N-Acetylaspartylglutamate (NAAG) Pretreatment Reduces Hypoxic-Ischemic Brain Damage and Oxidative Stress in Neonatal Rats



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethics Approval and Consent for Participation
2.2. Experimental Hypoxia-Ischemia
2.3. Drug Application
2.4. Evaluation of Brain Damage
2.5. Tissue Preparation for Biochemical Analysis
2.6. Determination of ROS Level
2.7. Determination of Glutathione Concentration
2.8. Determination of Antioxidant Enzyme Activity
2.8.1. Superoxide Dismutase
2.8.2. Glutathione Peroxidase (GPx)
2.8.3. Catalase
2.9. Determination of TGF-β Concentration
2.10. Statistical Analysis
3. Results
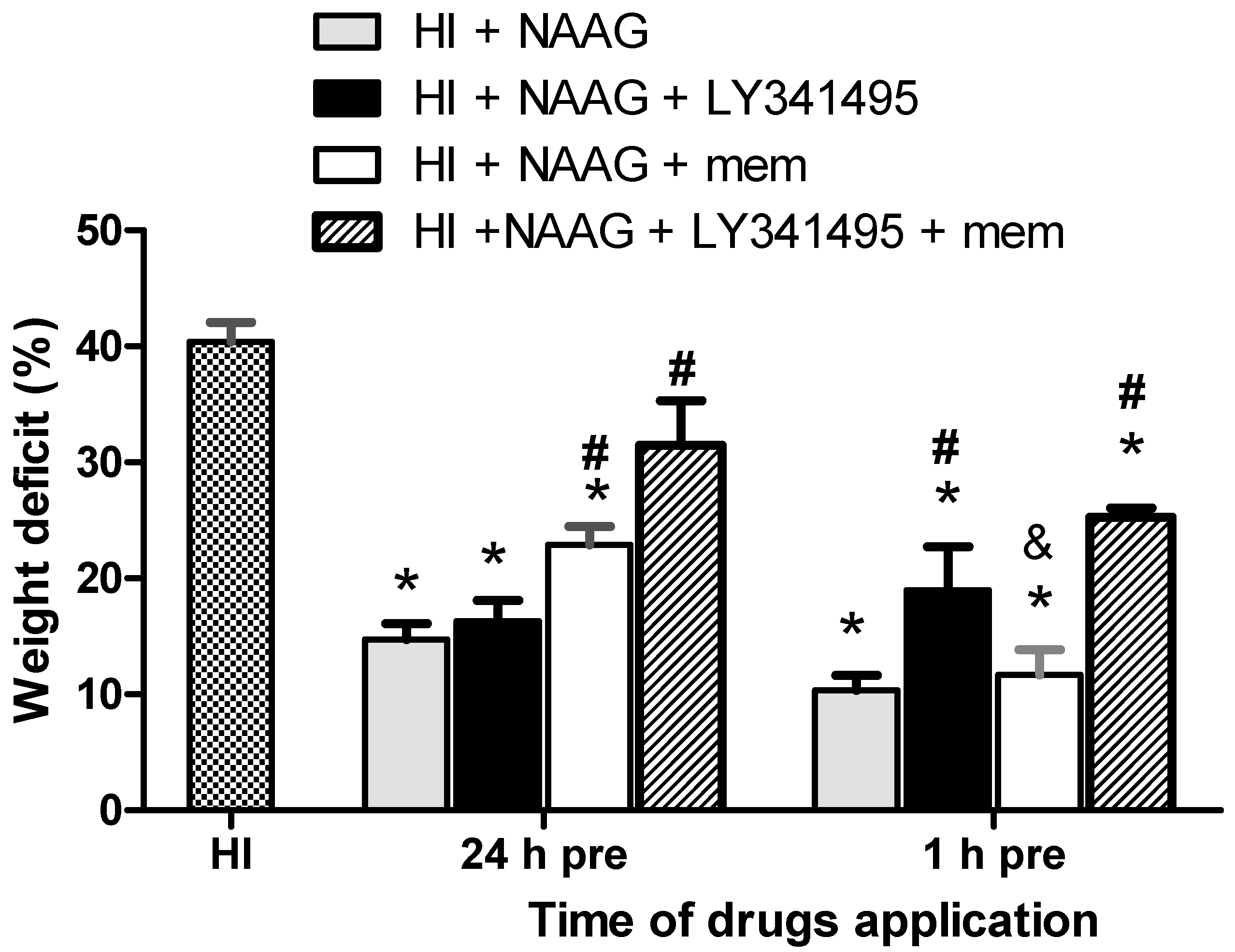
3.1. The Effect of NAAG Application on HI-Induced Brain Damage
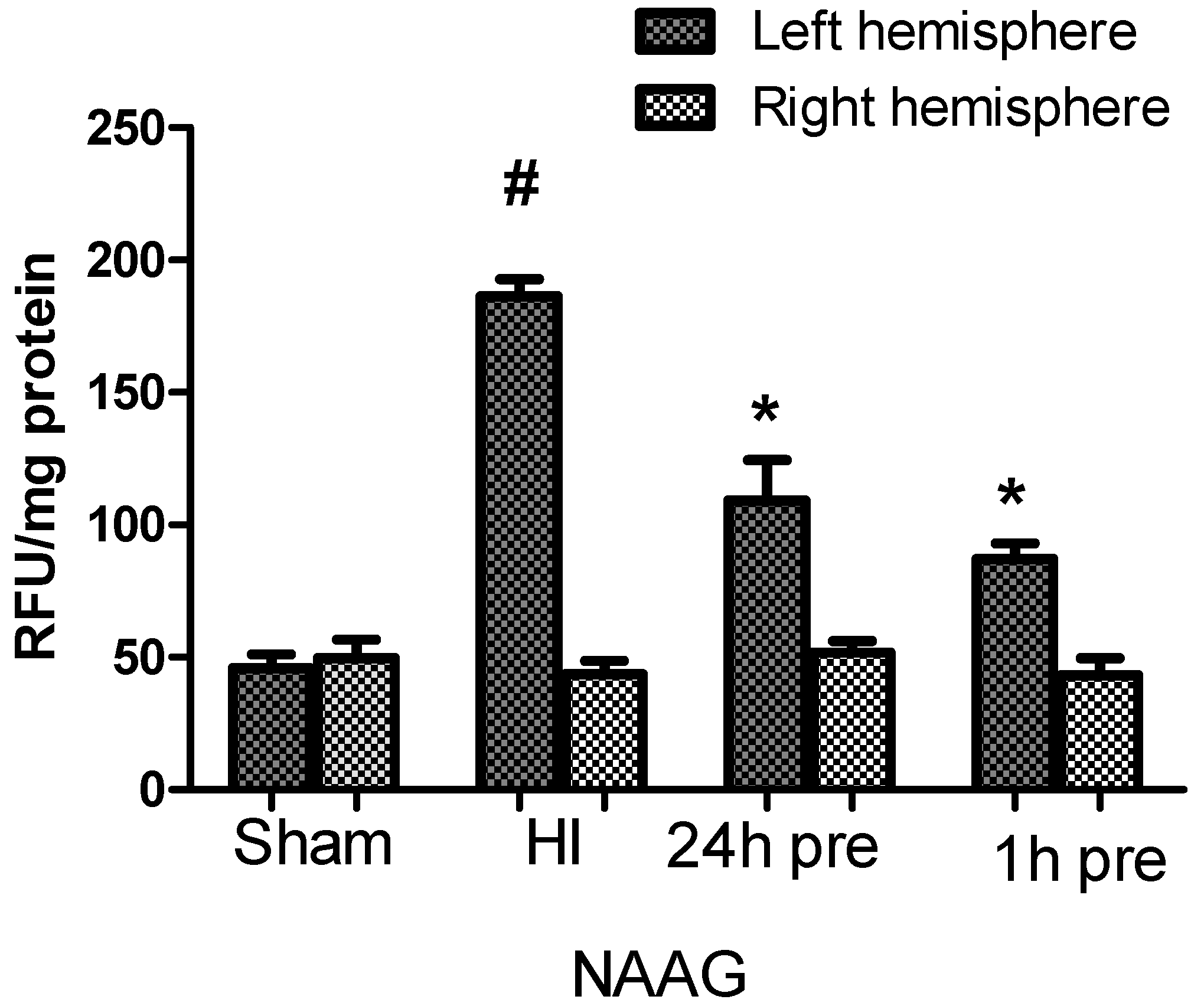
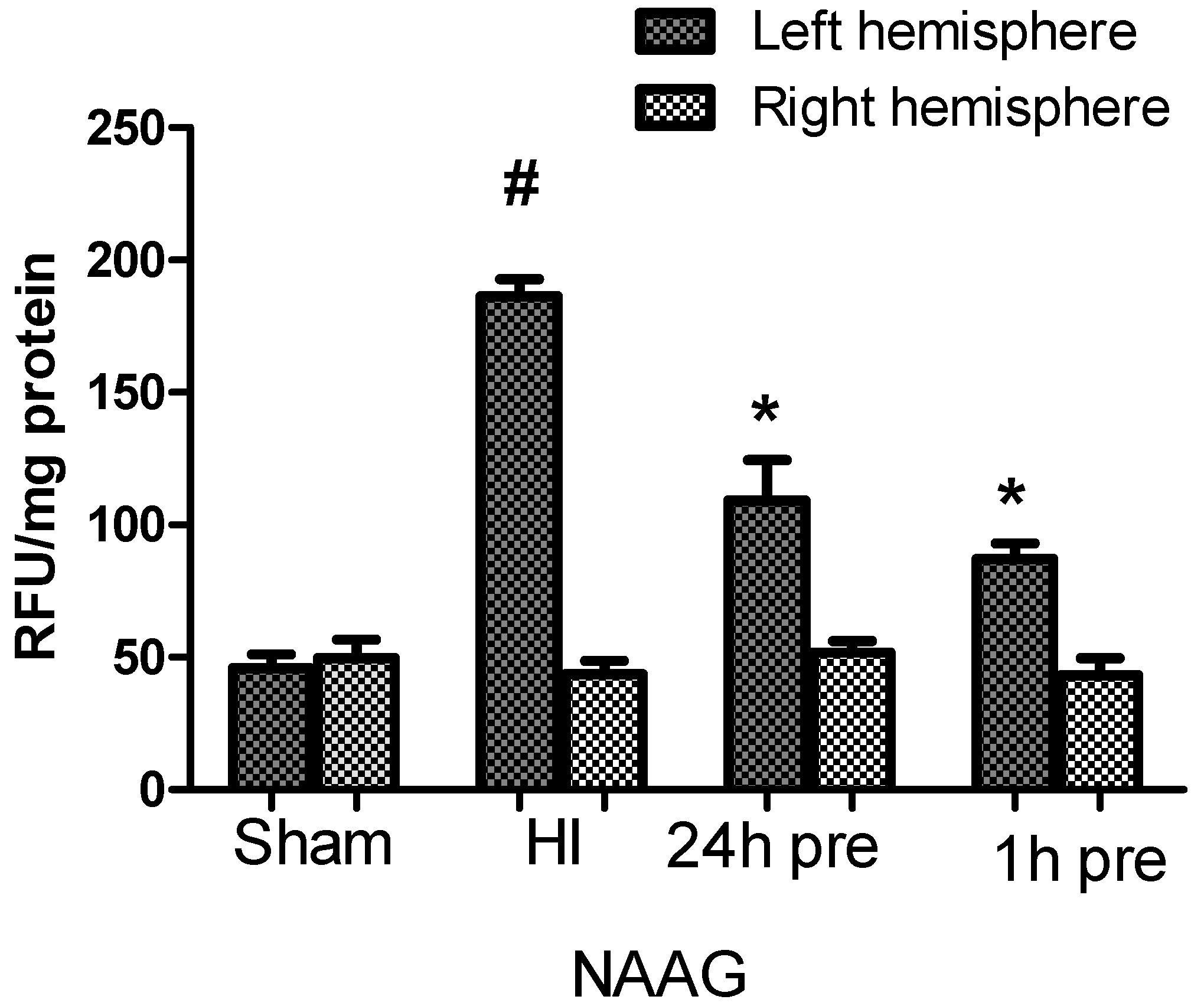
3.2. The Effect of NAAG Application on Changes in ROS Level in Rat Brain after HI
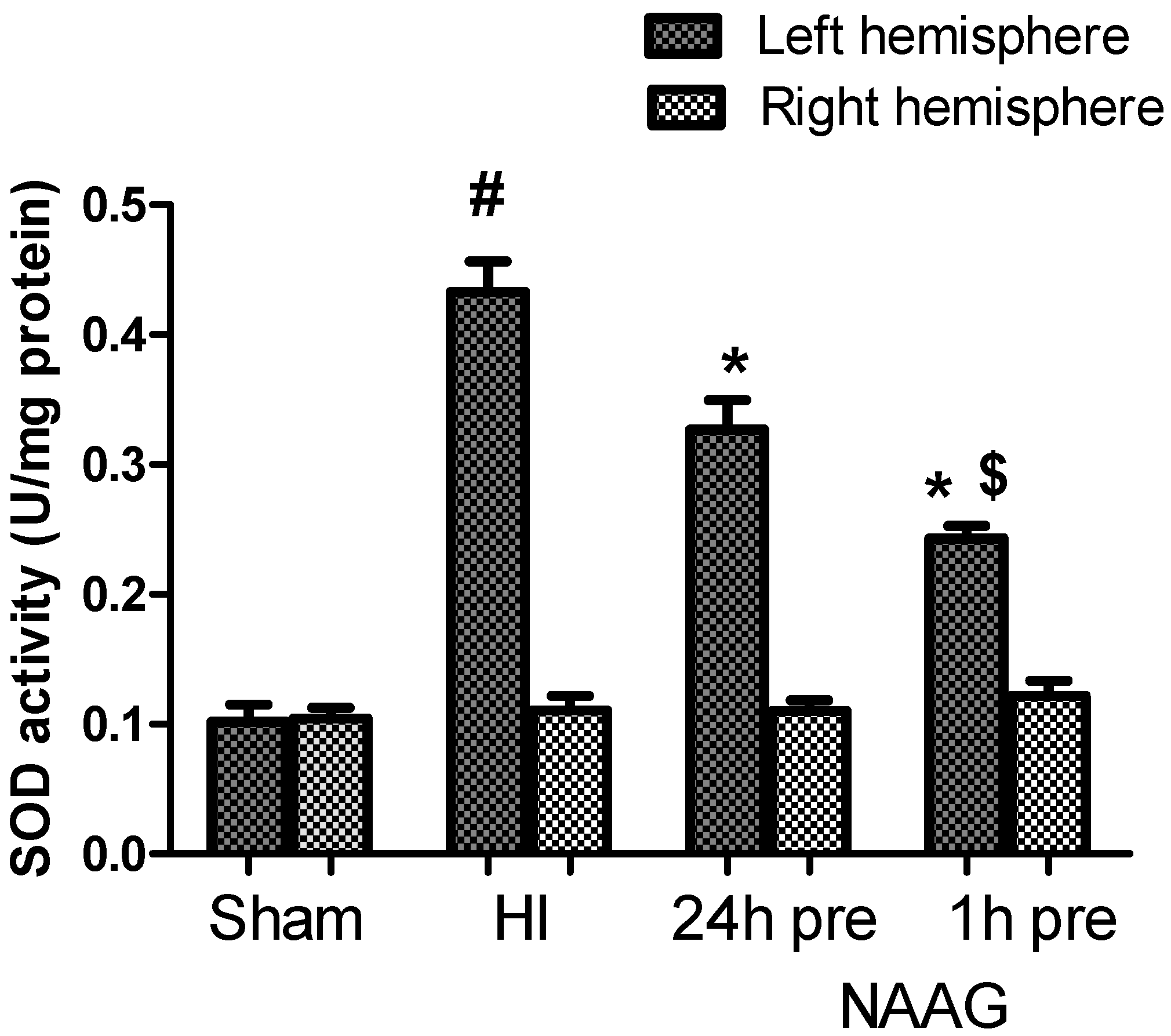
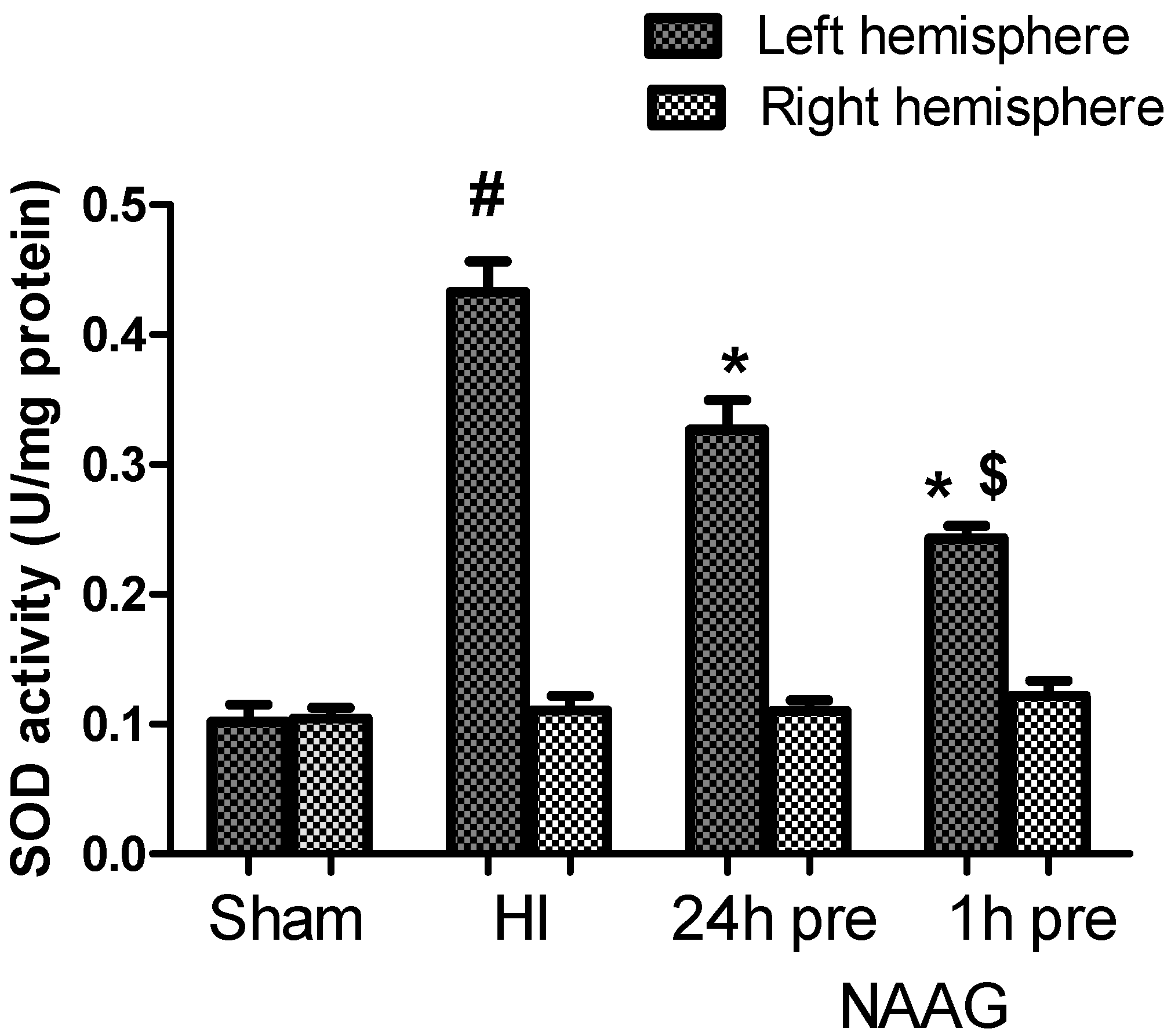
3.3. The Effect of NAAG Application on HI Induced Changes in SOD Activity
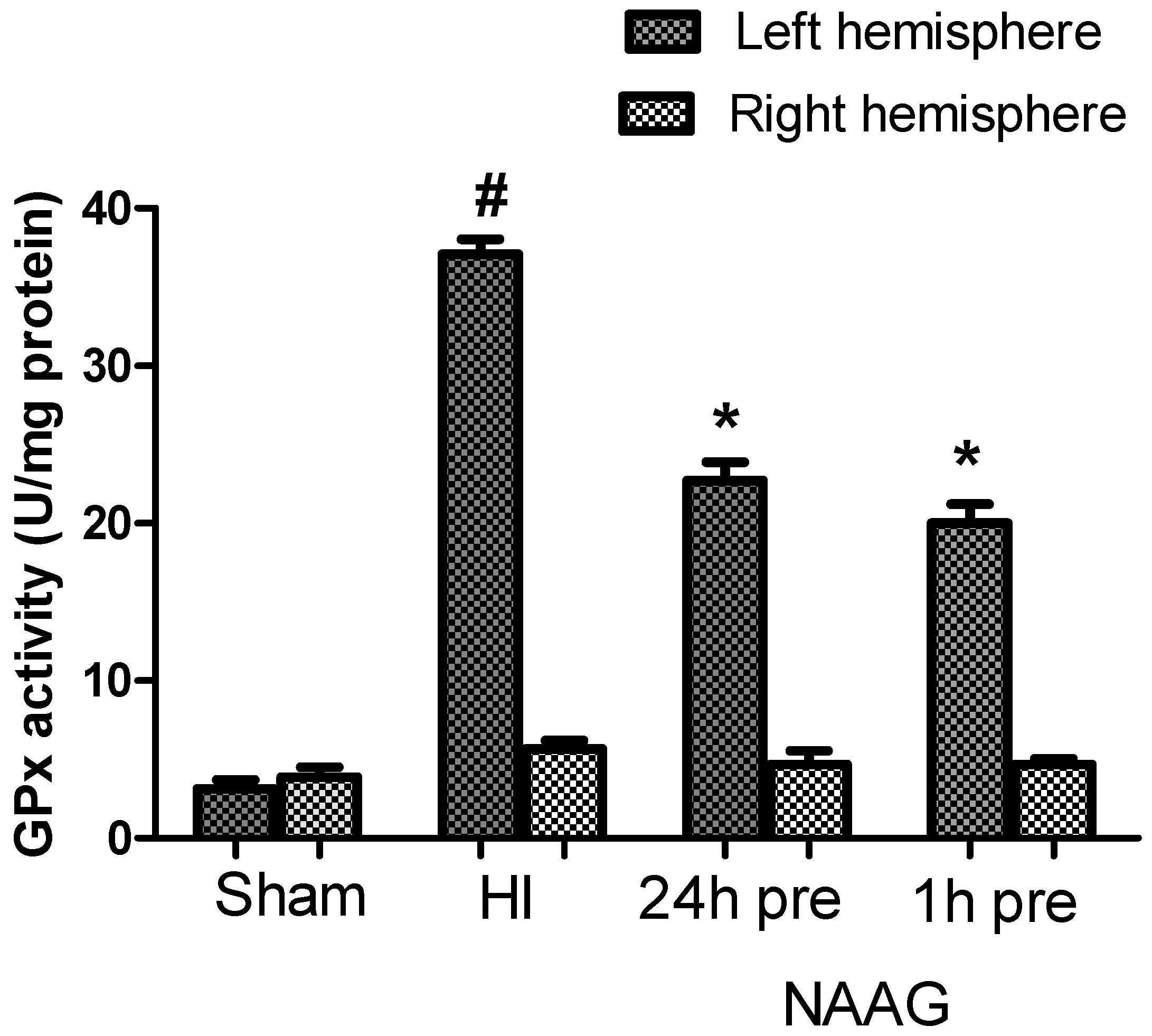
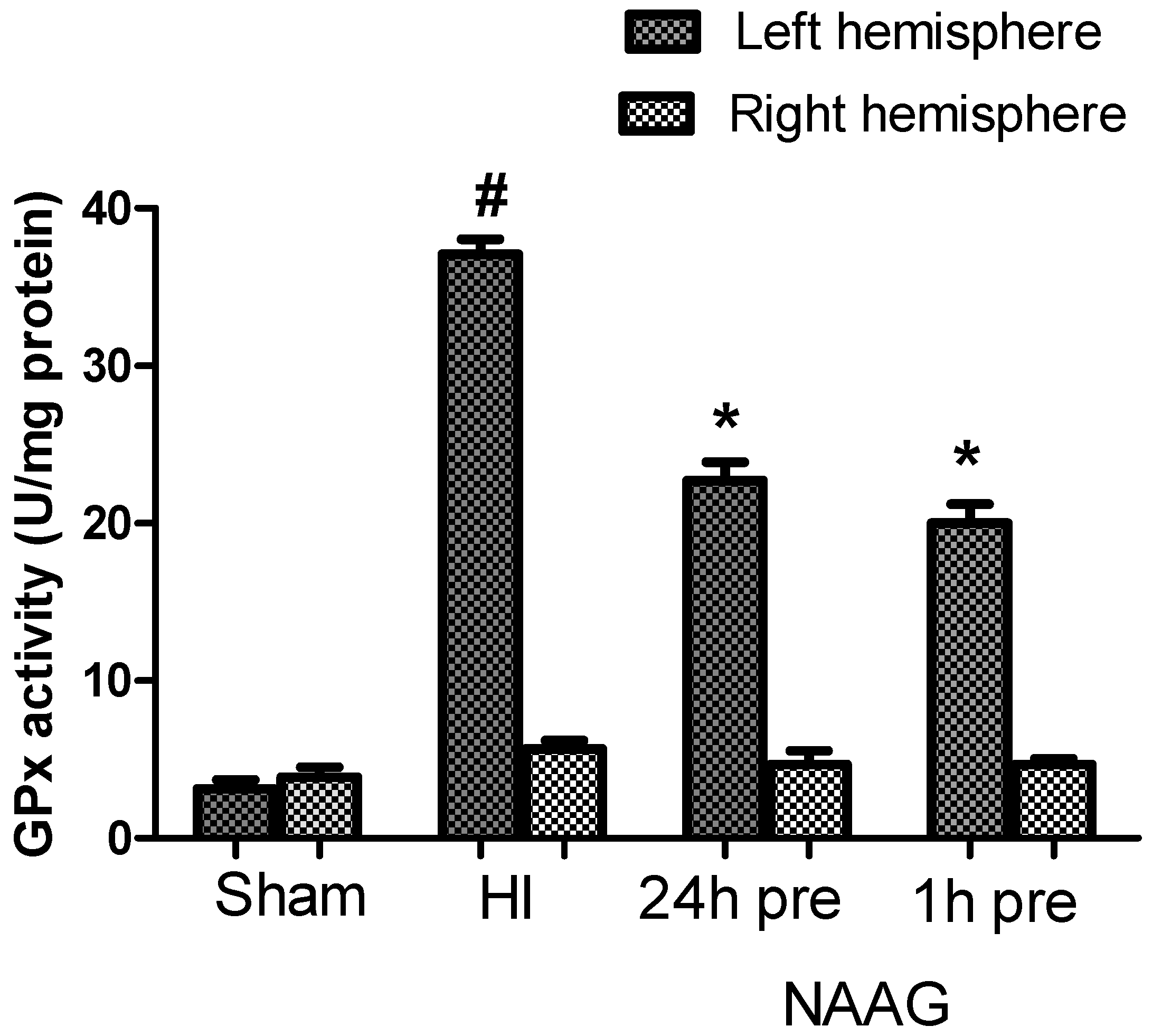
3.4. The Effect of NAAG Application on Changes in GPx Activity after HI
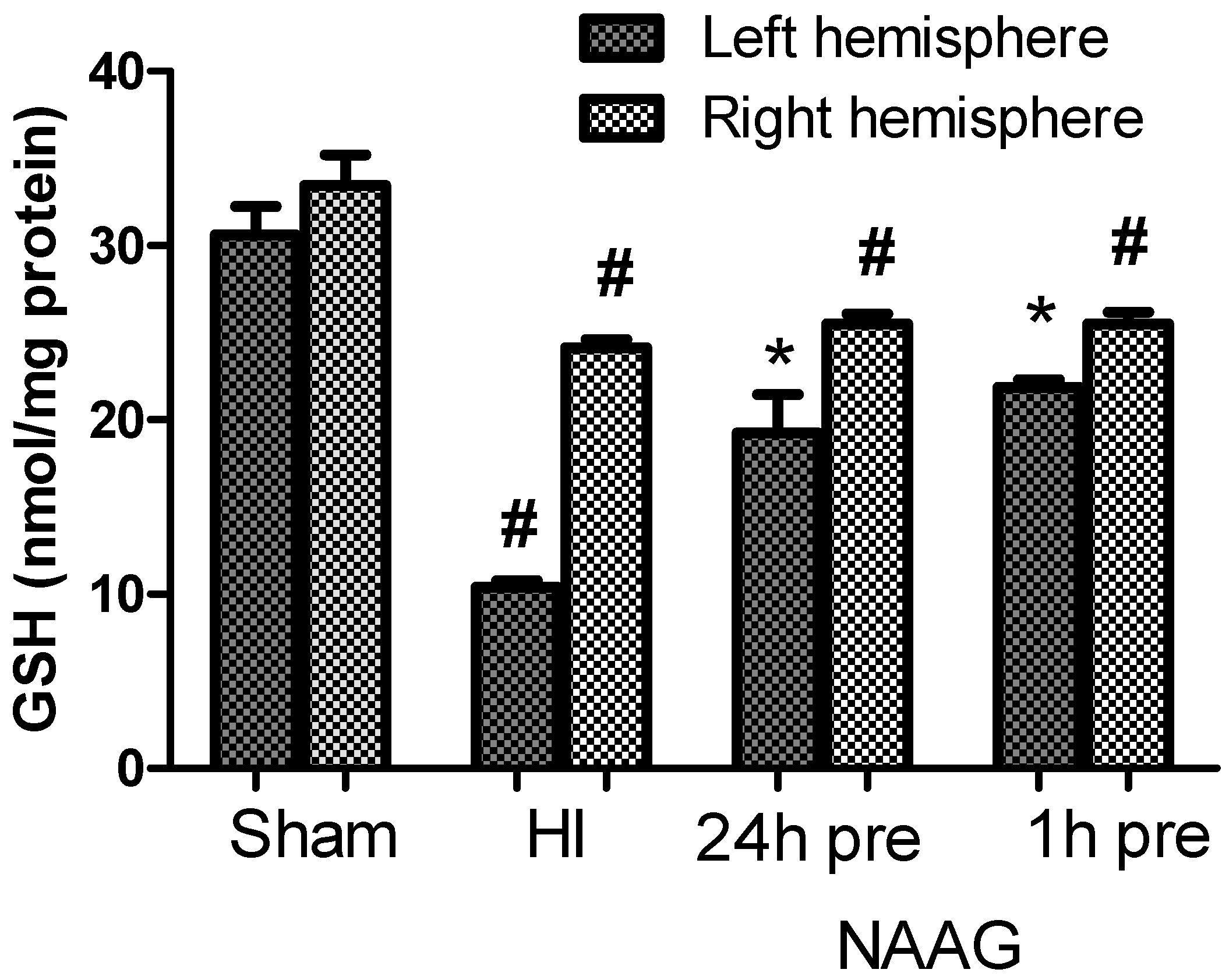
3.5. The Effect of the NAAG Application on Changes in GSH Level after HI
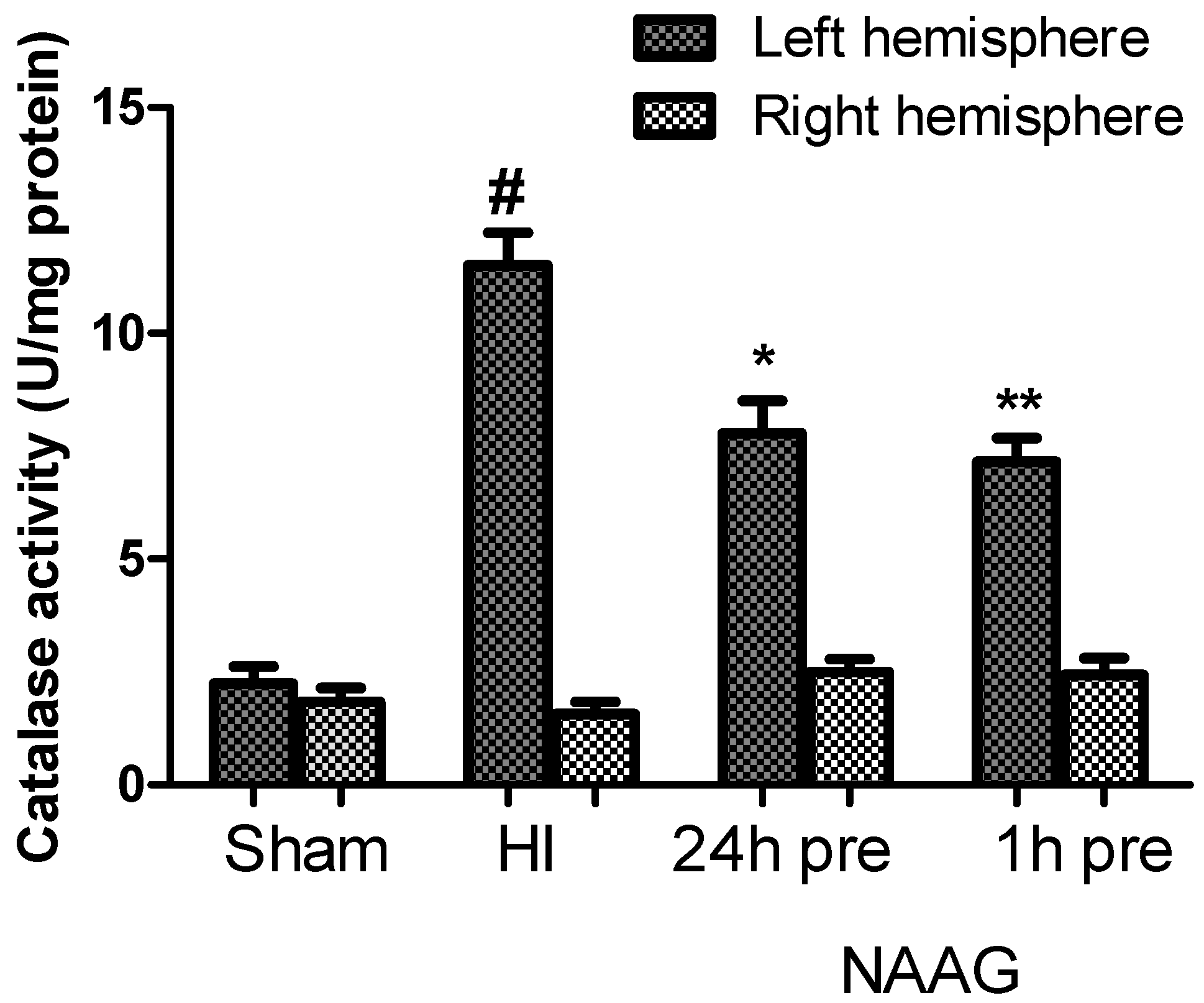
3.6. The Effect of NAAG Application on the Changes in Catalase Activity Observed after HI
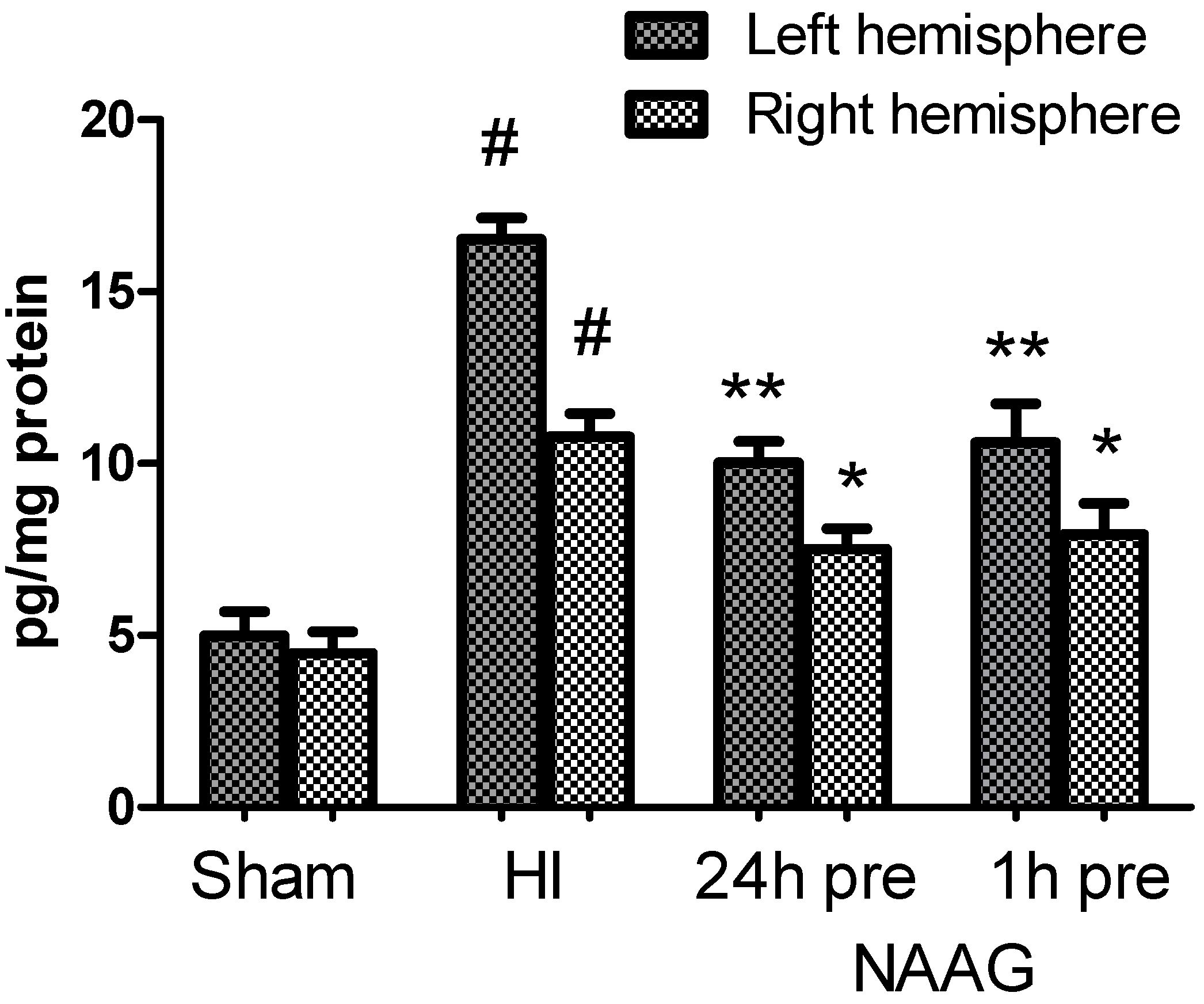
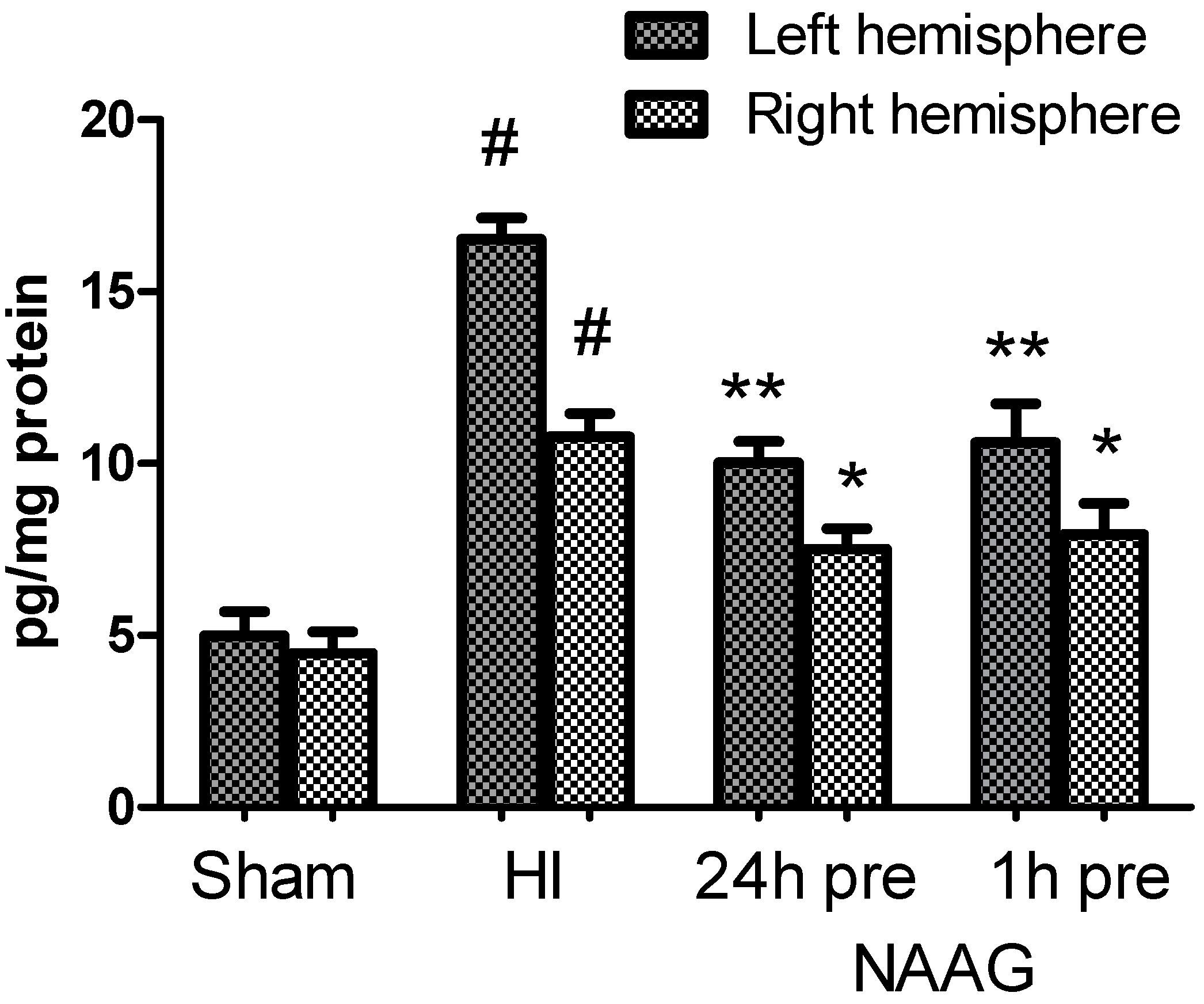
3.7. The Effect of NAAG Application on Changes in the TGF-β Concentration after HI
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Loidl, C.; Gavilanes, A.W.; Van Dijk, E.H.; Vreuls, W.; Blokland, A.; Vles, J.S.; Steinbusch, H.W.; Blanco, C.E. Effects of hypothermia and gender on survival and behavior after perinatal asphyxia in rats. Physiol. Behav. 2000, 68, 263–269. [Google Scholar] [CrossRef]
- Younkin, D.P. Hypoxic-Ischemic Brain Injury of the Newborn -Statement of the Problem and Overview. Brain Pathol. 1992, 2, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Perlman, J.M. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics 1997, 100, 1004–1114. [Google Scholar] [CrossRef] [PubMed]
