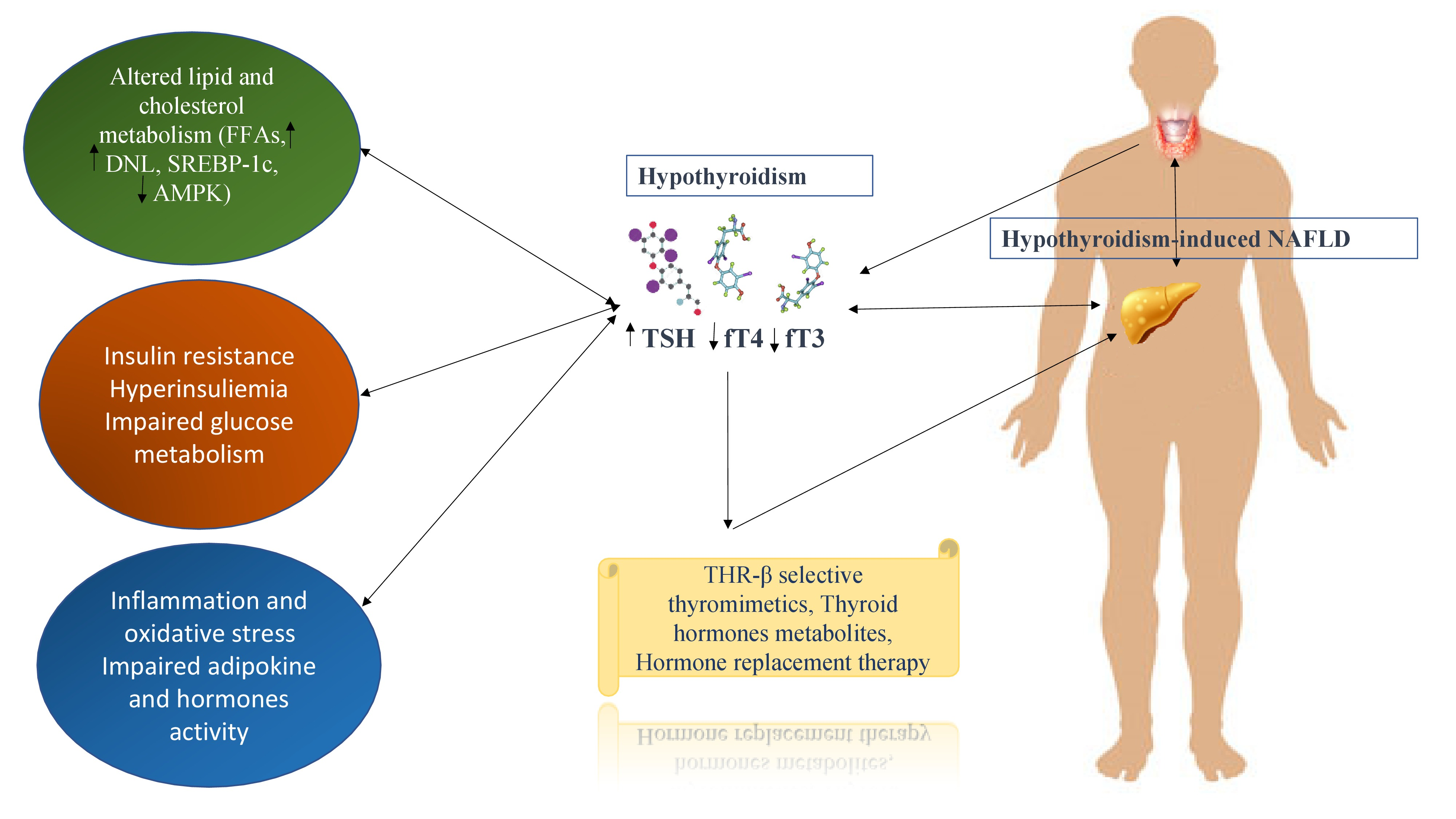
:
1. Introduction
Definitions and Epidemiological Discoveries
2. Immunopathogenesis
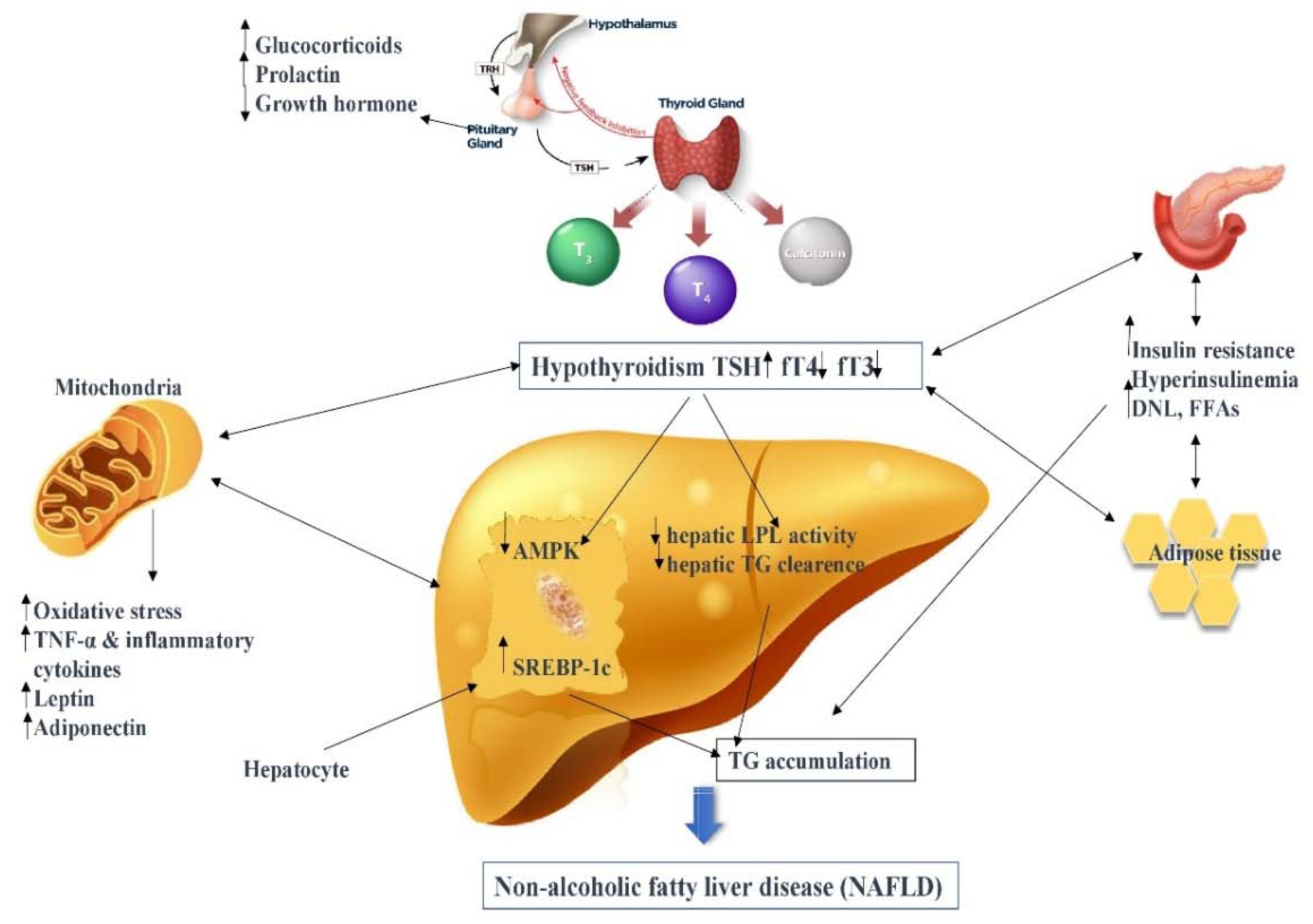
2.1. Thyroid Hormone–Liver Axis
2.2. Lipid and Cholesterol Metabolism
2.3. Glucose and Insulin Metabolism
2.4. Oxidative Stress and Inflammation
2.5. Adipokines and Hormones
3. Hypothyroidism-Induced NAFLD Treatment
3.1. Thyroid Hormones
3.2. Thyroid Hormone Metabolites
3.3. Alternative Treatment
4. THR-β Selective Thyromimetics and NAFLD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | Nonalcoholic fatty liver disease |
| NAFL | Nonalcoholic fatty liver |
| NASH | Nonalcoholic steatohepatitis |
| THs | Thyroid hormones |
| HCs | Hepatic cells |
| HCC | Hepatocellular carcinoma |
| HIN | Hypothyroidism-induced NAFLD |
| SCH | Subclinical hypothyroidism |
| IR | Insulin resistance |
| BMI | Body mass index |
| TSH | Thyroid-stimulating hormone |
| fT4 | Free Thyroxine |
| LT4 | Levothyroxine |
| AST | Aspartate aminotransferase |
| ALT | Alanine aminotransferase |
| TG | Triglyceride |
| HOMA-IR | Homeostatic Model Assessment for Insulin Resistance |
| TRs | Thyroid hormone receptors |
| T3 | 3,5,3′-triiodothyronine |
| T4 | 3,5,3′,5′-tetraiodothyronine |
| rT3 | Reverse T3 |
| TRH | Thyrotropin-releasing hormone |
| RXR | Retinoid X receptor |
| TREs | Thyroid hormone response elements |
| THR-α | Thyroid hormone receptor α1 |
| THR-β | Thyroid hormone receptor β1 |
| D1 | Deiodinases 1 |
| D2 | Deiodinases 2 |
| D3 | Deiodinases 3 |
| T2 | Diiodothyronine |
| TGF-β | Transforming growth factor-beta |
| IL6 | Interleukin-6 |
| FFAs | Free fatty acids |
| DNL | De novo lipogenesis |
| L-FABPs | Liver fatty acid binding proteins |
| SREBP-1c | Sterol regulatory element-binding transcription factor 1 |
| ChREBP | Carbohydrate- Responsive Element-Binding Protein |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| Cyp7A1 | Cholesterol 7 alpha-hydroxylase |
| T2DM | Type 2 diabetes mellitus |
| CKD | Chronic kidney disease |
| MRI | Magnetic Resonance Imaging |
| FLI | Fatty liver index |
| USG | Ultrasonography |
| LS | Longitudinal study |
| CSS | Cross-sectional study |
| CCS | Case-control study |
| WAT | White adipose tissue |
| mRNA | Messenger RNA |
| FAS | Fatty acid synthase |
| ACC | Acetyl-CoA carboxylase |
| VLDL | Very-low-density lipoprotein |
| LDL | Low-Density Lipoproteins |
| HDL | High-Density Lipoproteins |
| FATPs | Fatty acid transporter proteins |
| FAT | Fatty acid translocase |
| cAMP | Cyclic AMP |
| PKA | Protein kinase A |
| PPARα | Peroxisome proliferator-activated receptor-α |
| PPAR δ | Peroxisome proliferator-activated receptor- δ |
| AMPK | AMP-activated protein kinase |
| HMGCR | 3-hydroxy-3-methyl-glutaryl coenzyme A reductase |
| CETP | Cholesteryl ester transfer protein |
| LCAT | Lecithin-cholesterol acyltransferase |
| ApoA1 | Apolipoprotein A1 |
| ApoC3 | Apolipoprotein C3 |
| SRB1 | Scavenger receptor class B member 1 |
| SOAT2 | Sterol O-acyltransferase 2 |
| ROS | Reactive oxygen species |
| CAT | Catalase |
| CAMKK2 | Ca2+–calcium/calmodulin-dependent protein kinase kinase 2 |
| FGF-21 | Fibroblast growth factor-21 |
| TBA | Thyroxine-binding albumin |
| TBPA | Thyroxine-binding prealbumin |
| TBG | Thyroxine-binding globulin |
| L-FABP | Hepatic fatty acid-binding protein |
| CMD | Choline-methionine deficient |
| COX-2 | Cyclooxygenase-2 |
| AOX | Acyl-CoA oxidase |
| phosphor-SAPK/JNK | Phosphor-stress-activated protein kinase/c-Jun NH2-terminal kinase |
| phosphor-STAT3 | Phosphor-signal transducer and activator of transcription 3 |
| PGC-1α | Peroxisome proliferator-activated receptor γ coactivator-1α |
| CPT-1 | Carnitine palmitoyltransferase-1 |
| T1AM | 3-iodothyronamine |
| STIR1 | Hepatic nuclear sirtuin 1 |
| AKT | Protein kinase B |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular signal-regulated kinase |
| TAAR1 | Trace amine-associated receptor |
| TQ | Thymoquinone |
| NG | Nigella salvia |
| PTU | 6-propyl-2-thiouracil |
| α-SMA | Alpha-smooth muscle actin |
| HSCs | Hepatic stellate cells |
| GC-1 | Sobetirome |
| KB-2115 | Eprotirome |
| MGL-3196 | Resmetirom |
| MRI-PDFF | MRI-proton density fat fraction |
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Italian Association for the Study of the Liver (AISF). AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Hasanin, M.; Kaif, M.; Wiesner, R.; Kuo, Y.F. Nonalcoholic Steatohepatitis is the Most Rapidly Growing Indication for Simultaneous Liver Kidney Transplantation in the United States. Transplantation 2016, 100, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Li, A.A.; Ahmed, A.; Kim, D. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gut Liver 2020, 14, 168–178. [Google Scholar] [CrossRef]
- Wattacheril, J. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. N. Am. 2020, 49, 141–149. [Google Scholar] [CrossRef]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [Green Version]
- Hazlehurst, J.M.; Tomlinson, J.W. Non-alcoholic fatty liver disease in common endocrine disorders. Eur. J. Endocrinol. 2013, 169, R27–R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yang, W.; Karakas, S.; Sarkar, S. NASH in Nondiabetic Endocrine Disorders. Metab. Syndr. Relat. Disord. 2018, 16, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Lonardo, A. Extra-hepatic manifestations and complications of nonalcoholic fatty liver disease. Future Med. Chem. 2019, 11, 2171–2192. [Google Scholar] [CrossRef]
- Blum, M.R.; Gencer, B.; Adam, L.; Feller, M.; Collet, T.H.; da Costa, B.R. Impact of thyroid hormone therapy on atherosclerosis in the elderly with subclinical hypothyroidism: A randomized trial. J. Clin. Endocrinol. Metab. 2018, 103, 2988–2997. [Google Scholar] [CrossRef]
- Demir, Ş.; Ünübol, M.; Aypak, S.Ü.; İpek, E.; Aktaş, S.; Ekren, G.S.; Yılmaz, M.; Tunca, R.; Güney, E. Histopathologic Evaluation of Nonalcoholic Fatty Liver Disease in Hypothyroidism-Induced Rats. Int. J. Endocrinol. 2016, 2016, 5083746. [Google Scholar] [CrossRef]
- Jiang, L.; Du, J.; Wu, W.; Fang, J.; Wang, J.; Ding, J. Sex differences in subclinical hypothyroidism and associations with metabolic risk factors: A health examination-based study in mainland China. BMC Endocr. Disord. 2020, 20, 100. [Google Scholar] [CrossRef]
