CCL4 Inhibition in Atherosclerosis: Effects on Plaque Stability, Endothelial Cell Adhesiveness, and Macrophages Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
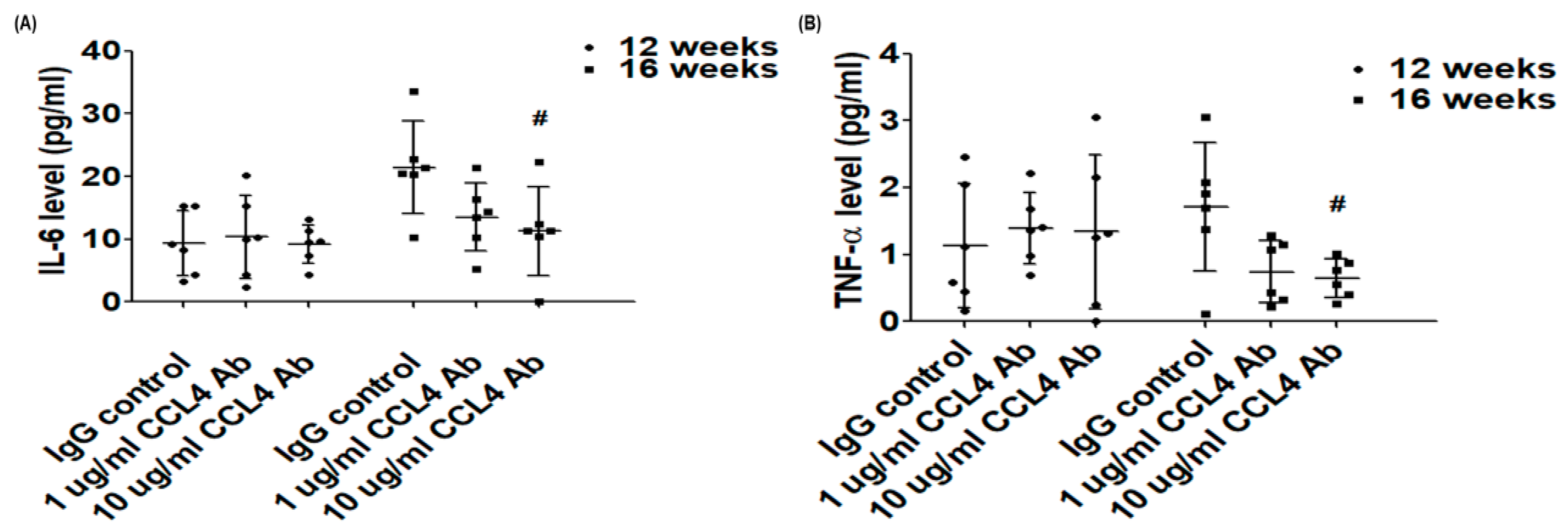
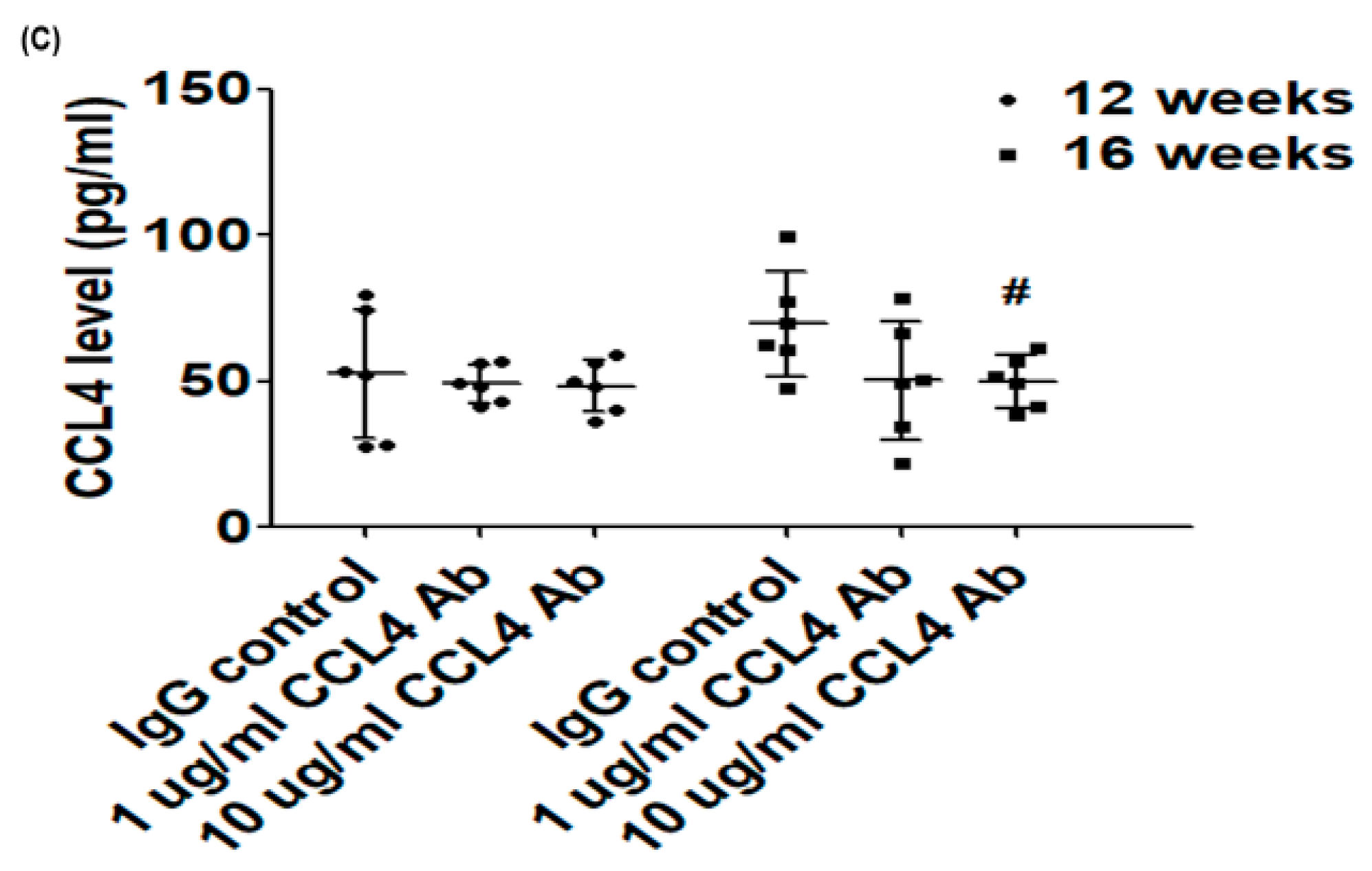
2.1. Direct Inhibition of CCL4 Attenuated Inflammatory Cytokines in Atherosclerotic Mice
2.2. Direct Inhibition of CCL4 Benefited Metabolic Parameters Might Be through the Upregulation of Liver X Receptors (LXRs) in Atherosclerotic Mice
2.3. Direct Inhibition of CCL4 Attenuated Plaque Development and Reduced Macrophage Infiltration in Atherosclerotic Mice
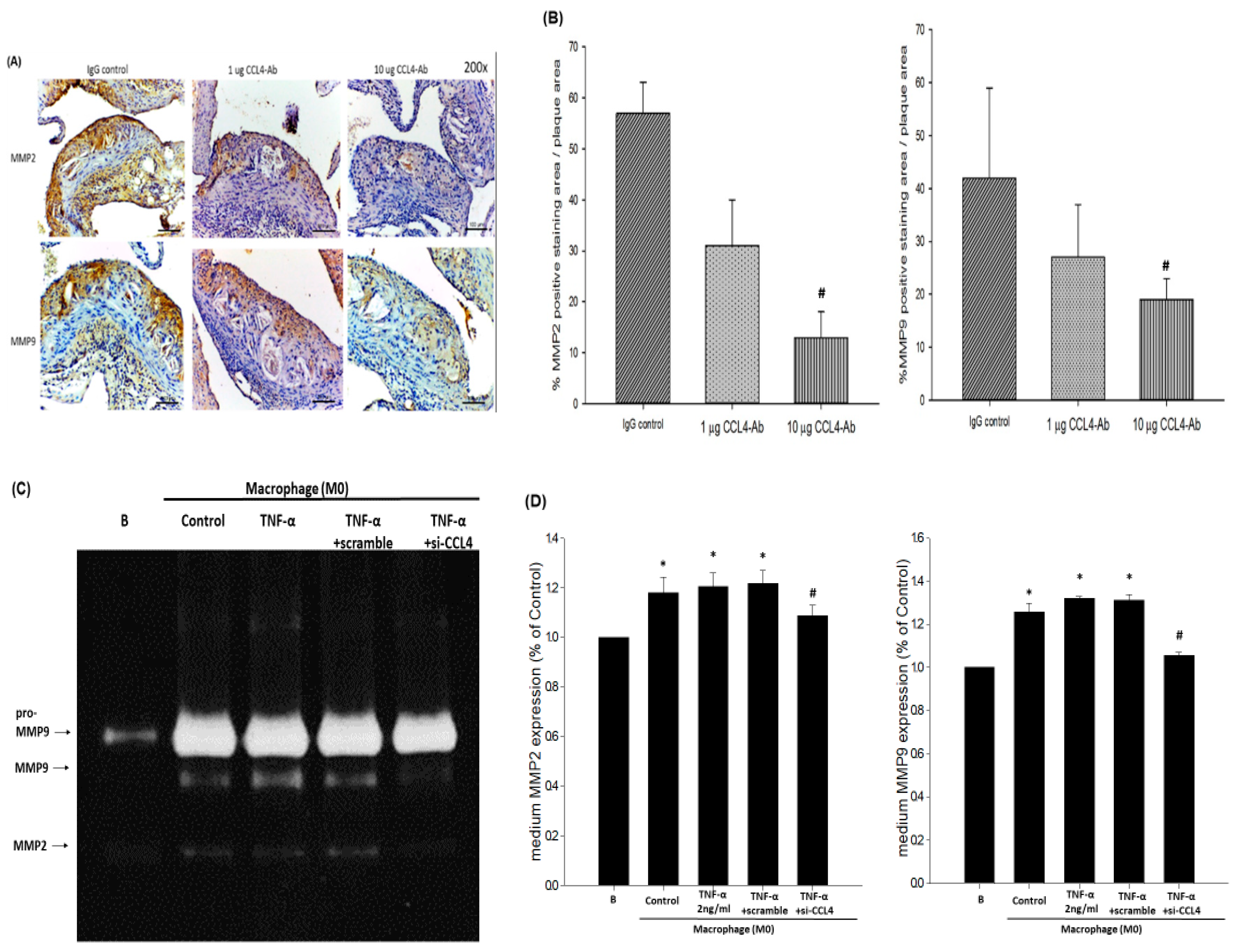
2.4. Blockade of CCL4 Reduced MMP2 and MMP9 Expression In Vivo and Their Expression and Activity in Macrophages In Vitro
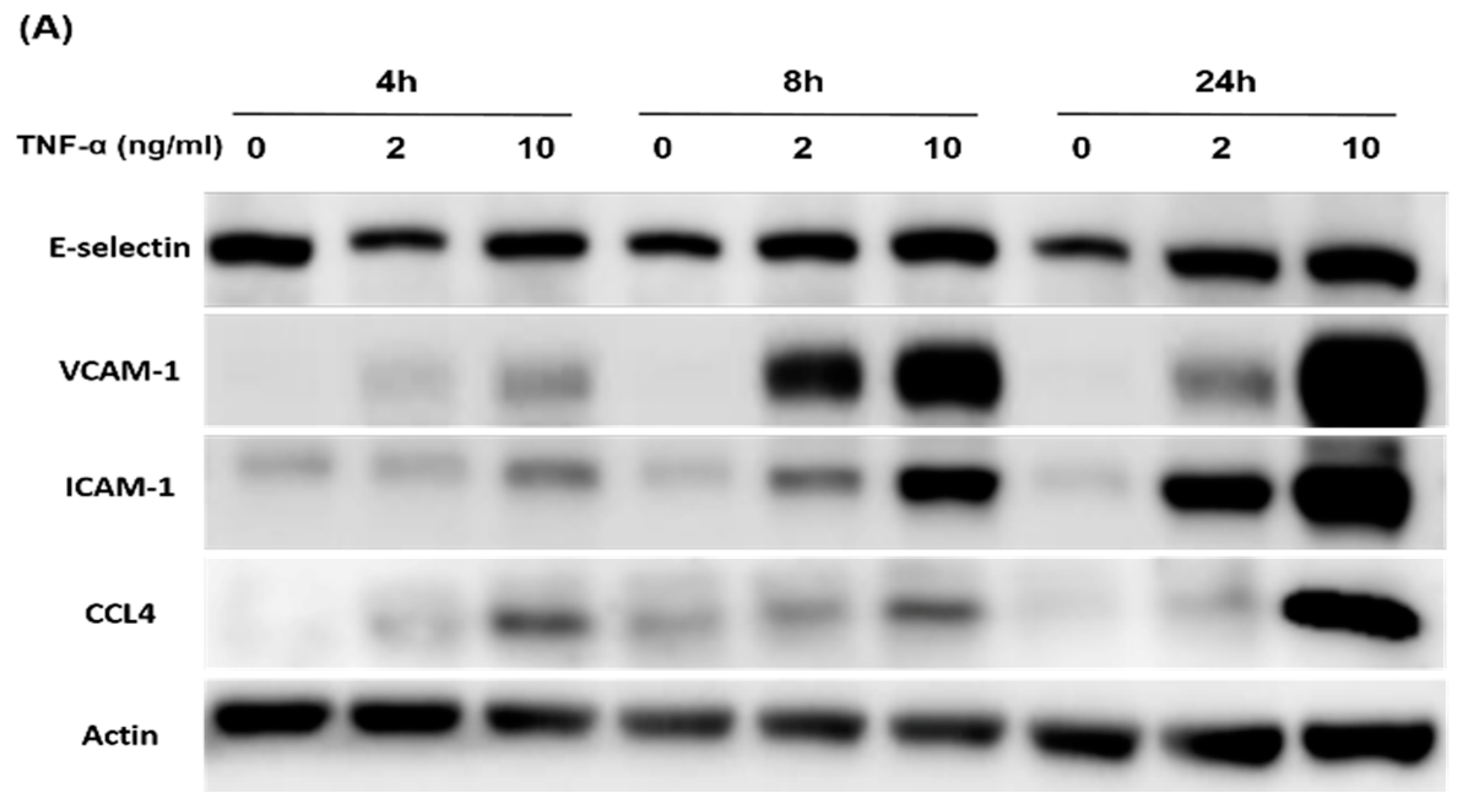
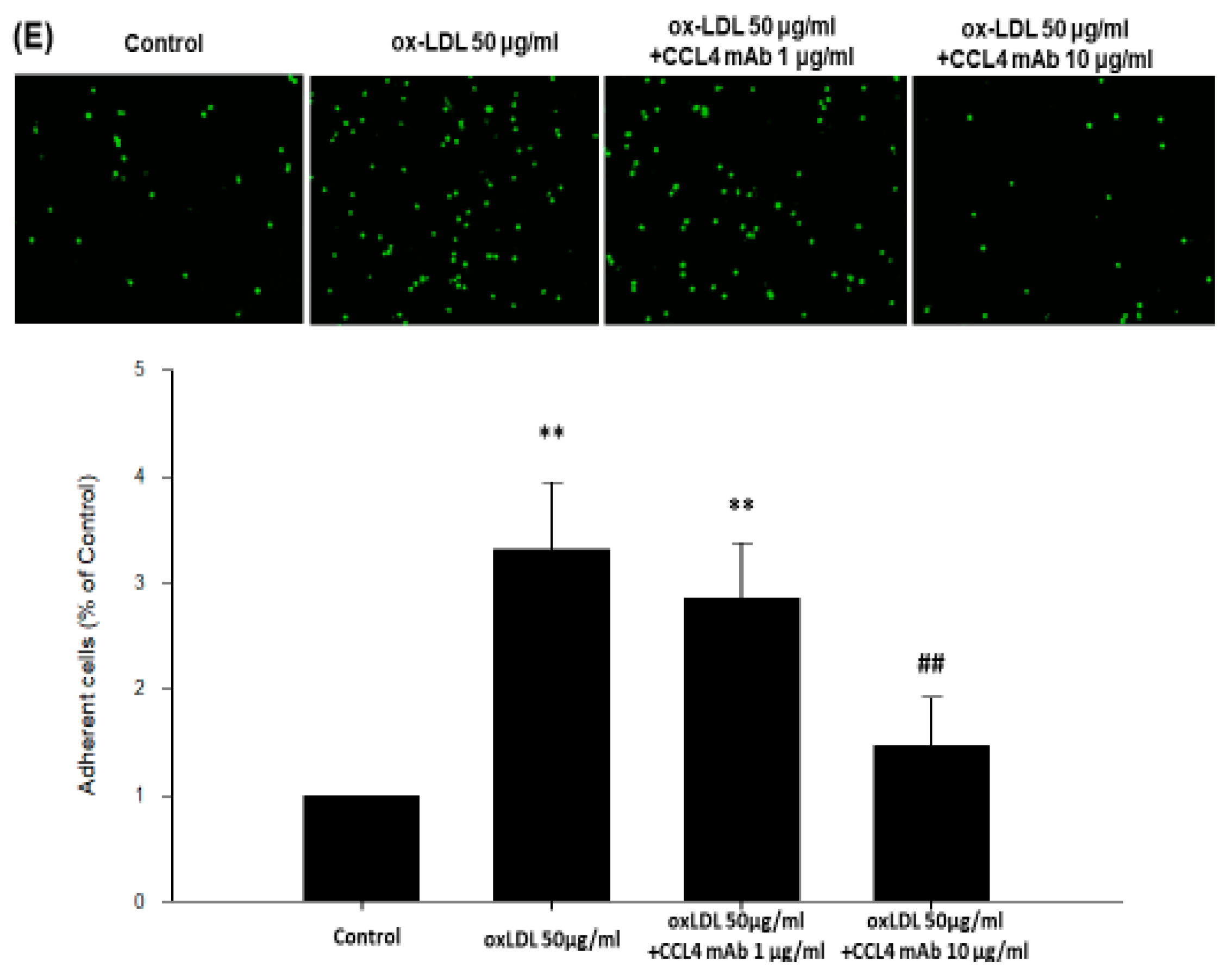
2.5. Direct Inhibition of CCL4 Decreased TNF-α- and ox-LDL-Induced Endothelial Adhesiveness and Adhesion Molecule Expression
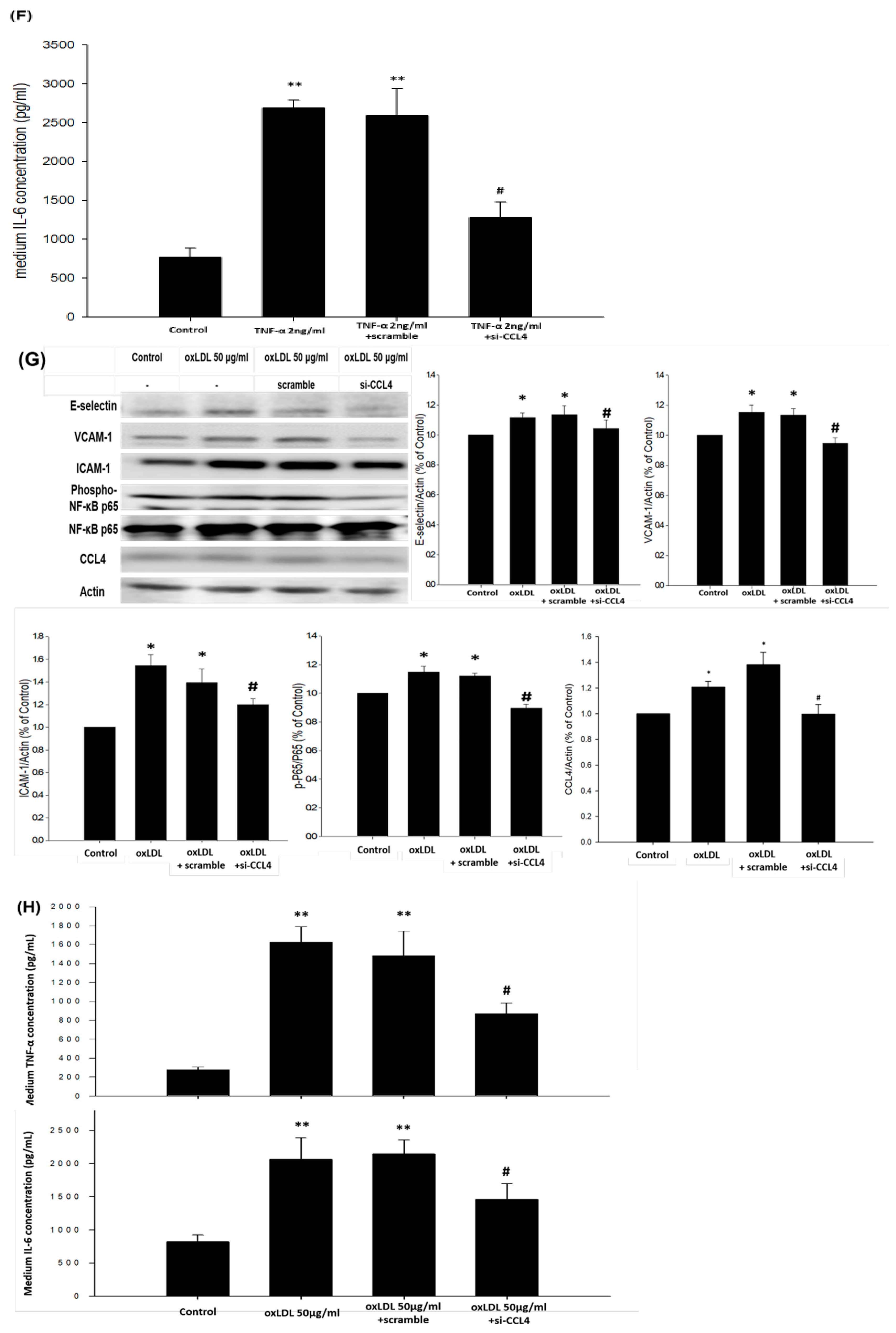
2.6. Silencing of Endogenous CCL4 Down-Regulated TNF-α- and ox-LDL-Induced Adhesion and Inflammation Molecules through the NFκB Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. In Vivo Study
4.1.1. Animal Model and Study Protocol
4.1.2. Tissue Harvesting
