
tRNAs as Antibiotic Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
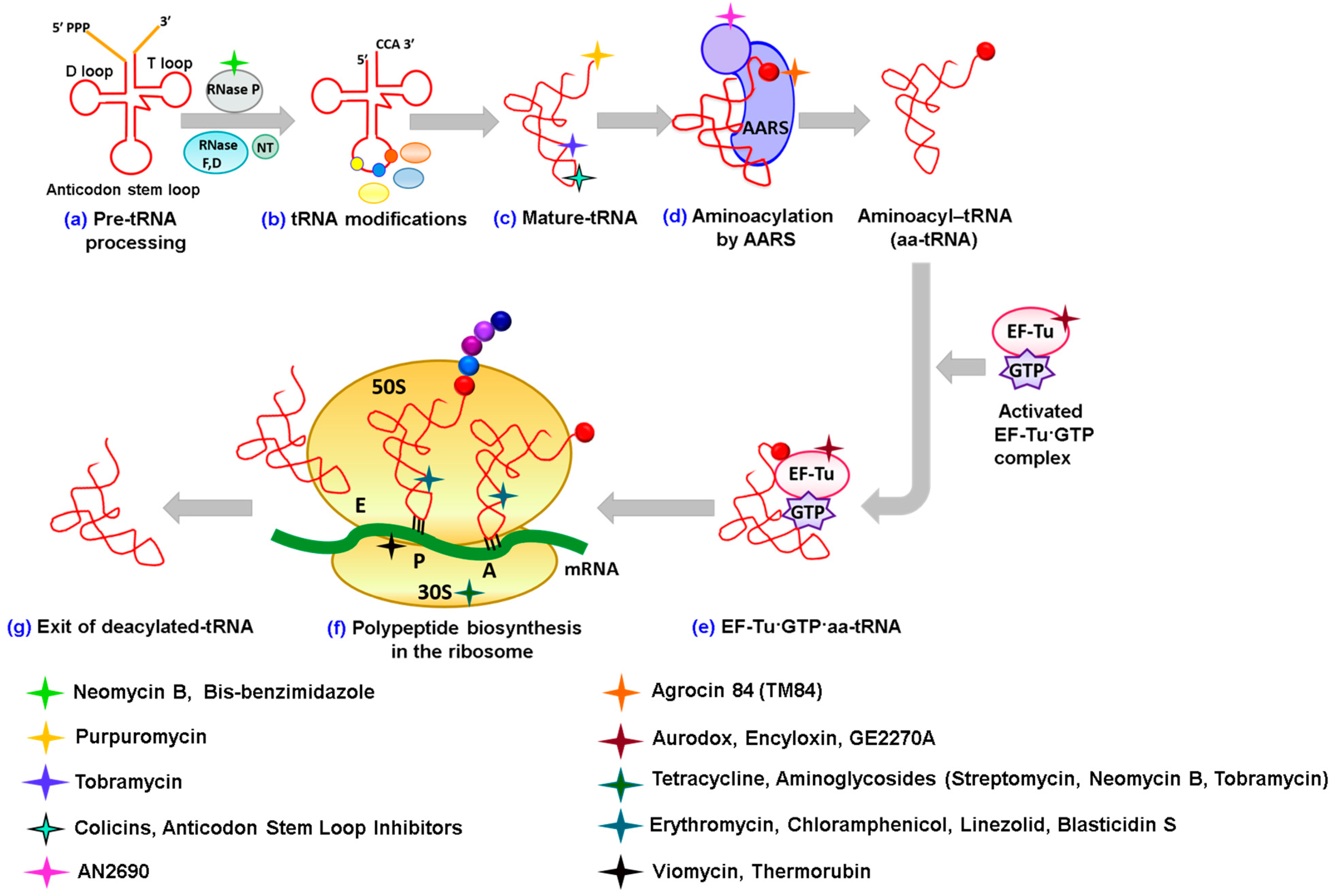
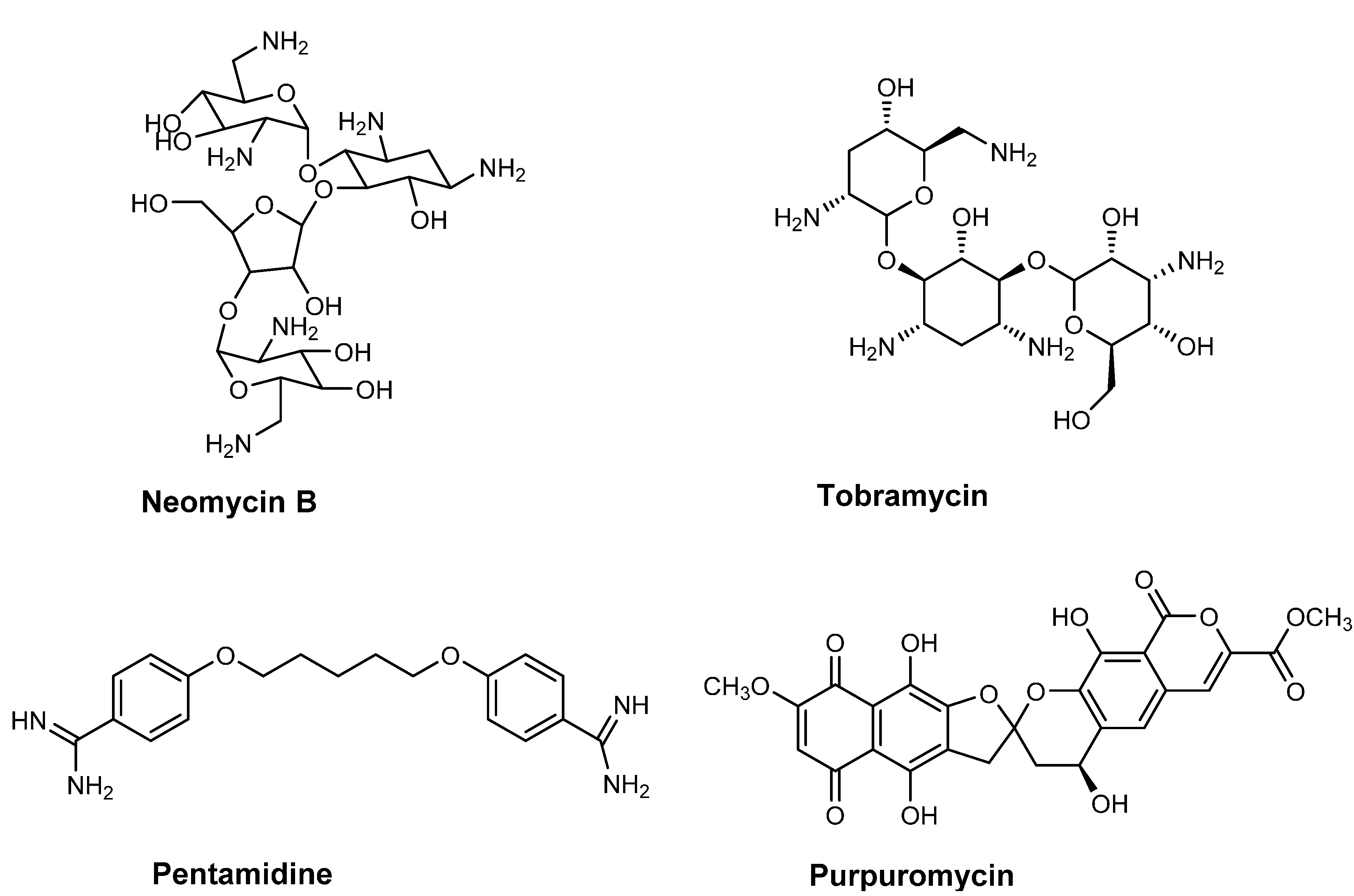
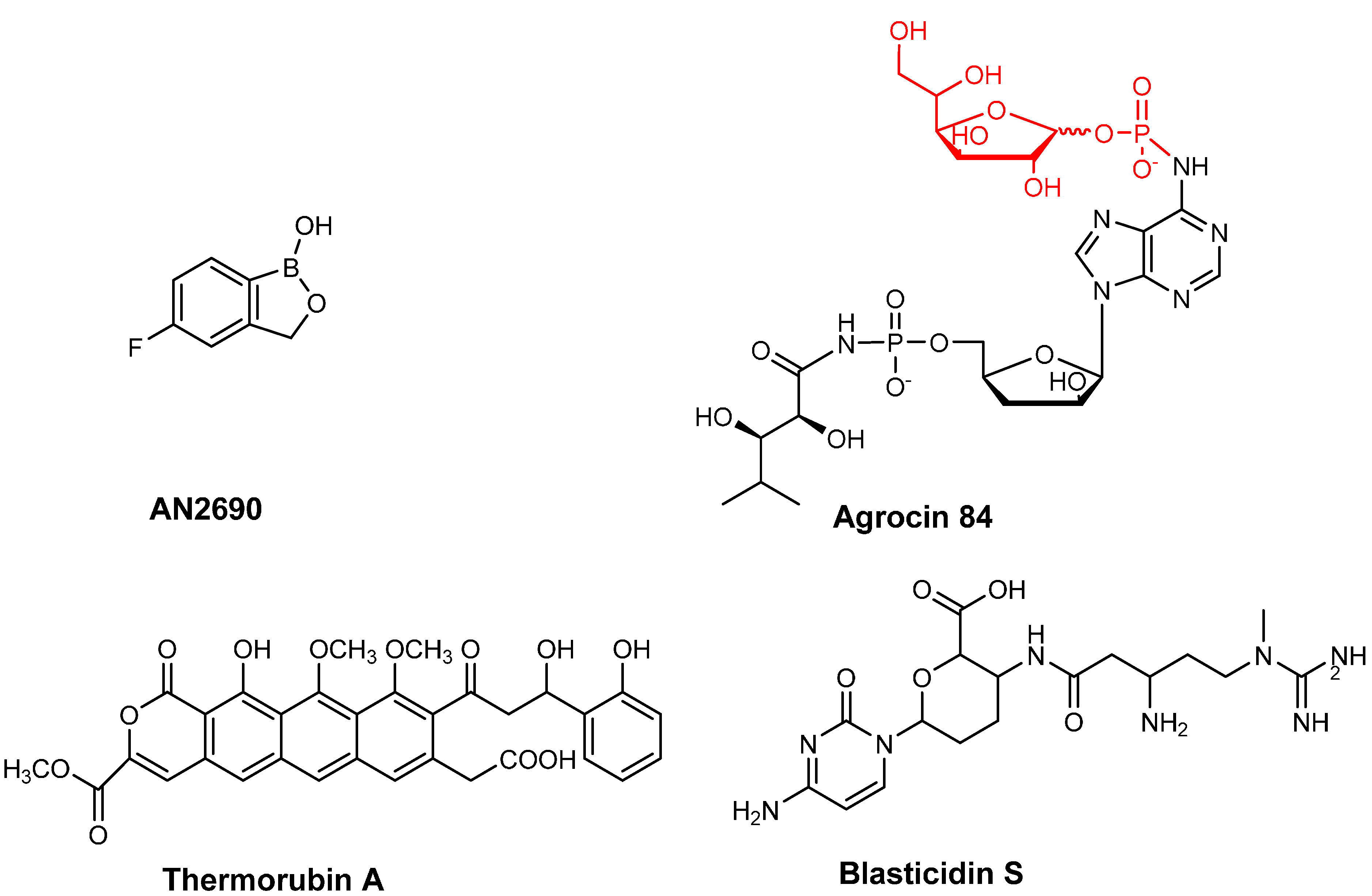
2. Antibiotics Affecting tRNA Biogenesis and Modification
2.1. Antibiotics Preventing tRNA Maturation
2.2. Antibiotics Affecting tRNA Modification
3. Antibiotics Having Direct Interactions with tRNAs
3.1. Alteration of tRNA Conformation

3.2. Blocking the CCA 3'-End of tRNA
3.3. Cleavage of tRNAs
3.4. Affecting the Binding of the Anticodon Stem Loop of tRNA
4. Antibiotics Inhibiting Aminoacylation of tRNA
4.1. Inhibition by Trapping tRNA in a LeuRS Editing Domain
4.2. Inhibition by Trapping tRNA in an “Aminoacylation-Like” Conformation

5. Antibiotics Affecting Elongation Factor EF-Tu
6. Targeting tRNAs in the Ribosome
7. Antibiotics Affecting Non-Canonical Roles of tRNA
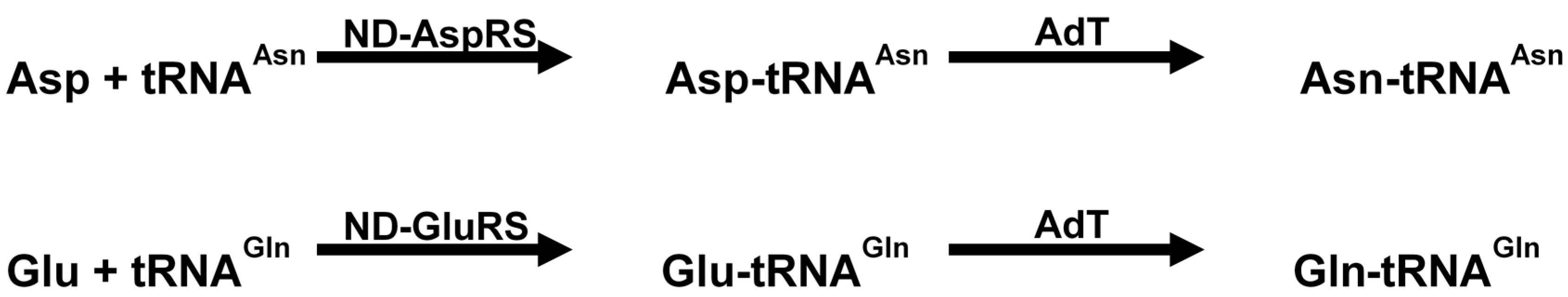
7.1. Inhibition of Amidotransferases
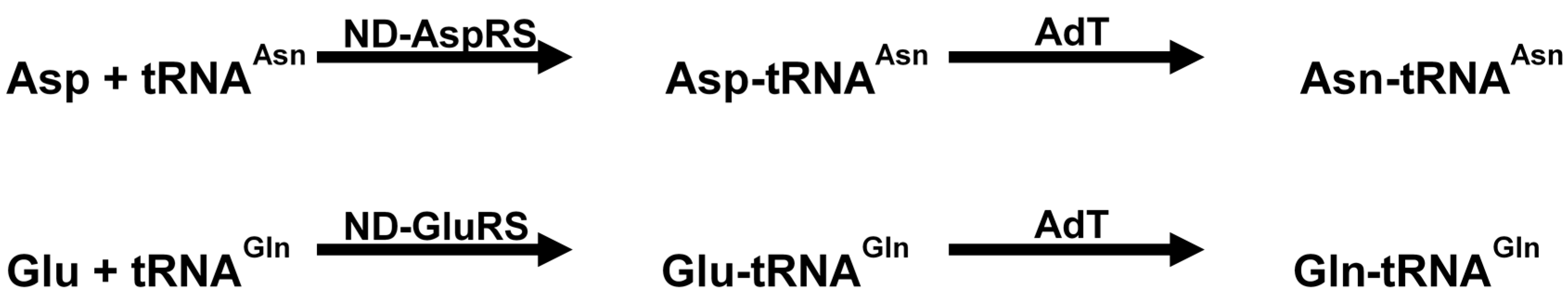
7.2. Affecting Cell Wall Biosynthesis and Cell Permeability
8. tRNAs and Antibiotic Persistence and Resistance
8.1. Deacylated tRNA and the Stringent Response
8.2. Bacterial Resistance Mechanisms
9. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Giege, R. Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol. 2008, 15, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Soll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- RajBhandary, U.L. Once there were twenty. Proc. Natl. Acad. Sci. USA 1997, 94, 11761–11763. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, K.; Kanai, A. tRNA gene diversity in the three domains of life. Front. Genet. 2014, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Raina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Sobala, A.; Hutvagner, G. Transfer RNA-derived fragments: Origins, processing, and functions. Wiley Interdiscip. Rev. RNA 2011, 2, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Korencic, D.; Ibba, M.; Soll, D. Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. USA 2003, 100, 15422–15427. [Google Scholar] [CrossRef] [PubMed]
- Critchley, I.A.; Young, C.L.; Stone, K.C.; Ochsner, U.A.; Guiles, J.; Tarasow, T.; Janjic, N. Antibacterial activity of REP8839, a new antibiotic for topical use. Antimicrob. Agents Chemother. 2005, 49, 4247–4252. [Google Scholar] [CrossRef] [PubMed]
- Giege, R.; Sissler, M.; Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef] [PubMed]
- Jarvest, R.L.; Armstrong, S.A.; Berge, J.M.; Brown, P.; Elder, J.S.; Brown, M.J.; Copley, R.C.; Forrest, A.K.; Hamprecht, D.W.; O’Hanlon, P.J.; et al. Definition of the heterocyclic pharmacophore of bacterial methionyl tRNA synthetase inhibitors: Potent antibacterially active non-quinolone analogues. Bioorg. Med. Chem. Lett. 2004, 14, 3937–3941. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Musier-Forsyth, K. tRANs-editing of Cys-tRNApro by Haemophilus influenzae YbaK protein. J. Biol. Chem. 2004, 279, 42359–42362. [Google Scholar] [CrossRef] [PubMed]
- Marintchev, A.; Wagner, G. Translation initiation: Structures, mechanisms and evolution. Q. Rev. Biophys. 2004, 37, 197–284. [Google Scholar] [CrossRef] [PubMed]