- Toh, V.C. Early predictors of adverse outcome in term infants with post-asphyxial hypoxic ischaemic encephalopathy. Acta Paediatr. 2000, 89, 343–347. [Google Scholar] [CrossRef]
- Newton, C.R. Global Burden of Pediatric Neurological Disorders. Semin. Pediatr. Neurol. 2018, 27, 10–15. [Google Scholar] [CrossRef]
- Aslam, S.; Strickland, T.; Molloy, E.J. Neonatal Encephalopathy: Need for Recognition of Multiple Etiologies for Optimal Management. Front. Pediatr. 2019, 7, 142. [Google Scholar] [CrossRef]
- Gopagondanahalli, K.R.; Li, J.; Fahey, M.C.; Hunt, R.W.; Jenkin, G.; Miller, S.L.; Malhotra, A. Preterm Hypoxic–Ischemic Encephalopathy. Front. Pediatr. 2016, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Salińska, E.; Danysz, W.; Lazarewicz, J.W. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005, 43, 322–339. [Google Scholar]
- Panfoli, I.; Candiano, G.; Malova, M.; De Angelis, L.; Cardiello, V.; Buonocore, G.; Ramenghi, L. Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns. Front. Pediatr. 2018, 6, 369. [Google Scholar] [CrossRef] [Green Version]
- Burd, I.; Welling, J.; Kannan, G.; Johnston, M.V. Excitotoxicity as a Common Mechanism for Fetal Neuronal Injury with Hypoxia and Intrauterine Inflammation. Adv. Pharmacol. 2016, 76, 85–101. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayama, Y.; Raaz, U.; Jagger, A.; Adam, M.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic Cardiovascular Disease Induced by Oxidative Stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals (Basel) 2019, 12, 126. [Google Scholar] [CrossRef] [Green Version]
- Baud, O.; Haynes, R.F.; Wang, H.; Folkerth, R.D.; Li, J.; Volpe, J.J.; Rosenberg, P.A. Developmental up-regulation of MnSOD in rat oligodendrocytes confers protection against oxidative injury. Eur. J. Neurosci. 2004, 20, 29–40. [Google Scholar] [CrossRef]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and Treatment Related to Oxidative Stress in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Mol. Neurosci. 2019, 12, 88. [Google Scholar] [CrossRef]