4.1.3. Histological Staining
4.1.4. Immunohistochemical Staining
4.1.5. Biochemical Indexes
4.1.6. ELISA
4.2. In Vitro Study
4.2.1. Human Coronary Artery Endothelial Cells (HCAEC) Culture
4.2.2. Adhesion Assay of HCAEC
4.2.3. Matrix Metalloproteinase Activity in the Culture Medium of Macrophages
4.2.4. Gelatin Zymography
4.2.5. Western Blotting
4.2.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seneviratne, A.; Hulsmans, M.; Holvoet, P.; Monaco, C. Biomechanical factors and macrophages in plaque stability. Cardiovasc. Res. 2013, 99, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.T.; Chen, J.W. Emerging role of chemokine CC motif ligand 4 related mechanisms in diabetes mellitus and cardiovascular disease: Friends or foes? Cardiovasc. Diabetol. 2016, 15, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, E.; Wang, J.; Norcross, M.A. Amino-terminal processing of MIP-1beta/CCL4 by CD26/dipeptidyl-peptidase IV. J Cell Biochem 2004, 92, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Guan, E.; Wang, J.; Roderiquez, G.; Norcross, M.A. Natural truncation of the chemokine MIP-1 beta /CCL4 affects receptor specificity but not anti-HIV-1 activity. J. Biol. Chem. 2002, 277, 32348–32352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proost, P.; Menten, P.; Struyf, S.; Schutyser, E.; De Meester, I.; Van Damme, J. Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood 2000, 96, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Bystry, R.S.; Aluvihare, V.; Welch, K.A.; Kallikourdis, M.; Betz, A.G. B cells and professional APCs recruit regulatory T cells via CCL4. Nat. Immunol. 2001, 2, 1126–1132. [Google Scholar] [CrossRef]
- Mallat, Z.; Taleb, S.; Ait-Oufella, H.; Tedgui, A. The role of adaptive T cell immunity in atherosclerosis. J. Lipid Res. 2009, 50, S364–S369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatara, Y.; Ohishi, M.; Yamamoto, K.; Shiota, A.; Hayashi, N.; Iwamoto, Y.; Takeda, M.; Takagi, T.; Katsuya, T.; Ogihara, T.; et al. Macrophage inflammatory protein-1beta induced cell adhesion with increased intracellular reactive oxygen species. J. Mol. Cell. Cardiol. 2009, 47, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, S.; Biscuola, M.; Patuzzo, C.; Trabetti, E.; Pasquali, A.; Laveder, P.; Faggian, G.; Iafrancesco, M.; Mazzucco, A.; Pignatti, P.F.; et al. Reconstruction and functional analysis of altered molecular pathways in human atherosclerotic arteries. BMC Genom. 2009, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reape, T.J.; Groot, P.H. Chemokines and atherosclerosis. Atherosclerosis 1999, 147, 213–225. [Google Scholar] [CrossRef]