- Laursen, B.S.; Sorensen, H.P.; Mortensen, K.K.; Sperling-Petersen, H.U. Initiation of protein synthesis in bacteria. Microbiol. Mol. Biol. Rev. 2005, 69, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Voigts-Hoffmann, F.; Klinge, S.; Ban, N. Structural insights into eukaryotic ribosomes and the initiation of translation. Curr. Opin. Struct. Biol. 2012, 22, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.Y.; Masquida, B.; Biswas, R.; Westhof, E.; Gopalan, V. Molecular modeling of the three-dimensional structure of the bacterial RNAse P holoenzyme. J. Mol. Biol. 2003, 325, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.; Marquez, S.M.; Pace, N.R. RNAse P: Interface of the RNA and protein worlds. Trends Biochem. Sci. 2006, 31, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Berchanski, A.; Lapidot, A. Bacterial RNAse P RNA is a drug target for aminoglycoside-arginine conjugates. Bioconjug. Chem. 2008, 19, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Biswas, R.; Jovanovic, M.; Litovchick, A.; Lapidot, A.; Gopalan, V. Inhibition of bacterial RNAse P by aminoglycoside-arginine conjugates. FEBS Lett. 2002, 511, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.A.; Sudhahar, C.G.; Hatfield, C.L.; Sun, J.; Behrman, E.J.; Gopalan, V. Studies on the mechanism of inhibition of bacterial ribonuclease P by aminoglycoside derivatives. Nucleic Acids Res. 2008, 36, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Tanaka, T.; Kikuchi, Y. Bacterial ribonuclease P reaction is affected by substrate shape and magnesium ion concentration. Nucleic Acids Symp. Ser. 2003, 3, 293–294. [Google Scholar] [CrossRef]
- Mikkelsen, N.E.; Brannvall, M.; Virtanen, A.; Kirsebom, L.A. Inhibition of RNAse P RNA cleavage by aminoglycosides. Proc. Natl. Acad. Sci. USA 1999, 96, 6155–6160. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Bichenkova, E.V.; Wilton, A.N.; Tanaka, T.; Douglas, K.T.; Kikuchi, Y. Porphyrins and porphines inhibit the ribonuclease P reaction in vitro. Nucleic Acids Symp. Ser. 2002, 2, 111–112. [Google Scholar] [CrossRef]
- Hori, Y.; Rogert, M.C.; Tanaka, T.; Kikuchi, Y.; Bichenkova, E.V.; Wilton, A.N.; Gbaj, A.; Douglas, K.T. Porphyrins and porphines bind strongly and specifically to tRNA, precursor tRNA and to M1 RNA and inhibit the ribonuclease P ribozyme reaction. Biochim. Biophys. Acta 2005, 1730, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Bichenkova, E.V.; Wilton, A.N.; El-Attug, M.N.; Sadat-Ebrahimi, S.; Tanaka, T.; Kikuchi, Y.; Araki, M.; Sugiura, Y.; Douglas, K.T. Synthetic inhibitors of the processing of pretransfer RNA by the ribonuclease P ribozyme: Enzyme inhibitors which act by binding to substrate. Biochemistry 2001, 40, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Vioque, A. Protein synthesis inhibitors and catalytic RNA. Effect of puromycin on tRNA precursor processing by the RNA component of Escherichia coli RNAse P. FEBS Lett. 1989, 246, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Kalavrizioti, D.; Vourekas, A.; Tekos, A.; Tsagla, A.; Stathopoulos, C.; Drainas, D. Kinetics of inhibition of ribonuclease P activity by peptidyltransferase inhibitors. Effect of antibiotics on RNAse P. Mol. Biol. Rep. 2003, 30, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gruegelsiepe, H.; Brandt, O.; Hartmann, R.K. Antisense inhibition of RNAse P: Mechanistic aspects and application to live bacteria. J. Biol. Chem. 2006, 281, 30613–30620. [Google Scholar] [CrossRef] [PubMed]
- Childs, J.L.; Poole, A.W.; Turner, D.H. Inhibition of Escherichia coli RNAse P by oligonucleotide directed misfolding of RNA. RNA 2003, 9, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Kirsebom, L.A.; Nilsson, L. Growth rate regulation of 4.5 s RNA and M1 RNA the catalytic subunit of Escherichia coli RNAse P. J. Mol. Biol. 1996, 261, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Willkomm, D.K.; Gruegelsiepe, H.; Goudinakis, O.; Kretschmer-Kazemi Far, R.; Bald, R.; Erdmann, V.A.; Hartmann, R.K. Evaluation of bacterial RNAse P RNA as a drug target. Chembiochem 2003, 4, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Y.; Fierke, C.A. A real-time fluorescence polarization activity assay to screen for inhibitors of bacterial ribonuclease P. Nucleic Acids Res. 2014, 42, e159. [Google Scholar] [CrossRef] [PubMed]