- Fang, M.; Jiang, H.; Ye, L.; Cai, C.; Hu, Y.; Pan, S.; Li, P.; Xiao, J.; Lin, Z. Metformin treatment after the hypoxia-ischemia attenuates brain injury in newborn rats. Oncotarget 2017, 8, 75308–75325. [Google Scholar] [CrossRef] [Green Version]
- Belousova, M.; Tokareva, O.; Gorodetskaya, E.; Kalenikova, E.I.; Medvedev, O. Intravenous treatment with coenzyme Q10 improves neurological outcome and reduces infarct volume after transient focal brain ischemia in rats. J. Cardiovasc. Pharmacol. 2016, 67, 103–109. [Google Scholar] [CrossRef]
- Hobbs, C.E.; Murphy, M.P.; Smith, R.A.; Oorschot, D. Neonatal rat hypoxia-ischemia: Effect of the anti-oxidant mitoquinol, and S-PBN. Pediatr. Int. 2008, 50, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fanjul, J.; Fernández-Feijóo, C.D.; Lopez-Abad, M.; Ramos, M.G.L.; Caballé, R.B.; Alcántara-Horillo, S.; Camprubí, M.C. Neuroprotection with hypothermia and allopurinol in an animal model of hypoxic-ischemic injury: Is it a gender question? PLoS ONE 2017, 12, e0184643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solevåg, A.L.; Schmölzer, G.M.; Cheung, P.Y. Novel interventions to reduce oxidative-stress related brain injury in neonatal asphyxia. Free Radic. Boil. Med. 2019, 142, 113–122. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Ziembowicz, A.; Bratek, E.; Salińska, E. Combining hypobaric hypoxia or hyperbaric oxygen postconditioning with memantine reduces neuroprotection in 7-day-old rat hypoxia-ischemia. Pharmacol. Rep. 2016, 68, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670. [Google Scholar] [CrossRef]
- Perez, M.; Robbins, M.E.; Revhaug, C.; Saugstad, O.D. Oxygen radical disease in the newborn, revisited: Oxidative stress and disease in the newborn period. Free Radic. Boil. Med. 2019, 142, 61–72. [Google Scholar] [CrossRef]
- West, T.; Atzeva, M.; Holtzman, D.M. Pomegranate polyphenols and resveratrol protect the neonatal brain against hypoxic-ischemic injury. Dev. Neurosci. 2007, 29, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, S.; Naylor, M.; Kapur, J. Nitric oxide alters GABAergic synaptic transmission in cultured hippocampal neurons. Brain Res. 2009, 1297, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Bruno, V.; Caraci, F.; Copani, A.; Matrisciano, F.; Nicoletti, F.; Battaglia, G. The impact of metabotropic glutamate receptors into active neurodegenerative processes: A “dark side” in the development of new symptomatic treatments for neurologic and psychiatric disorders. Neuropharmacology 2017, 115, 180–192. [Google Scholar] [CrossRef]