- Montecucco, F.; Lenglet, S.; Gayet-Ageron, A.; Bertolotto, M.; Pelli, G.; Palombo, D.; Pane, B.; Spinella, G.; Steffens, S.; Raffaghello, L.; et al. Systemic and intraplaque mediators of inflammation are increased in patients symptomatic for ischemic stroke. Stroke 2010, 41, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Schecter, A.D.; Calderon, T.M.; Berman, A.B.; McManus, C.M.; Fallon, J.T.; Rossikhina, M.; Zhao, W.; Christ, G.; Berman, J.W.; Taubman, M.B. Human vascular smooth muscle cells possess functional CCR5. J. Biol. Chem. 2000, 275, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Meyrelles, S.S.; Peotta, V.A.; Pereira, T.M.; Vasquez, E.C. Endothelial dysfunction in the apolipoprotein E-deficient mouse: Insights into the influence of diet, gender and aging. Lipids Health Dis. 2011, 10, 211. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J. Lipid Res. 2009, 50, S352–S357. [Google Scholar] [CrossRef] [Green Version]
- Newby, A.C.; George, S.J.; Ismail, Y.; Johnson, J.L.; Sala-Newby, G.B.; Thomas, A.C. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb. Haemost. 2009, 101, 1006–1011. [Google Scholar]