- Urbonavicius, J.; Qian, Q.; Durand, J.M.; Hagervall, T.G.; Bjork, G.R. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001, 20, 4863–4873. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.C.; Gustafsson, C.; Berg, D.E.; Bjork, G.R. The gene for a tRNA modifying enzyme, m5U54-methyltransferase, is essential for viability in Escherichia coli. Proc. Natl. Acad. Sci. USA 1992, 89, 3995–3998. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, A.S.; Bjork, G.R. Chromosomal location and cloning of the gene (trmD) responsible for the synthesis of tRNA (m1G) methyltransferase in Escherichia coli K-12. Mol. Gen. Genet. 1982, 188, 440–446. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, K.; Watts, J.M.; Biswas, S.; Ambrad, J.; Barber, M.; Brule, H.; Petit, C.; Holmes, D.J.; Zalacain, M.; Holmes, W.M. Characterization of Streptococcus pneumoniae trmD, a tRNA methyltransferase essential for growth. J. Bacteriol. 2004, 186, 2346–2354. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Ehrlich, S.D.; Albertini, A.; Amati, G.; Andersen, K.K.; Arnaud, M.; Asai, K.; Ashikaga, S.; Aymerich, S.; Bessieres, P.; et al. Essential Bacillussubtilis genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4678–4683. [Google Scholar] [CrossRef] [PubMed]
- Masuda, I.; Sakaguchi, R.; Liu, C.; Gamper, H.; Hou, Y.M. The temperature sensitivity of a mutation in the essential tRNA modification enzyme tRNA methyltransferase d (trmD). J. Biol. Chem. 2013, 288, 28987–28996. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Kim, H.W.; Yoon, H.J.; Lee, B.I.; Suh, S.W.; Yang, J.K. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J. 2003, 22, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, R.; Giessing, A.; Dai, Q.; Lahoud, G.; Liutkeviciute, Z.; Klimasauskas, S.; Piccirilli, J.; Kirpekar, F.; Hou, Y.M. Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 2012, 18, 1687–1701. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.P.; Chheda, G.B.; Baczynskyj, L.; Hall, R.H. Aminoacyl nucleosides. VII. N-(Purin-6-ylcarbamoyl)threonine. A new component of transfer ribonucleic acid. Biochemistry 1969, 8, 3283–3289. [Google Scholar]
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, C.; el Yacoubi, B.; de Crecy-Lagard, V.; Iwata-Reuyl, D. Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 2012, 287, 13666–13673. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Lyons, B.; Cruz, Y.; Reddy, R.; Nordin, B.; Agnelli, F.; Williamson, J.R.; Schimmel, P.; Swairjo, M.A.; de Crecy-Lagard, V. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009, 37, 2894–2909. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Fadl, A.A. tRNA modification enzymes GidA and MnmE: Potential role in virulence of bacterial pathogens. Int. J. Mol. Sci. 2014, 15, 18267–18280. [Google Scholar] [CrossRef] [PubMed]