- Zhang, X.; Gong, P.; Sheng, L.; Lin, Y.; Fan, Q.; Zhang, Y.; Bao, Y.; Li, S.; Du, H.; Chen, Z.; et al. Prognostic value of subclinical thyroid dysfunction in ischemic stroke patients treated with intravenous thrombolysis. Aging 2019, 11, 6839–6850. [Google Scholar] [CrossRef]
- Iwen, K.A.; Oelkrug, R.; Kalscheuer, H.; Brabant, G. Metabolic Syndrome in Thyroid Disease. Front. Horm. Res. 2018, 49, 48–66. [Google Scholar] [CrossRef]
- George, K.M.; Lutsey, P.L.; Selvin, E.; Palta, P.; Windham, B.G.; Folsom, A.R. Association Between Thyroid Dysfunction and Incident Dementia in the Atherosclerosis Risk in Communities Neurocognitive Study. J. Endocrinol. Metab. 2019, 9, 82–89. [Google Scholar] [CrossRef]
- Alfadhel, M.; Al Othaim, A.; Al Saif, S.; Al Mutairi, F.; Alsayed, M.; Rahbeeni, Z.; Alzaidan, H.; Alowain, M.; Al-Hassnan, Z.; Saeedi, M.; et al. Expanded Newborn Screening Program in Saudi Arabia: Incidence of screened disorders. J. Paediatr. Child Health 2017, 53, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Krashin, E.; Piekiełko-Witkowska, A.; Ellis, M.; Ashur-Fabian, O. Thyroid hormones and cancer: A comprehensive review of preclinical and clinical studies. Front. Endocrinol. 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Li, P.; Mi, Y.; Liu, Y.; Liu, Y.; Zhang, P. Thyroid-stimulating hormone is associated with nonalcoholic steatohepatitis in patients with chronic hepatitis B. Medicine 2019, 98, e17945. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Targher, G. Pathogenesis of hypothyroidism-induced NAFLD: Evidence for a distinct disease entity? Dig. Liver Dis. 2019, 51, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Okosieme, O.; Gilbert, J.; Abraham, P.; Boelaert, K.; Dayan, C.; Gurnell, M.; Leese, G.; McCabe, C.; Perros, P.; Smith, V.; et al. Management of primary hypothyroidism: Statement by the British ThyroidAssociation Executive Committee. Clin. Endocrinol. 2016, 84, 799–808. [Google Scholar] [CrossRef]
- Miyake, T.; Matsuura, B.; Furukawa, S.; Todo, Y.; Yamamoto, S.; Yoshida, O.; Imai, Y.; Watanabe, T.; Yamamoto, Y.; Hirooka, M.; et al. Hyperthyroidism Improves the Pathological Condition of Nonalcoholic Steatohepatitis: A Case of Nonalcoholic Steatohepatitis with Graves’ Disease. Intern. Med. 2016, 55, 2019–2023. [Google Scholar] [CrossRef] [Green Version]
- Feisa, S.V.; Chopei, I.V. Subclinical hypothyroidism in patients with non-alcoholic fatty liver disease at the background of carbohydrate metabolism disorders. Wiad. Lek. 2018, 71, 261–264. [Google Scholar]
- Chiovato, L.; Magri, F.; Carlé, A. Hypothyroidism in Context: Where We’ve Been and Where We’re Going. Adv. Ther. 2019, 36, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Biondi, B.; Cappola, A.R.; Cooper, D.S. Subclinical Hypothyroidism: A Review. JAMA 2019, 322, 153–160. [Google Scholar] [CrossRef]
- Marchesini, G.; Roden, M.; Vettor, R. Response to: Comment to “EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease”. J. Hepatol. 2017, 66, 466–467. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Sait, H.; Jindal, A.; Juneja, M.; Gupta, S.; Thelma, B.K.; Kapoor, S. Factors Associated with Transient Neonatal Hyperthyrotropinemia. Indian J. Pediatr. 2020, 87, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.J.; Cines, B.; Pogrebniak, E.; Sherafat-Kazemzadeh, R.; Demidowich, A.P.; Galescu, O.A.; Brady, S.M.; Reynolds, J.C.; Hubbard, V.S.; Yanovski, J.A. Associations between adiposity and indicators of thyroid status in children and adolescents. Pediatr. Obes. 2016, 11, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.W.; Tsai, M.C.; Yang, Y.J.; Chen, M.Y.; Chen, S.Y.; Chou, Y.Y. The relationship between nonalcoholic fatty liver disease and pediatric congenital hypothyroidism patients. Kaohsiung J. Med. Sci. 2019, 35, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.L.; Golshan, S.; Harlow, K.E.; Angeles, J.E.; Durelle, J.; Goyal, N.P.; Newton, K.P.; Sawh, M.C.; Hooker, J.; Sy, E.Z.; et al. Prevalence of Nonalcoholic Fatty Liver Disease in Children with Obesity. J. Pediatr. 2019, 207, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, T.E.; Graeter, T.; Oeztuerk, S.; Holzner, D.; Kratzer, W.; Wabitsch, M.; Denzer, C. Thyroid dysfunction and hepatic steatosis in overweight children and adolescents. Pediatr. Obes. 2017, 12, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Wassner, A.J. Pediatric Hypothyroidism: Diagnosis and Treatment. Paediatr. Drugs 2017, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Chalasani, N. Is hypothyroidism a risk factor for non-alcoholic steatohepatitis? J. Clin. Gastroenterol. 2003, 37, 340–343. [Google Scholar] [CrossRef]
- Targher, G.; Montagnana, M.; Salvagno, G.; Moghetti, P.; Zoppini, G.; Muggeo, M.; Lippi, G. Association between serum TSH, free T4 and serum liver enzyme activities in a large cohort of unselected outpatients. Clin. Endocrinol. 2008, 68, 481–484. [Google Scholar] [CrossRef]
- Silveira, M.G.; Mendes, F.D.; Diehl, N.N.; Enders, F.T.; Lindor, K.D. Thyroid dysfunction in primary biliary cirrhosis, primary sclerosing cholangitis and non-alcoholic fatty liver disease. Liver Int. 2009, 29, 1094–1100. [Google Scholar] [CrossRef]
- Moustafa, A.H.; Ali, E.M.; Mohamed, T.M.; Abdou, H.I. Oxidative stress and thyroid hormones in patients with liver diseases. Eur. J. Intern. Med. 2009, 20, 703–708. [Google Scholar] [CrossRef]
- Xu, C.; Xu, L.; Yu, C.; Miao, M.; Li, Y. Association between thyroid function and nonalcoholic fatty liver disease in euthyroid elderly Chinese. Clin. Endocrinol. 2011, 75, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Chung, G.E.; Kim, D.; Kim, W.; Yim, J.Y.; Park, M.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J. Hepatol. 2012, 57, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.R.; Zein, C.O.; Dasarathy, S.; Yerian, L.M.; Lopez, R.; McCullough, A.J. Prevalence of hypothyroidism in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2012, 57, 528–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carulli, L.; Ballestri, S.; Lonardo, A.; Lami, F.; Violi, E.; Losi, L.; Bonilauri, L.; Verrone, A.M.; Odoardi, M.R.; Scaglioni, F.; et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern. Emerg. Med. 2013, 8, 297–305. [Google Scholar] [CrossRef]
- Tao, Y.; Gu, H.; Wu, J.; Sui, J. Thyroid function is associated with non-alcoholic fatty liver disease in euthyroid subjects. Endocr. Res. 2015, 40, 74–78. [Google Scholar] [CrossRef]