- Caraci, F.; Battaglia, G.; Sortino, M.A.; Spampinato, S.F.; Molinaro, G.; Copani, A.; Nicoletti, F.; Bruno, V. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem. Int. 2012, 61, 559–565. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Therapeutic Promise and Principles: Metabotropic Glutamate Receptors. Oxidative Med. Cell. Longev. 2008, 1, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, F.; Bruno, V.; Ngomba, R.; Gradini, R.; Battaglia, G. Metabotropic glutamate receptors as drug targets: What’s new? Curr. Opin. Pharmacol. 2015, 20, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Imre, G. The Preclinical Properties of a Novel Group II Metabotropic Glutamate Receptor Agonist LY379268. CNS Drug Rev. 2007, 13, 444–464. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.S.; Mulle, C. Presynaptic glutamate receptors: Physiological functions and mechanisms of action. Nat. Rev. Neurosci. 2008, 9, 423–436. [Google Scholar] [CrossRef]
- Pin, J.-P.; Duvoisin, R. The metabotropic glutamate receptors: Structure and functions. Neuropharmacology 1995, 34, 1–26. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Casabona, G.; Copani, A.; Caciagli, F.; Nicoletti, F. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J. Neurosci. 1998, 18, 9594–9600. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.G.; Liu, W.; Olkowski, J.L.; Tang, Z.; Lin, Q.; Lu, X.C.; Slusher, B.S. Neuroprotection mediated by glutamate carboxypeptidase II (NAALADase) inhibition requires TGF-beta. Eur. J. Pharmacol. 2001, 430, 33–40. [Google Scholar] [CrossRef]
- Bratek, E.; Ziembowicz, A.; Bronisz, A.; Salińska, E. The activation of group II metabotropic glutamate receptors protects neonatal rat brains from oxidative stress injury after hypoxia-ischemia. PLoS ONE 2018, 13, e0200933. [Google Scholar] [CrossRef]