- Moukaddem, M.; Tangy, F.; Capmau, M.L.; le Goffic, F. Effects of cations, polyamines and other aminoglycosides on gentamicin C2. Binding to ribosomes from sensitive and resistant Escherichia coli strains. J. Antibiot. 1986, 39, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; Vicens, Q.; Westhof, E. Aminoglycoside-RNA interactions. Curr. Opin. Chem. Biol. 1999, 3, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Westhof, E. Crystal structure of a complex between the aminoglycoside tobramycin and an oligonucleotide containing the ribosomal decoding a site. Chem. Biol. 2002, 9, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; Putz, J.; Giege, R.; Westhof, E. Binding of tobramycin leads to conformational changes in yeast tRNAAsp and inhibition of aminoacylation. EMBO J. 2002, 21, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wilkinson, K.A.; Weeks, K.M. Complex ligand-induced conformational changes in tRNAAsp revealed by single-nucleotide resolution shape chemistry. Biochemistry 2008, 47, 3454–3461. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, N.E.; Johansson, K.; Virtanen, A.; Kirsebom, L.A. Aminoglycoside binding displaces a divalent metal ion in a tRNA-neomycin B complex. Nat. Struct. Biol. 2001, 8, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.; Kron, M.A.; Brown, R.B. Pentamidine: A review. Rev. Infect. Dis. 1985, 7, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res. 2008, 36, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, S.; Vitali, L.A.; Goldstein, B.P.; Monti, F.; Semenkov, Y.; Makhno, V.; Ripa, S.; Pon, C.L.; Gualerzi, C.O. Purpuromycin: An antibiotic inhibiting tRNA aminoacylation. RNA 1997, 3, 905–913. [Google Scholar] [PubMed]
- Landini, P.; Corti, E.; Goldstein, B.P.; Denaro, M. Mechanism of action of purpuromycin. Biochem. J. 1992, 284, 935. [Google Scholar] [PubMed]
- Coronelli, C.; Pagani, H.; Bardone, M.R.; Lancini, G.C. Purpuromycin, a new antibiotic isolated from Actinoplanes ianthinogenes N. sp. J. Antibiot. 1974, 27, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Trani, A.; Dallanoce, C.; Ferrari, P.; Goldstein, B.; Ripamonti, F.; Ciabatti, R. Synthesis and antimicrobial activities of 4-purpuromycin derivatives. Farmaco 1996, 51, 503–512. [Google Scholar] [PubMed]
- Trani, A.; Dallanoce, C.; Panzone, G.; Ripamonti, F.; Goldstein, B.P.; Ciabatti, R. Semisynthetic derivatives of purpuromycin as potential topical agents for vaginal infections. J. Med. Chem. 1997, 40, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Trani, A.; Kettenring, J.; Ripamonti, F.; Goldstein, B.; Ciabatti, R. Chemical modifications of the antibiotic purpuromycin. Farmaco 1993, 48, 637–651. [Google Scholar] [PubMed]
- Goldstein, B.P.; King, A.; Ripamonti, F.; Trani, A.; Phillips, I. In vitro activity of purpuromycin and MDL 63,604 against microorganisms that cause vaginitis and vaginosis. J. Antimicrob. Chemother. 1995, 36, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Pilsl, H.; Gross, P. Colicins: Structures, modes of action, transfer through membranes, and evolution. Arch. Microbiol. 1994, 161, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Ogawa, T.; Uozumi, T.; Watanabe, K.; Masaki, H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Natl. Acad. Sci. USA 2000, 97, 8278–8283. [Google Scholar] [CrossRef] [PubMed]
- Masaki, H.; Ogawa, T. The modes of action of colicins E5 and D, and related cytotoxic tRNAses. Biochimie 2002, 84, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, G. Anticodon nucleases. Trends Biochem. Sci. 2000, 25, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Guenther, R.H.; Yenne, S.P. Screening Methods for Identifying Specific staphylococcus aureus Inhibitors. US 2011 0229920 (A1), 22 September 2011. [Google Scholar]
- Agris, P.F.; Ashraf, S. Antibacterial Agents and Methods of Screening for the Same. US 6461815 (B1), 8 October 2002. [Google Scholar]
- TRANA Discovery. TRANA discovery Staphylococcus aureus 201 HTS-assay white paper. Available online: http://www.tranadiscovery.com/assays/staph-aureus-hts-assay/ (accessed on 17 December 2014).