- Liu, G.; Zheng, X.; Guan, L.; Jiang, Z.; Lin, H.; Jiang, Q.; Zhang, N.; Zhang, Y.; Zhang, X.; Yu, C.; et al. Free triiodothyronine levels are positively associated with non-alcoholic fatty liver disease in euthyroid middle-aged subjects. Endocr. Res. 2015, 40, 188–193. [Google Scholar] [CrossRef]
- Ding, W.J.; Wang, M.M.; Wang, G.S.; Shen, F.; Qin, J.J.; Fan, J.G. Thyroid function is associated with non-alcoholic fatty liver disease in chronic hepatitis B-infected subjects. J. Gastroenterol. Hepatol. 2015, 30, 1753–1758. [Google Scholar] [CrossRef]
- Gökmen, F.Y.; Ahbab, S.; Ataoğlu, H.E.; Türker, B.Ç.; Çetin, F.; Türker, F.; Mamaç, R.Y.; Yenigün, M. FT3/FT4 ratio predicts non-alcoholic fatty liver disease independent of metabolic parameters in patients with euthyroidism and hypothyroidism. Clinics 2016, 71, 221–225. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.; Joo, S.K.; Bae, J.M.; Kim, J.H.; Ahmed, A. Subclinical hypothyroidism and low-normal thyroid function are associated with nonalcoholic steatohepatitis and fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 123–131. [Google Scholar] [CrossRef]
- Uribe, M.; Roman-Sandoval, J.D.J.; Ramos-Ostos, M.; Alfaro-Lara, R.; Mendez-Sanchez, N.; Chavez-Tapia, N.C. Role of subclinical hypothyroidism in nonalcoholic fatty liver disease. J. Diabetes 2011, 3, 202. [Google Scholar]
- Xu, L.; Ma, H.; Miao, M.; Li, Y. Impact of subclinical hypothyroidism on the development of non-alcoholic fatty liver disease: A prospective case-control study. J. Hepatol. 2012, 57, 1153–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, P.; Phadke, A.; Sawant, P. Prevalence of hypothyroidism in nonalcoholic fatty liver disease in patients attending a tertiary hospital in western India. Indian J. Gastroenterol. 2015, 34, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, R.; Das, S.N.; Jena, A.K.; Behera, S.; Sahu, N.C.; Mohanty, B.; Suna, S.P.; Thatoi, P.K. Prevalence of non-alcoholic fatty liver disease in hypothyroidism in a tertiary care hospital in eastern India. J. Evol. Med. Dent. Sci. 2017, 6, 5589–5593. [Google Scholar] [CrossRef]
- Mazo, D.F.; Lima, V.M.; Stefano, J.T.; Rabelo, F.; Faintuch, J.; Oliveira, C.P. Gluco-lipidic indices in treated hypothyroidism associated with nonalcoholic fatty liver disease. Arq. Gastroenterol. 2011, 48, 186–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bano, A.; Chaker, L.; Plompen, E.P.; Hofman, A.; Dehghan, A.; Franco, O.H.; Janssen, H.L.; Darwish Murad, S.; Peeters, R.P. Thyroid Function and the Risk of Nonalcoholic Fatty Liver Disease: The Rotterdam Study. J. Clin. Endocrinol. Metab. 2016, 101, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, E.H.; van Tienhoven-Wind, L.J.; Amini, M.; Schreuder, T.C.; Faber, K.N.; Blokzijl, H.; Dullaart, R.P. Higher free triiodothyronine is associated with non-alcoholic fatty liver disease in euthyroid subjects: The Lifelines Cohort Study. Metabolism 2017, 67, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Manka, P.; Bechmann, L.; Best, J.; Sydor, S.; Claridge, L.C.; Coombes, J.D.; Canbay, A.; Moeller, L.; Gerken, G.; Wedemeyer, H.; et al. Low Free Triiodothyronine Is Associated with Advanced Fibrosis in Patients at High Risk for Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2019, 64, 2351–2358. [Google Scholar] [CrossRef]
- Jaruvongvanich, V.; Sanguankeo, A.; Upala, S. Nonalcoholic fatty liver disease is not associated with thyroid hormone levels and hypothyroidism: A systematic review and meta-analysis. Eur. Thyroid J. 2017, 6, 208–215. [Google Scholar] [CrossRef] [Green Version]
- He, W.; An, X.; Li, L.; Shao, X.; Li, Q.; Yao, Q.; Zhang, J.A. Relationship between hypothyroidism and non-alcoholic fatty liver disease: A systematic review and meta-analysis. Front. Endocrinol. 2017, 8, 335. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, M.; Han, B.; Qi, X. Association of non-alcoholic fatty liver disease with thyroid function: A systematic review and meta-analysis. Dig. Liver Dis. 2018, 50, 1153–1162. [Google Scholar] [CrossRef]
- Bril, F.; Kadiyala, S.; Cusi, K. Hypothyroidism in Nonalcoholic Fatty Liver Disease: Causative Factor or Innocent Bystander? Thyroid. 2019, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Nascimbeni, F.; Lonardo, A.; Zoppini, G.; Bonora, E.; Mantzoros, C.S.; Targher, G. Association Between Primary Hypothyroidism and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Thyroid 2018, 28, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, H.; Chen, L.; Zheng, J.; Hu, X.; Wang, S.; Chen, T. Relationship between serum TSH level with obesity and NAFLD in euthyroid subjects. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ittermann, T.; Haring, R.; Wallaschofski, H.; Baumeister, S.E.; Nauck, M.; Dörr, M.; Lerch, M.M.; Meyer zu Schwabedissen, H.E.; Rosskopf, D.; Völzke, H. Inverse association between serum free thyroxine levels and hepatic steatosis: Results from the Study of Health in Pomerania. Thyroid 2012, 22, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Eshraghian, A.; Dabbaghmanesh, M.H.; Eshraghian, H.; Fattahi, M.R.; Omrani, G.R. Nonalcoholic fatty liver disease in a cluster of Iranian population: Thyroid status and metabolic risk factors. Arch. Iran. Med. 2013, 16, 584–589. [Google Scholar] [PubMed]
- Posadas-Romero, C.; Jorge-Galarza, E.; Posadas-Sánchez, R.; Acuña-Valerio, J.; Juárez-Rojas, J.G.; Kimura-Hayama, E.; Medina-Urrutia, A.; Cardoso-Saldaña, G.C. Fatty liver largely explains associations of subclinical hypothyroidism with insulin resistance, metabolic syndrome, and subclinical coronary atherosclerosis. Eur. J. Endocrinol. 2014, 171, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.F.; Zhao, J.J. The Relationship between the Broad Spectrum of Subclinical Hypothyroidism and NAFLD in Elderly Subjects. Master’s Thesis, Shandong University, Jinan, China, 2014. [Google Scholar]
- Ludwig, U.; Holzner, D.; Denzer, C.; Greinert, A.; Haenle, M.M.; Oeztuerk, S.; Koenig, W.; Boehm, B.O.; Mason, R.A.; Kratzer, W.; et al. EMIL-Study. Subclinical and clinical hypothyroidism and non-alcoholic fatty liver disease: A cross-sectional study of a random population sample aged 18 to 65 years. BMC Endocr. Disord. 2015, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Bang, K.B.; Rhee, E.J.; Kwon, H.J.; Lee, M.Y.; Cho, Y.K. Impact of hypothyroidism on the development of non-alcoholic fatty liver disease: A 4-year retrospective cohort study. Clin. Mol. Hepatol. 2015, 21, 372–378. [Google Scholar] [CrossRef]