- Cai, Z.; Lin, S.; Rhodes, P.G. Neuroprotective effects of N-acetylaspartylglutamate in a neonatal rat model of hypoxia-ischemia. Eur. J. Pharmacol. 2002, 437, 139–145. [Google Scholar] [CrossRef]
- Lu, X.-C.M.; Tang, Z.; Liu, W.; Lin, Q.; Slusher, B.S. N-acetylaspartylglutamate protects against transient focal cerebral ischemia in rats. Eur. J. Pharmacol. 2000, 408, 233–239. [Google Scholar] [CrossRef]
- Cartmell, J.; Schoepp, D. Regulation of neurotransmitter release by metabotropic glutamate receptors. J. Neurochem. 2000, 75, 889–907. [Google Scholar] [CrossRef] [PubMed]
- Neale, J.H.; Olszewski, R.; Gehl, L.; Wroblewska, B.; Bzdega, T. The neurotransmitter N-acetylaspartylglutamate in models of pain, ALS, diabetic neuropathy, CNS injury and schizophrenia. Trends Pharmacol. Sci. 2005, 26, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-H.; Ding, J.-H.; Zhou, F.; Wang, F.; Hu, L.-F.; Sun, T.; Hu, G. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J. Neurochem. 2005, 92, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Van Hemelrijck, A.; Hachimi-Idrissi, S.; Sarre, S.; Ebinger, G.; Michotte, Y. Neuroprotective effect of N-acetyl-aspartyl-glutamate in combination with mild hypothermia in the endothelin-1 rat model of focal cerebral ischaemia. J. Neurochem. 2005, 95, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Lyeth, B. Application of Novel Therapeutic Agents for CNS Injury: NAAG Peptidase Inhibitors. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2015; Chapter 38. [Google Scholar]
- Cassidy, M.; Neale, J. N-Acetylaspartylglutamate catabolism is achieved by an enzyme on the cell surface of neurons and glia. Neuropeptides 1993, 24, 271–278. [Google Scholar] [CrossRef]
- Neale, J.H.; Yamamoto, T. N-acetylaspartylglutamate (NAAG) and glutamate carboxypeptidase II: An abundant peptide neurotransmitter-enzyme system with multiple clinical applications. Prog. Neurobiol. 2020, 184, 101722. [Google Scholar] [CrossRef]
- Blakely, R.D.; Robinson, M.B.; Thompson, R.C.; Coyle, J.T. Hydrolysis of the Brain Dipeptide N-Acetyl-l-Aspartyl-l-Glutamate: Subcellular and Regional Distribution, Ontogeny, and the Effect of Lesions on N-Acetylated-?-Linked Acidic Dipeptidase Activity. J. Neurochem. 1988, 50, 1200–1209. [Google Scholar] [CrossRef]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Neale, J.H.; Bzdega, T.; Wroblewska, B. N-Acetylaspartylglutamate: The most abundant peptide neurotransmitter in the mammalian central nervous system. J. Neurochem. 2000, 75, 443–452. [Google Scholar] [CrossRef]