- Agris, P.F.; Ashraf, S. Antibacterial and Antiviral Agents and Methods of Screening for the Same. US 20020212905, 9 January 2003. [Google Scholar]
- Agris, P.F.; Guenther, R.H. Compositions and Methods for the Inhibitors of Retroviral Infection. US 20110033850 (A1), 10 Febrerury 2011. [Google Scholar]
- Agris, P.F.; Guenther, R.; Ingram, P.C.; Basti, M.M.; Stuart, J.W.; Sochacka, E.; Malkiewicz, A. Unconventional structure of tRNA(Lys)SUU anticodon explains tRNAs role in bacterial and mammalian ribosomal frameshifting and primer selection by HIV-1. RNA 1997, 3, 420–428. [Google Scholar] [PubMed]
- Dewan, V.; Reader, J.; Forsyth, K.M. Role of aminoacyl-tRNA synthetases in infectious diseases and targets for therapeutic development. Top. Curr. Chem. 2014, 344, 293–329. [Google Scholar] [PubMed]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Tao, J.; Hill, J. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 1998, 12, 1599–1609. [Google Scholar] [PubMed]
- Lv, P.C.; Zhu, H.L. Aminoacyl-tRNA synthetase inhibitors as potent antibacterials. Curr. Med. Chem. 2012, 19, 3550–3563. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Sun, X.; Jarvis, T.; Critchley, I.; Janjic, N. Aminoacyl-tRNA synthetases: Essential and still promising targets for new anti-infective agents. Expert Opin. Investig. Drugs 2007, 16, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.; van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Guo, N.N.; Li, T.; Wang, E.D.; Wang, Y.L. CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry 2000, 39, 6726–6731. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, R.; Fukai, S.; Ishitani, R.; Nureki, O.; Yokoyama, S. Crystal structures of the CP1 domain from Thermus thermophilus isoleucyl-tRNA synthetase and its complex with l-valine. J. Biol. Chem. 2004, 279, 8396–8402. [Google Scholar] [CrossRef] [PubMed]
- Betha, A.K.; Williams, A.M.; Martinis, S.A. Isolated CP1 domain of Escherichia coli leucyl-tRNA synthetase is dependent on flanking hinge motifs for amino acid editing activity. Biochemistry 2007, 46, 6258–6267. [Google Scholar] [CrossRef] [PubMed]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Mao, W.; Lincecum, T.L., Jr.; Alley, M.R.; Martinis, S.A. Characterization of benzoxaborole-based antifungal resistance mutations demonstrates that editing depends on electrostatic stabilization of the leucyl-tRNA synthetase editing cap. FEBS Lett. 2011, 585, 2986–2991. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Wozniak, A.; Cyranski, M.K.; Jakubczyk, M.; Klimentowska, P.; Koll, A.; Kolodziejczak, J.; Pojmaj, G.; Zubrowska, A.; Zukowska, G.Z.; Sporzynski, A. Influence of the substituents on the structure and properties of benzoxaboroles. J. Phys. Chem. A 2010, 114, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Zhang, Y.K.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M.R.; Sanders, V.; Plattner, J.J. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 2006, 49, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Lipner, S.; Scher, R.K. Onychomycosis: Current and future therapies. Cutis 2014, 93, 60–63. [Google Scholar] [PubMed]
- Hui, X.; Baker, S.J.; Wester, R.C.; Barbadillo, S.; Cashmore, A.K.; Sanders, V.; Hold, K.M.; Akama, T.; Zhang, Y.K.; Plattner, J.J.; et al. In vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J. Pharm. Sci. 2007, 96, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Daigle, D. Tavaborole (AN2690) for the treatment of onychomycosis of the toenail in adults. Expert Rev. Anti-Infect. Ther. 2014, 12, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.T.; Tomsho, J.W.; Benkovic, S.J. The unique chemistry of benzoxaboroles: Current and emerging applications in biotechnology and therapeutic treatments. Bioorg. Med. Chem. 2014, 22, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Cao, K.; Zhou, Y.; Alley, M.R.; Rock, F.; Mohan, M.; Meewan, M.; Baker, S.J.; Lux, S.; Ding, C.Z.; et al. Synthesis and sar of novel benzoxaboroles as a new class of β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2533–2536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Gut, J.; Rosenthal, P.J.; Waterson, D.; Gamo, F.J.; Angulo-Barturen, I.; et al. Synthesis and structure-activity relationships of novel benzoxaboroles as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2011, 21, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Ye, L.; Zhou, H.; Waterson, D.; Gamo, F.J.; Sanz, L.M.; et al. Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. Bioorg. Med. Chem. Lett. 2012, 22, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Plattner, J.J.; Nare, B.; Wring, S.A.; Chen, D.; Freund, Y.; Gaukel, E.G.; Orr, M.D.; Perales, J.B.; Jenks, M.; et al. Benzoxaboroles: A new class of potential drugs for human African trypanosomiasis. Future Med. Chem. 2011, 3, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.S.; Dawson, K.L.; Jackson, K.E.; Lim, E.E.; Pasaje, C.F.; Turner, K.E.; Ralph, S.A. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol. 2014, 4, 1–13. [Google Scholar]
- Ding, D.; Meng, Q.; Gao, G.; Zhao, Y.; Wang, Q.; Nare, B.; Jacobs, R.; Rock, F.; Alley, M.R.; Plattner, J.J.; et al. Design, synthesis, and structure-activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J. Med. Chem. 2011, 54, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhao, Y.; Meng, Q.; Xie, D.; Nare, B.; Chen, D.; Bacchi, C.J.; Yarlett, N.; Zhang, Y.K.; Hernandez, V.; et al. Discovery of novel benzoxaborole-based potent antitrypanosomal agents. ACS Med. Chem. Lett. 2010, 1, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Simpson, F.C. New therapeutic options for onychomycosis. Expert Opin. Pharmacother. 2012, 13, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wang, Q.; Zhang, F.; Wang, Z.; Bowling, T.; Nare, B.; Jacobs, R.T.; Zhang, J.; Ding, D.; Liu, Y.; et al. Chalcone-benzoxaborole hybrid molecules as potent antitrypanosomal agents. J. Med. Chem. 2012, 55, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Tyrrell, K.L.; Merriam, C.V. Comparative in vitro activities of GSK2251052, a novel boron-containing leucyl-tRNA synthetase inhibitor, against 916 anaerobic organisms. Antimicrob. Agents Chemother. 2013, 57, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.; Tate, M. Agrocin 84 and the biological control of crown gall. In Agrobacterium Tumefaciens: From Plant Pathology to Biotechnology; Nester, E., Gordon, M.P., Kerr, A., Eds.; APS Press: St. Paul, MN, USA, 2004. [Google Scholar]
- Gelvin, S.B. Agrobacterium and plant genes involved in T-DNA transfer and integration. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 223–256. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.W.; Chilton, M.D. T-DNA of the agrobacterium Ti and Ri plasmids. Annu. Rev. Genet. 1982, 16, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Chilton, W.S.; Farrand, S.K. A Ti plasmid-encoded enzyme required for degradation of mannopine is functionally homologous to the T-region-encoded enzyme required for synthesis of this opine in crown gall tumors. J. Bacteriol. 1996, 178, 3285–3292. [Google Scholar] [PubMed]
- Ryder, M.H.; Tate, M.E.; Jones, G.P. Agrocinopine a, a tumor-inducing plasmid-coded enzyme product, is a phosphodiester of sucrose and l-arabinose. J. Biol. Chem. 1984, 259, 9704–9710. [Google Scholar] [PubMed]
- Ellis, J.G.; Kerr, A.; van Montagu, M.; Schell, J. Agrobacterium: Genetic studies on agrocin 84 production and the biological control of crown gall. Physiol. Plant Pathol. 1979, 15, 311–319. [Google Scholar] [CrossRef]
- Ellis, J.G.; Kerr, A. Transfer of agrocin 84 production from strain 84 to pathogenic recipients: A comment on a previous paper. In Soil-Borne Plant Pathogens; Schiffers, B.a.G., W., Ed.; Academic Press: London, UK, 1979; pp. 579–583. [Google Scholar]