- Lingad-Sayas, R.C.; Montano, C.N.; Isidro, M.J.C. Prevalence of elevated TSH and its association with dyslipidemia and NAFLD among Filipino adult executive check-up patients in a tertiary hospital. Philipp. J. Intern. Med. 2017, 55, 1–8. [Google Scholar]
- Lee, J.; Ha, J.; Jo, K.; Lim, D.J.; Lee, J.M.; Chang, S.A.; Kang, M.I.; Cha, B.Y.; Kim, M.H. Male-specific association between subclinical hypothyroidism and the risk of non-alcoholic fatty liver disease estimated by hepatic steatosis index: Korea National Health and Nutrition Examination Survey 2013 to 2015. Sci. Rep. 2018, 8, 15145. [Google Scholar] [CrossRef] [Green Version]
- Escudé, A.M.; Pera, G.; Arteaga, I.; Expósito, C.; Rodríguez, L.; Torán, P.; Caballeria, L. Relationship between hypothyroidism and non-alcoholic fatty liver disease in the Spanish population. Med. Clin. 2020, 154, 1–6. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhaldak, D.A.; Melekhovets, O.K.; Orlovskyi, V.F. CYP7A1 gene polymorphism and the characteristics of dyslipidemias in patients with nonalcoholic fatty liver disease concurrent with hypothyroidism. Ter. Arkh. 2017, 89, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Notariza, K.R.; Wisnu, W. The Risk of Developing Non-Alcoholic Fatty Liver Disease in Adult Patients with Subclinical Hypothyroidism Compared to Euthyroid: An Evidence-based Case Report. Acta Med. Indones. 2019, 51, 179–188. [Google Scholar] [PubMed]
- Chaker, L.; Bianco, A.C.; Jonklaas, J.; Peeters, R.P. Hypothyroidism. Lancet 2017, 390, 1550–1562. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.; Jung, J. Obese Subjects with Non-Alcoholic Fatty Liver Disease Have a Higher Risk of Thyroid Dysfunction. Kosin Med. J. 2019, 34, 117. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lv, X.; Yue, F.; Wei, D.; Liu, W.; Zhang, T. Subclinical hypothyroidism and the risk of metabolic syndrome: A meta-analysis of observational studies. Endocr. Res. 2016, 41, 158–165. [Google Scholar] [CrossRef]
- Nasr, P.; Blomdahl, J.; Kechagias, S.; Ekstedt, M. Modifiers of Liver-Related Manifestation in the Course of NAFLD. Curr. Pharm. Des. 2020, 26, 1062–1078. [Google Scholar] [CrossRef]
- Allelein, S.; Schott, M. Schilddrüsenfunktionsstörungen [Thyroid dysfunction]. MMW Fortschr. Med. 2016, 158, 45–52. [Google Scholar] [CrossRef]
- Bianco, A.C. We All Know We Need Them, We Hope They Are Coming, But When? Thyroid 2020, 30, 791–793. [Google Scholar] [CrossRef]
- Ortiga-Carvalho, T.M.; Chiamolera, M.I.; Pazos-Moura, C.C.; Wondisford, F.E. Hypothalamus-Pituitary-Thyroid Axis. Compr. Physiol. 2016, 6, 1387–1428. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.; Hollenberg, A.N. New insights into thyroid hormone action. Pharmacol. Ther. 2017, 173, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, M.J.; Amano, I.; Hollenberg, A.N. Thyroid Hormone Signaling and the Liver. Hepatology 2020. [Google Scholar] [CrossRef]
- Flamant, F.; Cheng, S.Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef]
- Hönes, G.S.; Rakov, H.; Logan, J.; Liao, X.H.; Werbenko, E.; Pollard, A.S.; Præstholm, S.M.; Siersbæk, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, N.; Jakobsson, T.; Fan, R.; Treuter, E. The Nuclear Receptor-Co-repressor Complex in Control of Liver Metabolism and Disease. Front. Endocrinol. 2019, 10, 411. [Google Scholar] [CrossRef] [Green Version]
- Krause, C.; Grohs, M.; El Gammal, A.T.; Wolter, S.; Lehnert, H.; Mann, O.; Mittag, J.; Kirchner, H. Reduced expression of thyroid hormone receptor β in human nonalcoholic steatohepatitis. Endocr. Connect. 2018, 7, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Briet, C.; Suteau-Courant, V.; Munier, M.; Rodien, P. Thyrotropin receptor, still much to be learned from the patients. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 155–164. [Google Scholar] [CrossRef]
- Cioffi, F.; Lanni, A.; Goglia, F. Thyroid hormones, mitochondrial bioenergetics and lipid handling. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 402–407. [Google Scholar] [CrossRef]
- Perra, A.; Plateroti, M.; Columbano, A. T3/TRs axis in hepatocellular carcinoma: New concepts for an old pair. Endocr. Relat. Cancer 2016, 23, R353–R369. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Tang, H.Y.; Leinung, M.; Mousa, S.A.; Hercbergs, A.; Davis, P.J. Action of reverse T3 on cancer cells. Endocr. Res. 2019, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.L.; Fernandes, G.W.; McAninch, E.A.; Bocco, B.M.; Abdalla, S.M.; Ribeiro, M.O.; Mohácsik, P.; Fekete, C.; Li, D.; Xing, X.; et al. Perinatal deiodinase 2 expression in hepatocytes defines epigenetic susceptibility to liver steatosis and obesity. Proc. Natl. Acad. Sci. USA 2015, 112, 14018–14023. [Google Scholar] [CrossRef] [Green Version]
- Luongo, C.; Dentice, M.; Salvatore, D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat. Rev. Endocrinol. 2019, 15, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Gionfra, F.; De Vito, P.; Pallottini, V.; Lin, H.Y.; Davis, P.J.; Pedersen, J.Z.; Incerpi, S. The Role of Thyroid Hormones in Hepatocyte Proliferation and Liver Cancer. Front. Endocrinol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentice, M.; Marsili, A.; Ambrosio, R.; Guardiola, O.; Sibilio, A.; Paik, J.H.; Minchiotti, G.; DePinho, R.A.; Fenzi, G.; Larsen, P.R.; et al. FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J. Clin. Investig. 2010, 120, 4021–4030. [Google Scholar] [CrossRef]
- Goemann, I.M.; Romitti, M.; Meyer, E.; Wajner, S.M.; Maia, A.L. Role of thyroid hormones in the neoplastic process: An overview. Endocr. Relat. Cancer 2017, 24, R367–R385. [Google Scholar] [CrossRef]
- Kester, M.H.; Toussaint, M.J.; Punt, C.A.; Matondo, R.; Aarnio, A.M.; Darras, V.M.; Everts, M.E.; de Bruin, A.; Visser, T.J. Large induction of type III deiodinase expression after partial hepatectomy in the regenerating mouse and rat liver. Endocrinology 2009, 150, 540–545. [Google Scholar] [CrossRef] [Green Version]
- De Pergola, G.; Ciampolillo, A.; Paolotti, S.; Trerotoli, P.; Giorgino, R. Free triiodothyronine and thyroid stimulating hormone are directly associated with waist circumference, independently of insulin resistance, metabolic parameters and blood pressure in overweight and obese women. Clin. Endocrinol. 2007, 67, 265–269. [Google Scholar] [CrossRef]
- Bruck, R.; Weiss, S.; Traister, A.; Zvibel, I.; Aeed, H.; Halpern, Z.; Oren, R. Induced hypothyroidism accelerates the regression of liver fibrosis in rats. J. Gastroenterol. Hepatol. 2007, 22, 2189–2194. [Google Scholar] [CrossRef]