- Khacho, P.; Wang, B.; Bergeron, R. The Good and Bad Sides of NAAG. Adv. Pharmacol. 2016, 76, 311–349. [Google Scholar] [CrossRef]
- Makarewicz, D.; Sulejczak, D.; Duszczyk, M.; Małek, M.; Słomka, M.; Lazarewicz, J.W. Original article Delayed preconditioning with NMDA receptor antagonists in a rat model of perinatal asphyxia. Folia Neuropathol. 2014, 3, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Puka-Sundvall, M.; Hagberg, H.; Weber, S.G.; Sandberg, M. Alterations in glutathione and amino acid concentrations after hypoxia–ischemia in the immature rat brain. Dev. Brain Res. 2000, 125, 51–60. [Google Scholar] [CrossRef]
- Gülcan, H.; Ozturk, I.; Arslan, S. Alterations in Antioxidant Enzyme Activities in Cerebrospinal Fluid Related with Severity of Hypoxic Ischemic Encephalopathy in Newborns. Neonatology 2005, 88, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Berent-Spillson, A.; Russell, J.W. Metabotropic glutamate receptor 3 protects neurons from glucose-induced oxidative injury by increasing intracellular glutathione concentration. J. Neurochem. 2007, 101, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.; Dunham, K.; Drager, U.C.; Grier, A.; Anderson, C.; Collura, J.; Coyle, J.T. Early embryonic death of glutamate carboxypeptidase II (NAALADase) homozygous mutants. Synapse 2003, 50, 285–292. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, S.; Cui, Z.; Zhang, M.; Lin, Y.; Cai, L.; Wang, Z.; Luo, X.; Zheng, Y.; Wang, Y.; et al. Mice lacking glutamate carboxypeptidase II develop normally, but are less susceptible to traumatic brain injury. J. Neurochem. 2015, 134, 340–353. [Google Scholar] [CrossRef]
- Bacich, D.J.; Wozniak, K.M.; Lu, X.-C.M.; O’Keefe, D.S.; Callizot, N.; Heston, W.D.W.; Slusher, B.S. Mice lacking glutamate carboxypeptidase II are protected from peripheral neuropathy and ischemic brain injury. J. Neurochem. 2005, 95, 314–323. [Google Scholar] [CrossRef]