- Murphy, P.J.; Tate, M.E.; Kerr, A. Substituents at N6 and C-5' control selective uptake and toxicity of the adenine-nucleotide bacteriocin, agrocin 84, in agrobacteria. Eur. J. Biochem. 1981, 115, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Farrand, S.K. Characterization of the acc operon from the nopaline-type Ti plasmid pTiC58, which encodes utilization of agrocinopines A and B and susceptibility to agrocin 84. J. Bacteriol. 1997, 179, 7559–7572. [Google Scholar] [PubMed]
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.G.; de Crecy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major biocontrol of plant tumors targets tRNA synthetase. Science 2005, 309, 1533. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat. Commun. 2013, 4, 1417. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.P.; Tate, M.E.; Kerr, A. Agrocin 84 is a 6-N-phosphoramidate of an adenine nucleotide analogue. Nature 1977, 265, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, H.; Reshetnikova, L.; Reiser, C.O.; Schirmer, N.K.; Sprinzl, M.; Hilgenfeld, R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature 1993, 365, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kjeldgaard, M.; Nissen, P.; Thirup, S.; Nyborg, J. The crystal structure of elongation factor EF-Tu from thermus aquaticus in the GTP conformation. Structure 1993, 1, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Abel, K.; Yoder, M.D.; Hilgenfeld, R.; Jurnak, F. An α to β conformational switch in EF-Tu. Structure 1996, 4, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; Thirup, S.; Kjeldgaard, M.; Nissen, P.; Lippmann, C.; Nyborg, J. Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure 1996, 4, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Chinali, G.; Parmeggiani, A. Mechanism of the inhibition of protein synthesis by kirromycin. Role of elongation factor Tu and ribosomes. Eur. J. Biochem. 1977, 75, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Chinali, G.; Parmeggiani, A. Kirromycin, an inhibitor of protein biosynthesis that acts on elongation factor Tu. Proc. Natl. Acad. Sci. USA 1974, 71, 4910–4914. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, A.; Nissen, P. Elongation factor Tu-targeted antibiotics: Four different structures, two mechanisms of action. FEBS Lett. 2006, 580, 4576–4581. [Google Scholar] [CrossRef] [PubMed]
- Anborgh, P.H.; Okamura, S.; Parmeggiani, A. Effects of the antibiotic pulvomycin on the elongation factor Tu-dependent reactions. Comparison with other antibiotics. Biochemistry 2004, 43, 15550–15556. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, A.; Krab, I.M.; Okamura, S.; Nielsen, R.C.; Nyborg, J.; Nissen, P. Structural basis of the action of pulvomycin and GE2270 A on elongation factor Tu. Biochemistry 2006, 45, 6846–6857. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Zahner, H. Metabolic products of microorganisms. 99. Kirromycin. Arch. Mikrobiol. 1972, 83, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Vogeley, L.; Palm, G.J.; Mesters, J.R.; Hilgenfeld, R. Conformational change of elongation factor Tu (EF-Tu) induced by antibiotic binding. Crystal structure of the complex between EF-Tu·GDP and aurodox. J. Biol. Chem. 2001, 276, 17149–17155. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Okubo, N.; Suzuki, T.; Izaki, K. New polyenic antibiotics active against Gram-positive and Gram-negative bacteria. VI. Non-lactonic polyene antibiotic, enacyloxin IIa, inhibits binding of aminoacyl-tRNA to a site of ribosomes. J. Antibiot. 1992, 45, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sugiyama, T.; Takahashi, M.; Shima, J.; Yamashita, K.; Izaki, K.; Furihata, K.; Seto, H. New polyenic antibiotics active against Gram-positive and Gram-negative bacteria. IV. Structural elucidation of enacyloxin IIa. J. Antibiot. 1992, 45, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Suzuki, T.; Izaki, K. New polyenic antibiotics active against Gram-positive and Gram-negative bacteria. V. Mode of action of enacyloxin IIa. J. Antibiot. 1991, 44, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Cetin, R.; Krab, I.M.; Anborgh, P.H.; Cool, R.H.; Watanabe, T.; Sugiyama, T.; Izaki, K.; Parmeggiani, A. Enacyloxin IIa, an inhibitor of protein biosynthesis that acts on elongation factor Tu and the ribosome. EMBO J. 1996, 15, 2604–2611. [Google Scholar] [PubMed]
- Parmeggiani, A.; Krab, I.M.; Watanabe, T.; Nielsen, R.C.; Dahlberg, C.; Nyborg, J.; Nissen, P. Enacyloxin IIa pinpoints a binding pocket of elongation factor Tu for development of novel antibiotics. J. Biol. Chem. 2006, 281, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Heffron, S.E.; Jurnak, F. Structure of an EF-Tu complex with a thiazolyl peptide antibiotic determined at 2.35 Å resolution: Atomic basis for GE2270 A inhibition of EF-Tu. Biochemistry 2000, 39, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Selva, E.; Beretta, G.; Montanini, N.; Saddler, G.S.; Gastaldo, L.; Ferrari, P.; Lorenzetti, R.; Landini, P.; Ripamonti, F.; Goldstein, B.P.; et al. Antibiotic GE2270 A: A novel inhibitor of bacterial protein synthesis. I. Isolation and characterization. J. Antibiot. 1991, 44, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Anborgh, P.H.; Parmeggiani, A. New antibiotic that acts specifically on the GTP-bound form of elongation factor Tu. EMBO J. 1991, 10, 779–784. [Google Scholar] [PubMed]
- Tor, Y. The ribosomal A-site as an inspiration for the design of RNA binders. Biochimie 2006, 88, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Poehlsgaard, J.; Douthwaite, S. The bacterial ribosome as a target for antibiotics. Nat. Rev. 2005, 3, 870–881. [Google Scholar]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. 2014, 12, 35–48. [Google Scholar]
- Yonath, A. Antibiotics targeting ribosomes: Resistance, selectivity, synergism and cellular regulation. Annu. Rev. Biochem. 2005, 74, 649–679. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Kanabar, P.; Schryer, D.; Florin, T.; Oh, E.; Bahroos, N.; Tenson, T.; Weissman, J.S.; Mankin, A.S. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2014, 111, 15958–15963. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, O.N.; Papadopoulos, G.; Kouvela, E.C.; Kalpaxis, D.L. Clindamycin binding to ribosomes revisited: Foot printing and computational detection of two binding sites within the peptidyl transferase center. Pharmazie 2013, 68, 616–621. [Google Scholar] [PubMed]
- Ungureanu, V. Macrolides, lincosamides, streptogramines (MLS): Mechanisms of action and resistance. Bacteriol. Virusol. Parazitol. Epidemiol. 2010, 55, 131–138. [Google Scholar] [PubMed]
- Lambert, T. Antibiotics that affect the ribosome. Rev. Sci. Tech. 2012, 31, 57–64. [Google Scholar] [PubMed]
- Aoki, H.; Ke, L.; Poppe, S.M.; Poel, T.J.; Weaver, E.A.; Gadwood, R.C.; Thomas, R.C.; Shinabarger, D.L.; Ganoza, M.C. Oxazolidinone antibiotics target the P-site on Escherichia coli ribosomes. Antimicrob. Agents Chemother. 2002, 46, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Occi, J.; Jayasuriya, H.; Herath, K.; Motyl, M.; Dorso, K.; Gill, C.; Hickey, E.; Overbye, K.M.; Barrett, J.F.; et al. Antibacterial evaluations of thiazomycin—A potent thiazolyl peptide antibiotic from Amycolatopsis fastidiosa. J. Antibiot. 2007, 60, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N.; Schluenzen, F.; Harms, J.M.; Starosta, A.L.; Connell, S.R.; Fucini, P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc. Natl. Acad. Sci. USA 2008, 105, 13339–13344. [Google Scholar] [CrossRef] [PubMed]