- Abdalla, S.M.; Bianco, A.C. Defending plasma T3 is a biological priority. Clin. Endocrinol. 2014, 81, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbers, L.P.; Kastelein, J.J.; Sjouke, B. Thyroid Hormone Mimetics: The Past, Current Status and Future Challenges. Curr. Atheroscler. Rep. 2016, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohinc, B.N.; Michelotti, G.; Xie, G.; Pang, H.; Suzuki, A.; Guy, C.D.; Piercy, D.; Kruger, L.; Swiderska-Syn, M.; Machado, M.; et al. Repair-related activation of hedgehog signaling in stromal cells promotes intrahepatic hypothyroidism. Endocrinology 2014, 155, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Yamamoto, N.; Inoue, A.; Morisawa, S. Fatty acyl-CoAs are potent inhibitors of the nuclear thyroid hormone receptor in vitro. J. Biochem. 1990, 107, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Hou, S.; Zhang, D.; Xia, H.; Wang, Y.C.; Jiang, J.; Yin, H.; Ying, H. Regulation of fatty acid composition and lipid storage by thyroid hormone in mouse liver. Cell Biosci. 2014, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int. J. Mol. Sci. 2017, 18, 744. [Google Scholar] [CrossRef]
- Kechagias, S.; Nasr, P.; Blomdahl, J.; Ekstedt, M. Established and emerging factors affecting the progression of nonalcoholic fatty liver disease. Metabolism 2020, 154183. [Google Scholar] [CrossRef]
- Teixeira, P.; Dos Santos, P.B.; Pazos-Moura, C.C. The role of thyroid hormone in metabolism and metabolic syndrome. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820917869. [Google Scholar] [CrossRef]
- Coppola, M.; Cioffi, F.; Moreno, M.; Goglia, F.; Silvestri, E. 3,5-diiodo-L-thyronine: A Possible Pharmacological Agent? Curr. Drug Deliv. 2016, 13, 330–338. [Google Scholar] [CrossRef]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Grasselli, E.; Voci, A.; Demori, I.; Vecchione, G.; Compalati, A.D.; Gallo, G.; Goglia, F.; De Matteis, R.; Silvestri, E.; Vergani, L. Triglyceride Mobilization from Lipid Droplets Sustains the Anti-Steatotic Action of Iodothyronines in Cultured Rat Hepatocytes. Front. Physiol. 2016, 6, 418. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302. [Google Scholar] [CrossRef] [PubMed]
- Pandrc, M.S.; Ristić, A.; Kostovski, V.; Stanković, M.; Antić, V.; Milin-Lazović, J.; Ćirić, J. The Effect of Early Substitution of Subclinical Hypothyroidism on Biochemical Blood Parameters and the Quality of Life. J. Med. Biochem. 2017, 36, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Efstathiadou, Z.A.; Kita, M.D.; Polyzos, S.A. Thyroid dysfunction and non-alcoholic fatty liver disease. Minerva Endocrinol. 2018, 43, 367–376. [Google Scholar] [CrossRef]
- Wahrenberg, H.; Wennlund, A.; Arner, P. Adrenergic regulation of lipolysis in fat cells from hyperthyroid and hypothyroid patients. J. Clin. Endocrinol. Metab. 1994, 78, 898–903. [Google Scholar] [CrossRef]
- Song, Y.; Xu, C.; Shao, S.; Liu, J.; Xing, W.; Xu, J.; Qin, C.; Li, C.; Hu, B.; Yi, S.; et al. Thyroid-stimulating hormone regulates hepatic bile acid homeostasis via SREBP-2/HNF-4α/CYP7A1 axis. J. Hepatol. 2015, 62, 1171–1179. [Google Scholar] [CrossRef]
- Duntas, L.H.; Brenta, G. Renewed Focus on the Association Between Thyroid Hormones and Lipid Metabolism. Front. Endocrinol. 2018, 9, 511. [Google Scholar] [CrossRef]
- Jain, R.B. Associations between the levels of thyroid hormones and lipid/lipoprotein levels: Data from National Health and Nutrition Examination Survey 2007–2012. Environ. Toxicol. Pharmacol. 2017, 53, 133–144. [Google Scholar] [CrossRef]
- Incerpi, S.; Davis, P.J.; Pedersen, J.Z.; Lanni, A. Nongenomic Actions of Thyroid Hormones. Endocrinology 2018, 259–284. [Google Scholar] [CrossRef]
- Zhang, X.; Song, Y.; Feng, M.; Zhou, X.; Lu, Y.; Gao, L.; Yu, C.; Jiang, X.; Zhao, J. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J. Lipid Res. 2015, 56, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, G.; Eder, K.; Ringseis, R. Sterol regulatory element-binding proteins are transcriptional regulators of the thyroglobulin gene in thyroid cells. Biochim. Biophys. Acta 2016, 1859, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sánchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buqué, X.; Aurrekoetxea, I.; Delgado, T.C.; Vázquez-Martínez, R.; et al. Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab. 2017, 26, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Fazaeli, M.; Khoshdel, A.; Shafiepour, M.; Rohban, M. The influence of subclinical hypothyroidism on serum lipid profile, PCSK9 levels and CD36 expression on monocytes. Diabetes Metab. Syndr. 2019, 13, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 2019, 26, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanicki, T.; Balcerzyk, A.; Niemiec, P.; Trautsolt, W.; Grzeszczak, W.; Ochalska-Tyka, A.; Krauze, J.; Nowak, T.; Żak, I. The relationship between CYP7A1 polymorphisms, coronary artery disease & serum lipid markers. Biomark. Med. 2019, 13, 1199–1208. [Google Scholar] [CrossRef]
- Sharma, P.; Levesque, T.; Boilard, E.; Park, E.A. Thyroid hormone status regulates the expression of secretory phospholipases. Biochem. Biophys. Res. Commun. 2014, 444, 56–62. [Google Scholar] [CrossRef]
- Araki, O.; Ying, H.; Zhu, X.G.; Willingham, M.C.; Cheng, S.Y. Distinct dysregulation of lipid metabolism by unliganded thyroid hormone receptor isoforms. Mol. Endocrinol. 2009, 23, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Lee, H.Y.; Jurczak, M.J.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.A.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid hormone receptor-α gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 2012, 153, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Iannucci, L.F.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Yen, P.M.; Sinha, R.A. Metabolomic analysis shows differential hepatic effects of T2 and T3 in rats after short-term feeding with high fat diet. Sci. Rep. 2017, 7, 2023. [Google Scholar] [CrossRef] [Green Version]