- Osborn, E.A.; Jaffer, F.A. Imaging atherosclerosis and risk of plaque rupture. Curr. Atheroscler. Rep. 2013, 15, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafont, A. Basic aspects of plaque vulnerability. Heart Br. Card. Soc. 2003, 89, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucukguven, A.; Khalil, R.A. Matrix metalloproteinases as potential targets in the venous dilation associated with varicose veins. Curr. Drug Targets 2013, 14, 287–324. [Google Scholar] [PubMed]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorner, B.G.; Scheffold, A.; Rolph, M.S.; Huser, M.B.; Kaufmann, S.H.; Radbruch, A.; Flesch, I.E.; Kroczek, R.A. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 6181–6186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossakowska, A.E.; Edwards, D.R.; Prusinkiewicz, C.; Zhang, M.C.; Guo, D.; Urbanski, S.J.; Grogan, T.; Marquez, L.A.; Janowska-Wieczorek, A. Interleukin-6 regulation of matrix metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood 1999, 94, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Chen, W.C.; Hsieh, H.L.; Chi, P.L.; Hsiao, L.D.; Yang, C.M. TNF-alpha induces matrix metalloproteinase-9-dependent soluble intercellular adhesion molecule-1 release via TRAF2-mediated MAPKs and NF-kappaB activation in osteoblast-like MC3T3-E1 cells. J. Biomed. Sci. 2014, 21, 12. [Google Scholar] [CrossRef] [Green Version]