- Vornov, J.; Hollinger, K.; Jackson, P.; Wozniak, K.; Farah, M.; Majer, P.; Rais, R.; Slusher, B.S. Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. Adv. Pharmacol. 2016, 76, 215–255. [Google Scholar] [CrossRef]
- Zhang, Z.; Bassam, B.; Thomas, A.G.; Williams, M.; Liu, J.; Nance, E.; Rojas, C.; Slusher, B.S.; Kannan, S. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiol. Dis. 2016, 94, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Dobolyi, A.; Vincze, C.; Pál, G.; Lovas, G. The Neuroprotective Functions of Transforming Growth Factor Beta Proteins. Int. J. Mol. Sci. 2012, 13, 8219–8258. [Google Scholar] [CrossRef]
- Slusher, B.S.; Vornov, J.J.; Thomas, A.G.; Hurn, P.D.; Harukuni, I.; Bhardwaj, A.; Traystman, R.J.; Robinson, M.B.; Britton, P.; Lu, X.-C.M.; et al. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999, 5, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Tortella, F.C.; Lin, Y.; Ved, H.; Slusher, B.S.; Dave, J.R. Neuroprotection produced by the NAALADase inhibitor 2-PMPA in rat cerebellar neurons. Eur. J. Pharmacol. 2000, 402, 31–37. [Google Scholar] [CrossRef]
- Lou, Z.; Wang, A.-P.; Duan, X.-M.; Hu, G.-H.; Song, G.-L.; Zuo, M.-L.; Yang, Z. Upregulation of NOX2 and NOX4 Mediated by TGF-β Signaling Pathway Exacerbates Cerebral Ischemia/Reperfusion Oxidative Stress Injury. Cell. Physiol. Biochem. 2018, 46, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Z.; Li, Y.; Ji, Q. NG25, an inhibitor of transforming growth factorbetaactivated kinase 1, ameliorates neuronal apoptosis in neonatal hypoxicischemic rats. Mol. Med. Rep. 2018, 17, 1710–1716. [Google Scholar] [PubMed]
- Dhandapani, K.M.; Brann, D.W. Transforming Growth Factor-betaβ: A Neuroprotective Factor in Cerebral Ischemia. Cell Biochem. Biophys. 2003, 39, 13–22. [Google Scholar] [CrossRef]
- Khacho, P.; Wang, B.; Ahlskog, N.; Hristova, E.; Bergeron, R. Differential effects of N-acetyl-aspartyl-glutamate on synaptic and extrasynaptic NMDA receptors are subunit- and pH-dependent in the CA1 region of the mouse hippocampus. Neurobiol. Dis. 2015, 82, 580–592. [Google Scholar] [CrossRef]