- Beukhof, C.M.; Massolt, E.T.; Visser, T.J.; Korevaar, T.; Medici, M.; de Herder, W.W.; Roeters van Lennep, J.E.; Mulder, M.T.; de Rijke, Y.B.; Reiners, C.; et al. Effects of Thyrotropin on Peripheral Thyroid Hormone Metabolism and Serum Lipids. Thyroid 2018, 28, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Socarrás, J.F.A.; Bedi, M.; Ness, G.C. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim. Biophys. Acta 2007, 1771, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.S.; Sinha, R.A.; Ota, S.; Katsuki, M.; Yen, P.M. Thyroid hormone negatively regulates CDX2 and SOAT2 mRNA expression via induction of miRNA-181d in hepatic cells. Biochem. Biophys. Res. Commun. 2013, 440, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, K.; Bo, T.; Zhou, L.; Gao, L.; Zhou, X.; Chen, W. Integrated microRNA and proteome analysis reveal a regulatory module in hepatic lipid metabolism disorders in mice with subclinical hypothyroidism. Exp. Ther. Med. 2020, 19, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.A.; Yen, P.M. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Krotkiewski, M. Thyroid hormones and treatment of obesity. Int. J. Obes. Relat. Metab. Disord. 2000, 24, S116–S119. [Google Scholar] [CrossRef] [Green Version]
- Karbownik-Lewińska, M.; Stępniak, J.; Żurawska, A.; Lewiński, A. Less Favorable Lipid Profile and Higher Prevalence of Thyroid Antibodies in Women of Reproductive Age with High-Normal TSH-Retrospective Study. Int. J. Environ. Res. Public Health 2020, 17, 2122. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Priyadarshi, R.N.; Anand, U. Non-alcoholic Fatty Liver Disease: Growing Burden, Adverse Outcomes and Associations. J. Clin. Transl. Hepatol. 2020, 8, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xie, Z.; Shen, Y.; Xia, S.F. The Roles of Thyroid and Thyroid Hormone in Pancreas: Physiology and Pathology. Int. J. Endocrinol. 2018, 2018, 2861034. [Google Scholar] [CrossRef]
- Geisler, C.E.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.N. Update in lipid alterations in subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2012, 97, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, I.; Borawski, J.; Nikołajuk, A.; Budlewski, T.; Otziomek, E.; Górska, M.; Strączkowski, M. Insulin sensitivity, plasma adiponectin and sICAM-1 concentrations in patients with subclinical hypothyroidism: Response to levothyroxine therapy. Endocrine 2011, 40, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Velarde-Ruiz Velasco, J.A.; García-Jiménez, E.S.; García-Zermeño, K.R.; Morel-Cerda, E.C.; Aldana-Ledesma, J.M.; Castro-Narro, G.E.; Cerpa-Cruz, S.; Tapia-Calderón, D.K.; Mercado-Jauregui, L.A.; Contreras-Omaña, R. Extrahepatic complications of non-alcoholic fatty liver disease: Its impact beyond the liver. Complicaciones extrahepáticas de la enfermedad por hígado graso no alcohólico: Impacto más allá del hígado. Rev. Gastroenterol. Mex. 2019, 84, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Moro, L. Mitochondria at the Crossroads of Physiology and Pathology. J. Clin. Med. 2020, 9, 1971. [Google Scholar] [CrossRef]
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Currò, D. Thyroid Hormones, Oxidative Stress, and Inflammation. Mediat. Inflamm. 2016, 2016, 6757154. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Lanni, A.; Moreno, M.; Goglia, F. Mitochondrial Actions of Thyroid Hormone. Compr. Physiol. 2016, 6, 1591–1607. [Google Scholar] [CrossRef]
- Yamauchi, M.; Kambe, F.; Cao, X.; Lu, X.; Kozaki, Y.; Oiso, Y.; Seo, H. Thyroid hormone activates adenosine 5′-monophosphate-activated protein kinase via intracellular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase kinase-beta. Mol. Endocrinol. 2008, 22, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górska, M.; Dobrzyń, A.; Langfort, J.; Górski, J. Effect of hypothyreosis on the content of ceramides in rat tissues. J. Physiol. Pharmacol. 2003, 54, 89–97. [Google Scholar] [PubMed]
- Öztürk, Ü.; Vural, P.; Özderya, A.; Karadağ, B.; Doğru-Abbasoğlu, S.; Uysal, M. Oxidative stress parameters in serum and low density lipoproteins of Hashimoto’s thyroiditis patients with subclinical and overt hypothyroidism. Int. Immunopharmacol. 2012, 14, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Manka, P.; Coombes, J.D.; Boosman, R.; Gauthier, K.; Papa, S.; Syn, W.K. Thyroid hormone in the regulation of hepatocellular carcinoma and its microenvironment. Cancer Lett. 2018, 419, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int. J. Mol. Sci. 2020, 21, 4140. [Google Scholar] [CrossRef]
- Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int. J. Mol. Sci. 2019, 20, 1948. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Gao, C.; Chen, X.; Wang, Y.; Tian, L. Adipokine expression and endothelial function in subclinical hypothyroidism rats. Endocr. Connect. 2018, 7, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Gamberi, T.; Magherini, F.; Modesti, A.; Fiaschi, T. Adiponectin Signaling Pathways in Liver Diseases. Biomedicines 2018, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Nier, A.; Huber, Y.; Labenz, C.; Michel, M.; Bergheim, I.; Schattenberg, J.M. Adipokines and Endotoxemia Correlate with Hepatic Steatosis in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 699. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Ribaudo, M.C.; Zappaterreno, A.; Iannucci, C.V.; Leonetti, F. Relationship of thyroid function with body mass index, leptin, insulin sensitivity and adiponectin in euthyroid obese women. Clin. Endocrinol. 2005, 62, 487–491. [Google Scholar] [CrossRef]
- Johannsen, K.; Flechtner-Mors, M.; Kratzer, W.; Koenig, W.; Boehm, B.O.; Schmidberger, J.; EMIL-Study Group. Association Between Visfatin and Hepatic Steatosis in the General Population During Long-Term Follow-Up. Horm. Metab. Res. 2019, 51, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Elkabany, Z.A.; Hamza, R.T.; Ismail, E.; Elsharkawy, A.; Yosry, A.; Musa, S.; Khalaf, M.A.; Elgawesh, R.M.; Esmat, G. Serum visfatin level as a noninvasive marker for nonalcoholic fatty liver disease in children and adolescents with obesity: Relation to transient elastography with controlled attenuation parameter. Eur. J. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Li, H.; Long, X.; Rye, K.A.; Ong, K.L. Fibroblast growth factor 21 in non-alcoholic fatty liver disease. Metabolism 2019, 101, 153994. [Google Scholar] [CrossRef]
- Lee, Y.; Park, Y.J.; Ahn, H.Y.; Lim, J.A.; Park, K.U.; Choi, S.H.; Park, D.J.; Oh, B.C.; Jang, H.C.; Yi, K.H. Plasma FGF21 levels are increased in patients with hypothyroidism independently of lipid profile. Endocr. J. 2013, 60, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Adams, A.C.; Astapova, I.; Fisher, F.M.; Badman, M.K.; Kurgansky, K.E.; Flier, J.S.; Hollenberg, A.N.; Maratos-Flier, E. Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARalpha-dependent manner. J. Biol. Chem. 2010, 285, 14078–14082. [Google Scholar] [CrossRef] [Green Version]