- Champney, W.S.; Tober, C.L. Specific inhibition of 50s ribosomal subunit formation in Staphylococcus aureus cells by 16-membered macrolide, lincosamide, and streptogramin B antibiotics. Curr. Microbiol. 2000, 41, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.J.; Mukhtar, T.A.; Wright, G.D. Streptogramin antibiotics: Mode of action and resistance. Curr. Drug Targets 2002, 3, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Atherly, A.G. Specific inhibition of ribosomal RNA synthesis in Escherichia coli by tetracycline. Cell 1974, 3, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Day, L.E. Tetracycline inhibition of cell-free protein synthesis. II. Effect of the binding of tetracycline to the components of the system. J. Bacteriol. 1966, 92, 197–203. [Google Scholar] [PubMed]
- Reusser, F. Effect of spectinomycin on peptide chain initiation. J. Antibiot. 1976, 29, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.J.; Tai, P.C.; Davis, B.D. Selective inhibition of initiating ribosomes by spectinomycin. Proc. Natl. Acad. Sci. USA 1974, 71, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Borovinskaya, M.A.; Pai, R.D.; Zhang, W.; Schuwirth, B.S.; Holton, J.M.; Hirokawa, G.; Kaji, H.; Kaji, A.; Cate, J.H. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mol. Biol. 2007, 14, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.D. Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 1987, 51, 341. [Google Scholar] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 2008, 135, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.D.; Chen, L.L.; Tai, P.C. Misread protein creates membrane channels: An essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. USA 1986, 83, 6164–6168. [Google Scholar] [CrossRef] [PubMed]
- Busscher, G.F.; Rutjes, F.P.; van Delft, F.L. 2-Deoxystreptamine: Central scaffold of aminoglycoside antibiotics. Chem. Rev. 2005, 105, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Stanley, R.E.; Blaha, G.; Grodzicki, R.L.; Strickler, M.D.; Steitz, T.A. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat. Struct. Mol. Biol. 2010, 17, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Akbergenov, R.; Shcherbakov, D.; Matt, T.; Duscha, S.; Meyer, M.; Wilson, D.N.; Bottger, E.C. Molecular basis for the selectivity of antituberculosis compounds capreomycin and viomycin. Antimicrob. Agents Chemother. 2011, 55, 4712–4717. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Teshima, T.; Shiba, T.; Nierhaus, K.H. tRNA binding to programmed ribosomes increases the ribosomal affinity for tuberactinomycin O. FEBS Lett. 1985, 179, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Johnson, F.; Steitz, T.A. The antibiotic thermorubin inhibits protein synthesis by binding to inter-subunit bridge B2a of the ribosome. J. Mol. Biol. 2012, 416, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Cone, M.C.; Petrich, A.K.; Gould, S.J.; Zabriskie, T.M. Cloning and heterologous expression of blasticidin s biosynthetic genes from Streptomyces griseochromogenes. J. Antibiot. 1998, 51, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Kalpaxis, D.L.; Theocharis, D.A.; Coutsogeorgopoulos, C. Kinetic studies on ribosomal peptidyltransferase. The behaviour of the inhibitor blasticidin s. Eur. J. Biochem. 1986, 154, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Svidritskiy, E.; Ling, C.; Ermolenko, D.N.; Korostelev, A.A. Blasticidin s inhibits translation by trapping deformed tRNA on the ribosome. Proc. Natl. Acad. Sci. USA 2013, 110, 12283–12288. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Celic, I.; Curnow, A.; Kim, H.; Pelaschier, J.; Tumbula, D.; Vothknecht, U.; Woese, C.; Soll, D. Aminoacyl-tRNA synthesis in Archaea. Nucleic Acids Symp. Ser. 1997, 305–306. [Google Scholar]
- Tumbula-Hansen, D.; Feng, L.; Toogood, H.; Stetter, K.O.; Soll, D. Evolutionary divergence of the Archaeal aspartyl-tRNA synthetases into discriminating and nondiscriminating forms. J. Biol. Chem. 2002, 277, 37184–37190. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.L.; Balg, C.; Jahn, D.; Moser, J.; Emond, A.; Blais, S.P.; Chenevert, R.; Lapointe, J. Mechanism of a GatCAB amidotransferase: Aspartyl-tRNA synthetase increases its affinity for asp-tRNAAsn and novel aminoacyl-tRNA analogues are competitive inhibitors. Biochemistry 2007, 46, 13190–13198. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.; Akochy, P.M.; Salazar, J.C.; Soll, D. The Helicobacter pylori amidotransferase GatCAB is equally efficient in glutamine-dependent transamidation of asp-tRNAAsn and glu-tRNAGln. J. Biol. Chem. 2007, 282, 11866–11873. [Google Scholar] [CrossRef] [PubMed]
- Balg, C.; Huot, J.L.; Lapointe, J.; Chenevert, R. Inhibition of Helicobacter pylori aminoacyl-tRNA amidotransferase by puromycin analogues. J. Am. Chem. Soc. 2008, 130, 3264–3265. [Google Scholar] [CrossRef] [PubMed]
- Van Heijenoort, J. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat. Prod. Rep. 2001, 18, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Kopp, U.; Roos, M.; Wecke, J.; Labischinski, H. Staphylococcal peptidoglycan interpeptide bridge biosynthesis: A novel antistaphylococcal target? Microb. Drug Resist. 1996, 2, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Biarrotte-Sorin, S.; Maillard, A.P.; Delettre, J.; Sougakoff, W.; Arthur, M.; Mayer, C. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: Insights into FemABX family substrates recognition. Structure 2004, 12, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.S.; Shrader, T.E. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J. Biol. Chem. 2001, 276, 6998–7003. [Google Scholar] [CrossRef] [PubMed]
- Chemama, M.; Fonvielle, M.; Villet, R.; Arthur, M.; Valery, J.M.; Etheve-Quelquejeu, M. Stable analogues of aminoacyl-tRNA for inhibition of an essential step of bacterial cell-wall synthesis. J. Am. Chem. Soc. 2007, 129, 12642–12643. [Google Scholar] [CrossRef] [PubMed]
- Cressina, E.; Lloyd, A.J.; de Pascale, G.; James Mok, B.; Caddick, S.; Roper, D.I.; Dowson, C.G.; Bugg, T.D. Inhibition of tRNA-dependent ligase MurM from Streptococcus pneumoniae by phosphonate and sulfonamide inhibitors. Bioorg. Med. Chem. 2009, 17, 3443–3455. [Google Scholar] [CrossRef] [PubMed]
- Cashel, M.; Gallant, J. Two compounds implicated in the function of the RC gene of Escherichia coli. Nature 1969, 221, 838–841. [Google Scholar] [CrossRef]
- Haseltine, W.A.; Block, R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc. Natl. Acad. Sci. USA 1973, 70, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Sy, J.; Lipmann, F. Identification of the synthesis of guanosine tetraphosphate (MS I) as insertion of a pyrophosphoryl group into the 3'-position in guanosine 5'-diphosphate. Proc. Natl. Acad. Sci. USA 1973, 70, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, L.U.; Farewell, A.; Nystrom, T. ppGpp: A global regulator in Escherichia coli. Trends Microbiol. 2005, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Givens, R.M.; Lin, M.H.; Taylor, D.J.; Mechold, U.; Berry, J.O.; Hernandez, V.J. Inducible expression, enzymatic activity, and origin of higher plant homologues of bacterial RelA/SpoT stress proteins in Nicotiana tabacum. J. Biol. Chem. 2004, 279, 7495–7504. [Google Scholar] [CrossRef] [PubMed]
- Nanamiya, H.; Kasai, K.; Nozawa, A.; Yun, C.S.; Narisawa, T.; Murakami, K.; Natori, Y.; Kawamura, F.; Tozawa, Y. Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol. Microbiol. 2008, 67, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.; Vrentas, C.E.; Sanchez-Vazquez, P.; Gaal, T.; Gourse, R.L. The magic spot: A ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol. Cell 2013, 50, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Wexselblatt, E.; Oppenheimer-Shaanan, Y.; Kaspy, I.; London, N.; Schueler-Furman, O.; Yavin, E.; Glaser, G.; Katzhendler, J.; Ben-Yehuda, S. Relacin, a novel antibacterial agent targeting the stringent response. PLoS Pathog. 2012, 8, e1002925. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Kumar, M.; Chatterji, D. ppGpp: Stringent response and survival. J. Microbiol. 2006, 44, 1–10. [Google Scholar] [PubMed]