- Kołodziejczyk, K.; Hamilton, N.B.; Wade, A.; Karadottir, R.T.; Attwell, D. The effect of N-acetyl-aspartyl-glutamate and N-acetyl-aspartate on white matter oligodendrocytes. Brain 2009, 132 Pt 6, 1496–1508. [Google Scholar] [CrossRef]
- Papadia, S.; Soriano, F.X.; Léveillé, F.; Martel, M.-A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef]
- Palomino, J.; Echavarria, R.; Franco-Acevedo, A.; Moreno-Carranza, B.; Melo, Z. Opioids Preconditioning Upon Renal Function and Ischemia-Reperfusion Injury: A Narrative Review. Medicina (Kaunas) 2019, 55, 522. [Google Scholar] [CrossRef] [Green Version]
- Sprick, J.D.; Mallet, R.T.; Przyklenk, K.; Rickards, C.A. Ischaemic and hypoxic conditioning: Potential for protection of vital organs. Exp. Physiol. 2019, 104, 278–294. [Google Scholar] [CrossRef]
- Hu, S.; Feng, H.; Xi, G.-H. Hyperbaric oxygen therapy and preconditioning for ischemic and hemorrhagic stroke. Med. Gas Res. 2016, 6, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Gamdzyk, M.; Małek, M.; Bratek, E.; Koks, A.; Kaminski, K.; Ziembowicz, A.; Salińska, E. Hyperbaric oxygen and hyperbaric air preconditioning induces ischemic tolerance to transient forebrain ischemia in the gerbil. Brain Res. 2016, 1648 Pt A, 257–265. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bratek, E.; Ziembowicz, A.; Salinska, E. N-Acetylaspartylglutamate (NAAG) Pretreatment Reduces Hypoxic-Ischemic Brain Damage and Oxidative Stress in Neonatal Rats. Antioxidants 2020, 9, 877. https://doi.org/10.3390/antiox9090877
Bratek E, Ziembowicz A, Salinska E. N-Acetylaspartylglutamate (NAAG) Pretreatment Reduces Hypoxic-Ischemic Brain Damage and Oxidative Stress in Neonatal Rats. Antioxidants. 2020; 9(9):877. https://doi.org/10.3390/antiox9090877
Chicago/Turabian StyleBratek, Ewelina, Apolonia Ziembowicz, and Elzbieta Salinska. 2020. "N-Acetylaspartylglutamate (NAAG) Pretreatment Reduces Hypoxic-Ischemic Brain Damage and Oxidative Stress in Neonatal Rats" Antioxidants 9, no. 9: 877. https://doi.org/10.3390/antiox9090877
APA StyleBratek, E., Ziembowicz, A., & Salinska, E. (2020). N-Acetylaspartylglutamate (NAAG) Pretreatment Reduces Hypoxic-Ischemic Brain Damage and Oxidative Stress in Neonatal Rats. Antioxidants, 9(9), 877. https://doi.org/10.3390/antiox9090877