- Delitala, A.P.; Steri, M.; Pilia, M.G.; Dei, M.; Lai, S.; Delitala, G.; Schlessinger, D.; Cucca, F. Menopause modulates the association between thyrotropin levels and lipid parameters: The SardiNIA study. Maturitas 2016, 92, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Brenta, G.; Berg, G.; Miksztowicz, V.; Lopez, G.; Lucero, D.; Faingold, C.; Murakami, M.; Machima, T.; Nakajima, K.; Schreier, L. Atherogenic Lipoproteins in Subclinical Hypothyroidism and Their Relationship with Hepatic Lipase Activity: Response to Replacement Treatment with Levothyroxine. Thyroid 2016, 26, 365–372. [Google Scholar] [CrossRef]
- Jonklass, J.; Bianco, A.C.; Bauer, A.J.; Burman, K.D.; Cappola, A.R.; Celi, F.S.; Cooper, D.S.; Kim, B.W.; Peeters, R.P.; Rosenthal, M.S.; et al. Guidelines for the treatment of hypothyroidism. Thyroid 2014, 24, 1670–1751. [Google Scholar] [CrossRef] [Green Version]
- Garber, J.R.; Cobin, R.H.; Gharib, H. Clinical practice guidelines for hypothyroidism in adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr. Pract. 2012, 18, 988–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yu, Y.; Zhao, M.; Zheng, D.; Zhang, X.; Guan, Q.; Xu, C.; Gao, L.; Zhao, J.; Zhang, H. Benefits of levothyroxine replacement therapy on nonalcoholic fatty liver disease in subclinical hypothyroidism patients. Int. J. Endocrinol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Bruinstroop, E.; Dalan, R.; Yang, C.; Bee, Y.M.; Chandran, K.; Cho, L.W.; Soh, S.B.; Teo, E.K.; Toh, S.A.; Leow, M.K.S.; et al. Low dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD. J. Clin. Endocrinol. Metab. 2018, 103, 2698–2706. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Chen, C.Y.; Tsai, M.M.; Tsai, C.Y.; Lin, K.H. Molecular functions of thyroid hormones and their clinical significance in liver-related diseases. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Senese, R.; Cioffi, F.; de Lange, P.; Leanza, C.; Iannucci, L.F.; Silvestri, E.; Moreno, M.; Lombardi, A.; Goglia, F.; Lanni, A. Both 3,5-diiodo-L-thyronine and 3,5,3′-triiodo-L-thyronine prevent short-term hepatic lipid accumulation via distinct mechanisms in rats being fed a high-fat diet. Front. Physiol. 2017, 8, 706. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Columbano, A.; Perra, A. Thyroid hormones, thyromimetics and their metabolites in the treatment of liver disease. Front. Endocinol. 2018, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Perra, A.; Simbula, G.; Simbula, M.; Pibiri, M.; Kowalik, M.A.; Sulas, P.; Cocco, M.T.; Ledda-Columbano, G.M.; Columbano, A. Thyroid hormone (T3) and TRβ agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008, 22, 2981–2989. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clint. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Cable, E.E.; Finn, P.D.; Stebbins, J.W.; Hou, J.; Ito, B.R.; van Poelje, P.D.; Linemeyer, D.L.; Erion, M.D. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepayology 2009, 49, 407–417. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic fatty liver disease and hypercholesterolemia: Role of thyroid hormones, metabolites and agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Crespo, M.; Csikasz, R.I.; Martínez-Sánchez, N.; Diéguez, C.; Cannon, B.; Nedergaard, J.; López, M. Essential role of UCP1 modulating the central effects of thyroid hormones on energy balance. Mol. Metab. 2016, 5, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Arrojo, E.D.R.; Fonseca, T.L.; Werneck-de-Castro, J.P.; Bianco, A.C. Role of type 2 iodothyronine deiodinase (D2) in the control of thyroid hormone signaling. Biochim. Biophys. Acta 2013, 1830, 3956–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, A.; Navarrete-Ramírez, P.; Olvera, A.; García, G.C. 3,5-Diiodothyronine (T2) is on a role. A new hormone in search of recognition. Gen. Comp. Endocrinol. 2014, 203, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; De Matteis, R.; Goglia, F.; Cioffi, F.; Fugassa, E.; Gallo, G.; Vergani, L. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. J. Hepatol. 2011, 54, 1230–1236. [Google Scholar] [CrossRef]
- Jonas, W.; Lietzow, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-L-thyronine (3,5-t2) exerts thyromimetic effects on hypothalamus-pituitary-thyroid axis, body composition, and energy metabolism in male diet-induced obese mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, F.; Zambad, S.P.; Chhipa, L.; Senese, R.; Busiello, R.A.; Tuli, D.; Munshi, S.; Moreno, M.; Lombardi, A.; Gupta, R.C.; et al. TRC150094, a novel functional analog of iodothyronines, reduces adiposity by increasing energy expenditure and fatty acid oxidation in rats receiving a high-fat diet. FASEB J. 2010, 24, 3451–3461. [Google Scholar] [CrossRef]
- Da Silva Teixeira, S.; Filgueira, C.; Sieglaff, D.H.; Benod, C.; Villagomez, R.; Minze, L.J.; Zhang, A.; Webb, P.; Nunes, M.T. 3,5-diiodothyronine (3,5-T2) reduces blood glucose independently of insulin sensitization in obese mice. Acta Physiol. 2017, 220, 238–250. [Google Scholar] [CrossRef]
- Zucchi, R.; Chiellini, G.; Scanlan, T.S.; Grandy, D.K. Trace amine-associated receptors and their ligands. Br. J. Pharmacol. 2006, 149, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, R.; Onali, S.; Thorburn, D.; Davidson, B.R.; Gurusamy, K.S.; Tsochatzis, E. Pharmacological interventions for non-alcohol related fatty liver disease (NAFLD): An attempted network meta-analysis. Cochrane Database Syst. Rev. 2017, 3, CD011640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghelardoni, S.; Chiellini, G.; Frascarelli, S.; Saba, A.; Zucchi, R. Uptake and metabolic effects of 3-iodothyronamine in hepatocytes. J. Endocrinol. 2014, 221, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilà-Brau, A.; De Sousa-Coelho, A.L.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF2 expression in HepG2 cell line. J. Biol. Chem. 2011, 286, 20423–20430. [Google Scholar] [CrossRef] [Green Version]