- Ojha, A.; Anand, M.; Bhatt, A.; Kremer, L.; Jacobs, W.R., Jr.; Hatfull, G.F. GroEL1: A dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 2005, 123, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.K.; Baughn, A.D.; Sambandan, D.; Hsu, T.; Trivelli, X.; Guerardel, Y.; Alahari, A.; Kremer, L.; Jacobs, W.R., Jr.; Hatfull, G.F. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 2008, 69, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.K.; Trivelli, X.; Guerardel, Y.; Kremer, L.; Hatfull, G.F. Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J. Biol. Chem. 2010, 285, 17380–17389. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Van Delden, C.; Comte, R.; Bally, A.M. Stringent response activates quorum sensing and modulates cell density-dependent gene expression in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 5376–5384. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.L.; Lines, J.L.; Pesci, E.C.; Venturi, V.; Storey, D.G. Pseudomonas aeruginosa relA contributes to virulence in Drosophila melanogaster. Infect. Immun. 2004, 72, 5638–5645. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.K.; Swanson, M.S. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol. Microbiol. 1999, 33, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Haralalka, S.; Nandi, S.; Bhadra, R.K. Mutation in the relA gene of Vibrio cholerae affects in vitro and in vivo expression of virulence factors. J. Bacteriol. 2003, 185, 4672–4682. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerda, J.; Tedin, K. The bacterial signal molecule, ppGpp, regulates Salmonella virulence gene expression. Mol. Microbiol. 2004, 52, 1827–1844. [Google Scholar] [CrossRef] [PubMed]
- Chakraburtty, R.; Bibb, M. The ppGpp synthetase gene (relA) of Streptomyces coelicolor A3(2) plays a conditional role in antibiotic production and morphological differentiation. J. Bacteriol. 1997, 179, 5854–5861. [Google Scholar] [PubMed]
- Garza, A.G.; Pollack, J.S.; Harris, B.Z.; Lee, A.; Keseler, I.M.; Licking, E.F.; Singer, M. SdeK is required for early fruiting body development in Myxococcus xanthus. J. Bacteriol. 1998, 180, 4628–4637. [Google Scholar] [PubMed]
- Harris, B.Z.; Kaiser, D.; Singer, M. The guanosine nucleotide (p)ppGpp initiates development and A-factor production in Myxococcus xanthus. Genes Dev. 1998, 12, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Flores, A.; Du Pont, G.; Huerta-Saquero, A.; Merchant-Larios, H.; Servin-Gonzalez, L.; Duran, S. The stringent response is required for amino acid and nitrate utilization, Nod factor regulation, nodulation, and nitrogen fixation in Rhizobium etli. J. Bacteriol. 2005, 187, 5075–5083. [Google Scholar] [CrossRef] [PubMed]
- Moris, M.; Braeken, K.; Schoeters, E.; Verreth, C.; Beullens, S.; Vanderleyden, J.; Michiels, J. Effective symbiosis between Rhizobium etli and Phaseolus vulgaris requires the alarmone ppGpp. J. Bacteriol. 2005, 187, 5460–5469. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hipolito, C.J.; Maturana, A.D.; Ito, K.; Kuroda, T.; Higuchi, T.; Katoh, T.; Kato, H.E.; Hattori, M.; Kumazaki, K.; et al. Structural basis for the drug extrusion mechanism by a mate multidrug transporter. Nature 2013, 496, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Pages, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [PubMed]
- LaMarre, J.; Mendes, R.E.; Szal, T.; Schwarz, S.; Jones, R.N.; Mankin, A.S. The genetic environment of the cfr gene and the presence of other mechanisms account for the very high linezolid resistance of Staphylococcus epidermidis isolate 426-3147L. Antimicrobial Agents Chemother. 2013, 57, 1173–1179. [Google Scholar] [CrossRef]
- Bilgin, N.; Richter, A.A.; Ehrenberg, M.; Dahlberg, A.E.; Kurland, C.G. Ribosomal RNA and protein mutants resistant to spectinomycin. EMBO J. 1990, 9, 735–739. [Google Scholar] [PubMed]
- Chittum, H.S.; Champney, W.S. Ribosomal protein gene sequence changes in erythromycin-resistant mutants of Escherichia coli. J. Bacteriol. 1994, 176, 6192–6198. [Google Scholar] [PubMed]
- Gregory, S.T.; Dahlberg, A.E. Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23S ribosomal RNA. J. Mol. Biol. 1999, 289, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.H.; Slota, J.E.; Scarim, A.; Farrand, S.K. Genetic analysis of agrocin 84 production and immunity in Agrobacterium spp. J. Bacteriol. 1987, 169, 4184–4189. [Google Scholar] [PubMed]
- Beauclerk, A.A.; Cundliffe, E. Sites of action of two ribosomal RNA methylases responsible for resistance to aminoglycosides. J. Mol. Biol. 1987, 193, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin a antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Nguyen, F.; Bierschenk, M.; Sohmen, D.; Menzel, T.; Antes, I.; Wilson, D.N.; Bach, T. Amythiamicin d and related thiopeptides as inhibitors of the bacterial elongation factor EF-Tu: Modification of the amino acid at carbon atom C2 of ring C dramatically influences activity. Chem. Med. Chem. 2013, 8, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Donhofer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural basis for TetM-mediated tetracycline resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Atkinson, G.C.; Thakor, N.S.; Allas, U.; Lu, C.C.; Chan, K.Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V.; et al. Mechanism of tetracycline resistance by ribosomal protection protein tet(O). Nat. Commun. 2013, 4, 1477. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist Updat 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Mosher, R.H.; Camp, D.J.; Yang, K.; Brown, M.P.; Shaw, W.V.; Vining, L.C. Inactivation of chloramphenicol by O-phosphorylation. A novel resistance mechanism in Streptomyces venezuelae ISP5230, a chloramphenicol producer. J. Biol. Chem. 1995, 270, 27000–27006. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, T.; Sung, C.; Kim, H.; Song, E.; Park, H.Y.; Jeon, J.M.; Yoo, D.; Kim, H.J.; Kim, Y.H.; Choi, K.Y.; et al. Phosphorylation of chloramphenicol by a recombinant protein Yhr2 from Streptomyces avermitilis MA4680. Bioorg. Med. Chem. Lett. 2013, 23, 3614–3619. [Google Scholar] [CrossRef] [PubMed]
- Dhote, V.; Gupta, S.; Reynolds, K.A. An O-phosphotransferase catalyzes phosphorylation of hygromycin a in the antibiotic-producing organism Streptomyces hygroscopicus. Antimicrob. Agents Chemother. 2008, 52, 3580–3588. [Google Scholar] [CrossRef] [PubMed]
- Volkers, G.; Palm, G.J.; Weiss, M.S.; Wright, G.D.; Hinrichs, W. Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett. 2011, 585, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Nawa, K.; Tamura, Y.; Sato, K.; Hattori, J.; Shimotohno, K.W.; Endo, T. Inactivation of blasticidin S by Bacillus cereus. V. Purification and characterization of blasticidin S-deaminase mediated by a plasmid from blasticidin S resistant Bacillus cereus K55-S1. Biol. Pharm. Bull. 1995, 18, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Trana Discovery, Inc. Trana discovery technology-overview. Available online: http://www.tranadiscovery.com/wp-content/uploads/2013/08/Trana-Discovery-Company-and-Technology-Overview.pdf (accessed on 17 December 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chopra, S.; Reader, J. tRNAs as Antibiotic Targets. Int. J. Mol. Sci. 2015, 16, 321-349. https://doi.org/10.3390/ijms16010321
Chopra S, Reader J. tRNAs as Antibiotic Targets. International Journal of Molecular Sciences. 2015; 16(1):321-349. https://doi.org/10.3390/ijms16010321
Chicago/Turabian StyleChopra, Shaileja, and John Reader. 2015. "tRNAs as Antibiotic Targets" International Journal of Molecular Sciences 16, no. 1: 321-349. https://doi.org/10.3390/ijms16010321
APA StyleChopra, S., & Reader, J. (2015). tRNAs as Antibiotic Targets. International Journal of Molecular Sciences, 16(1), 321-349. https://doi.org/10.3390/ijms16010321