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: From mechanisms to evaluation and clinical implications. Biochem. Med. (Zagreb.) 2013, 23, 266–280. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef]
- Nieto-Vazquez, I.; Fernandez-Veledo, S.; de Alvaro, C.; Lorenzo, M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 2008, 57, 3211–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, C.; Netea, M.G.; van Riel, P.L.; van der Meer, J.W.; Stalenhoef, A.F. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 2007, 48, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, M.; Kopp, A.; Wurm, S.; Buchler, C.; Scholmerich, J.; Schaffler, A. Type 2 diabetes and lipoprotein metabolism affect LPS-induced cytokine and chemokine release in primary human monocytes. Exp. Clin. Endocrinol. Diabetes 2011, 119, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1513–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, N.; Bischoff, E.D.; Daige, C.L.; Thomas, D.; Vu, C.T.; Heyman, R.A.; Tangirala, R.K.; Schulman, I.G. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Taub, D.D.; Conlon, K.; Lloyd, A.R.; Oppenheim, J.J.; Kelvin, D.J. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science 1993, 260, 355–358. [Google Scholar] [CrossRef]
- Jones, K.L.; Maguire, J.J.; Davenport, A.P. Chemokine receptor CCR5: From AIDS to atherosclerosis. Br. J. Pharmacol. 2011, 162, 1453–1469. [Google Scholar] [CrossRef] [Green Version]
- Potteaux, S.; Combadiere, C.; Esposito, B.; Lecureuil, C.; Ait-Oufella, H.; Merval, R.; Ardouin, P.; Tedgui, A.; Mallat, Z. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1858–1863. [Google Scholar] [CrossRef] [Green Version]
- Mantani, P.T.; Dunér, P.; Bengtsson, E.; Ljungcrantz, I.; Sundius, L.; To, F.; Nilsson, J.; Björkbacka, H.; Fredrikson, G.N. Interleukin-25 (IL-25) has a protective role in atherosclerosis development in the aortic arch in mice. J. Biol. Chem. 2018, 293, 6791–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.T.; Lin, L.Y.; Chen, J.W. Inhibition of macrophage inflammatory protein-1β improves endothelial progenitor cell function and ischemia-induced angiogenesis in diabetes. Angiogenesis 2019, 22, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, S.; Gramanzini, M.; Mancini, M. Molecular imaging of vulnerable atherosclerotic plaques in animal models. Int. J. Mol. Sci. 2016, 17, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, T.; Bauler, T.J.; Malik-Kale, P.; Steele-Mortimer, O. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS ONE 2018, 13, e0193601. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Libby, P.; Everett, B.M. Novel atherosclerotic therapies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Abdolmaleki, F.; Gheibi Hayat, S.M.; Bianconi, V.; Johnston, T.P.; Sahebkar, A. Atherosclerosis and immunity: A prospective. Trends Cardiovasc. Med. 2019, 29, 363–371. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.-T.; Yang, H.-Y.; Chen, C.; Chen, J.-W. CCL4 Inhibition in Atherosclerosis: Effects on Plaque Stability, Endothelial Cell Adhesiveness, and Macrophages Activation. Int. J. Mol. Sci. 2020, 21, 6567. https://doi.org/10.3390/ijms21186567
Chang T-T, Yang H-Y, Chen C, Chen J-W. CCL4 Inhibition in Atherosclerosis: Effects on Plaque Stability, Endothelial Cell Adhesiveness, and Macrophages Activation. International Journal of Molecular Sciences. 2020; 21(18):6567. https://doi.org/10.3390/ijms21186567
Chicago/Turabian StyleChang, Ting-Ting, Hsin-Ying Yang, Ching Chen, and Jaw-Wen Chen. 2020. "CCL4 Inhibition in Atherosclerosis: Effects on Plaque Stability, Endothelial Cell Adhesiveness, and Macrophages Activation" International Journal of Molecular Sciences 21, no. 18: 6567. https://doi.org/10.3390/ijms21186567