- Haviland, J.A.; Reiland, H.; Butz, D.E.; Tonelli, M.; Porter, W.P.; Zucchi, R.; Scanlan, T.S.; Chiellini, G.; Assadi-Porter, F.M. NMR-based metabolomics and breath studies show lipid and protein catabolism during low dose chronic T(1)AM treatment. Obesity 2013, 21, 2538–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assadi-Porter, F.M.; Reiland, H.; Sabatini, M.; Lorenzini, L.; Carnicelli, V.; Rogowski, M.; Alpergin, E.S.S.; Tonelli, M.; Ghelardoni, S.; Saba, A.; et al. Metabolic Reprogramming by 3-Iodothyronamine (T1AM): A New Perspective to Reverse Obesity through Co-Regulation of Sirtuin 4 and 6 Expression. Int. J. Mol. Sci. 2018, 19, 1535. [Google Scholar] [CrossRef] [Green Version]
- Ayuob, N.N.; Abdel-Hamid, A.A.H.M.; Helal, G.M.M.; Mubarak, W.A. Thymoquinone reverses nonalcoholic fatty liver disease (NAFLD) associated with experimental hypothyroidism. Rom. J. Morphol. Embryol. 2019, 60, 479–486. [Google Scholar]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Mak, K.M.; Chen, L.L.; Lee, T.F. Codistribution of collagen type IV and laminin in liver fibrosis of elderly cadavers: Immune-histochemical marker of perisinusoidal basement membrane formation. Anat. Rec. 2013, 296, 953–964. [Google Scholar] [CrossRef]
- Luo, F.; Ishigami, M.; Achiwa, K.; Ishizu, Y.; Juzuya, T.; Honda, T.; Hayashi, K.; Ishikawa, T.; Katano, Y.; Goto, H. Raloxifene ameliorates liver fibrosis of nonalcoholic steatohepatitis induced by choline-deficient high-fat diet in ovariectomized mice. Dig. Dis. Sci. 2015, 60, 2730–2739. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Subudhi, U.; Chainy, G.B. Expression of hepatic antioxidant genes in L-thyroxine-induced hyperthyroid rats: Regulation by vitamin E and curcumin. Chem. Biol. Interact 2010, 183, 3014–3316. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, H.; Ahmad, A.; Ahmad, I.Z. Nigella sativa L. and its bioactive constituents as hepatoprotectant: A review. Curr. Pharm. Biotechnol. 2018, 19, 43–67. [Google Scholar] [CrossRef]
- Vatner, D.F.; Weismann, D.; Beddow, S.A.; Kumashiro, N.; Erion, D.M.; Liao, X.H.; Grover, G.J.; Webb, P.; Phillips, K.J.; Weiss, R.E.; et al. Thyroid hormone receptor-β agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am. J. Physiol. Endocrinol. Metab. 2013, 305, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Lammel Lindemann, J.; Webb, P. Sobetirome: The past, present and questions about the future. Expert. Opin. Ther. Targets 2016, 20, 145–149. [Google Scholar] [CrossRef]
- Kelly, M.J.; Pietranico-Cole, S.; Larigan, J.D. Discovery of 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridine-3-yloxy) phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,3]triazine-6-carbonitrile (MGL-3196), a highly selective thyroid hormone receptor β agonist in clinical trials for the treatment of dyslipidemia. J. Med. Chem. 2014, 57, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicenter, randomized, double-blind, placebo-controlled, phase 2 trail. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Taub, R.; Chiang, E.; Chabot-Blanchet, M. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted hormone receptor—β agonist. Atherosclerosis 2013, 230, 373–380. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Sanyal, A.J.; Neuschwander-Tetri, B.A. Improvements in histologic features and diagnosis associated with improvement in fibrosis in nonalcoholic steatohepatitis: Results from the Nonalcoholic Steatohepatitis Clinical Research Network treatment trials. Hepatology 2019, 70, 522–531. [Google Scholar] [CrossRef]
- Tsimikas, S. A test in context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Loomba, R.; Neutel, J.; Bernard, D.; Severance, R.; Mohseni, R.; Dao, M.; Saini, S.; Margaritescu, C.; Homer, K.; Tran, B.; et al. VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat in patients with non-alcoholic liver disease: A phase 2 randomized, placebo-controlled trial. Hepatology 2018, 68, 1447A. [Google Scholar]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Müller, L.; de Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell 2016, 26, 1422–1430. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Country | Year | Main Findings | NAFLD Impact | First Author (Reference) | |
|---|---|---|---|---|---|
| Thyroid hormones | |||||
| Levothyroxine (T4) | China | 2017 |
|
| Liu, L. [174] |
| Singapore | 2018 |
|
| Bruinstroop, E. [175] | |
| Triiodothyronine (T3) | Italy | 2008 |
|
| Perra, A. [179] |
| USA | 2009 |
|
| Cable, E.E. [181] | |
| China | 2014 |
|
| Yao, X. [106] | |
| Spain | 2016 |
| - | Alvarez-Crespo, M. [183] | |
| Italy | 2017 |
|
| Iannucci, L.F. [131] | |
| Thyroid hormones metabolites | |||||
| T2 (3,5-diiodo-L-thyronine) | Italy | 2017 |
|
| Grasselli, E. [187] |
| Italy | 2017 |
|
| Iannucci, L.F. [131] | |
| Italy | 2017 |
|
| Senese, R. [188] | |
| T1AM (3-iodothyronamine) | Italy | 2014 |
| - | Ghelardoni, S. [193] |
| USA | 2018 |
|
| Assadi-Porter, F.M. [196] | |
| THR-β Selective Thyromimetics | |||||
| Sobetirome (GC-1) | Italy | 2008 |
|
| Perra, A. [179] |
| Eprotirome (KB-2115) | USA | 2013 |
|
| Vatner, D.F. [204] |
| USA | 2015 |
| - | Lammel Lindemann, J. [205] | |
| MB07811 | USA | 2009 |
|
| Cable, E.E. [181] |
| Resmetirom (MGL-3196) | USA | 2019 |
|
| Harrison, S.A. [207] |
| VK2809 (MB 07811) | USA | 2018 |
|
| Loomba, R. [211] |
| Glucagon/T3 Conjugate | |||||
| Germany | 2016 |
|
| Finan, B. [212] | |
| Thymoquinone | |||||
| Saudi Arabia | 2019 |
|
| Ayuob, N.N. [197] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanase, D.M.; Gosav, E.M.; Neculae, E.; Costea, C.F.; Ciocoiu, M.; Hurjui, L.L.; Tarniceriu, C.C.; Floria, M. Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options. Int. J. Mol. Sci. 2020, 21, 5927. https://doi.org/10.3390/ijms21165927
Tanase DM, Gosav EM, Neculae E, Costea CF, Ciocoiu M, Hurjui LL, Tarniceriu CC, Floria M. Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options. International Journal of Molecular Sciences. 2020; 21(16):5927. https://doi.org/10.3390/ijms21165927
Chicago/Turabian StyleTanase, Daniela Maria, Evelina Maria Gosav, Ecaterina Neculae, Claudia Florida Costea, Manuela Ciocoiu, Loredana Liliana Hurjui, Claudia Cristina Tarniceriu, and Mariana Floria. 2020. "Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options" International Journal of Molecular Sciences 21, no. 16: 5927. https://doi.org/10.3390/ijms21165927
APA StyleTanase, D. M., Gosav, E. M., Neculae, E., Costea, C. F., Ciocoiu, M., Hurjui, L. L., Tarniceriu, C. C., & Floria, M. (2020). Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options. International Journal of Molecular Sciences, 21(16), 5927. https://doi.org/10.3390/ijms21165927