
The DDX23 Negatively Regulates Translation and Replication of Foot-and-Mouth Disease Virus and Is Degraded by 3C Proteinase

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Antibodies and Reagents
2.3. Biotinylated RNA Pull-Down Assay
2.4. Plasmid Construction and Transfection
2.5. In vitro Transcription
2.6. Knockdown and FMDV Infection
2.7. Western Blotting
2.8. TCID50 Assay
2.9. RNA Extraction and qRT–PCR Analysis
2.10. Confocal Microscopy
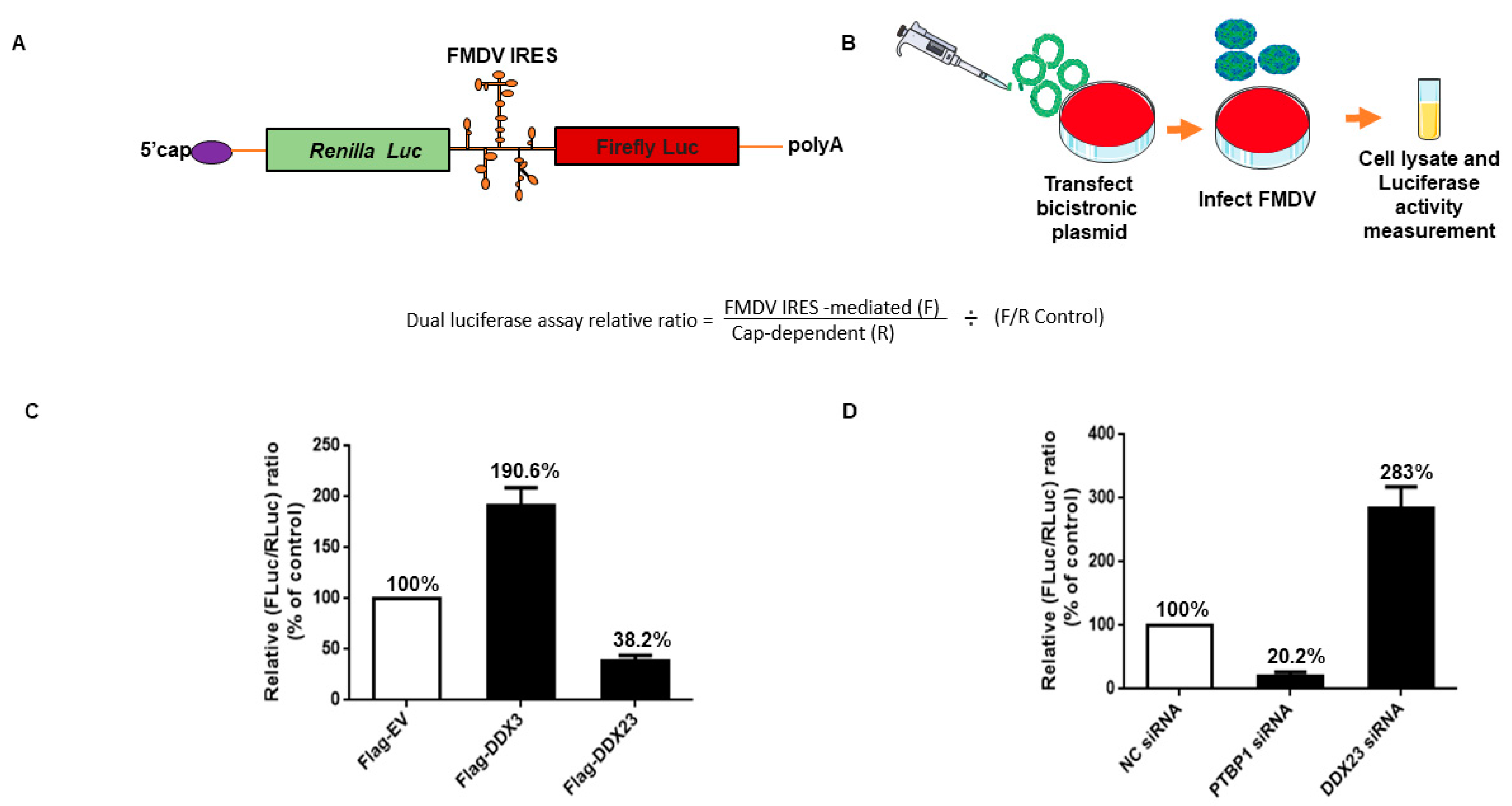
2.11. Luciferase Reporter Assay
2.12. Immunoprecipitation Assay
2.13. RNA Immunoprecipitation and RT–PCR
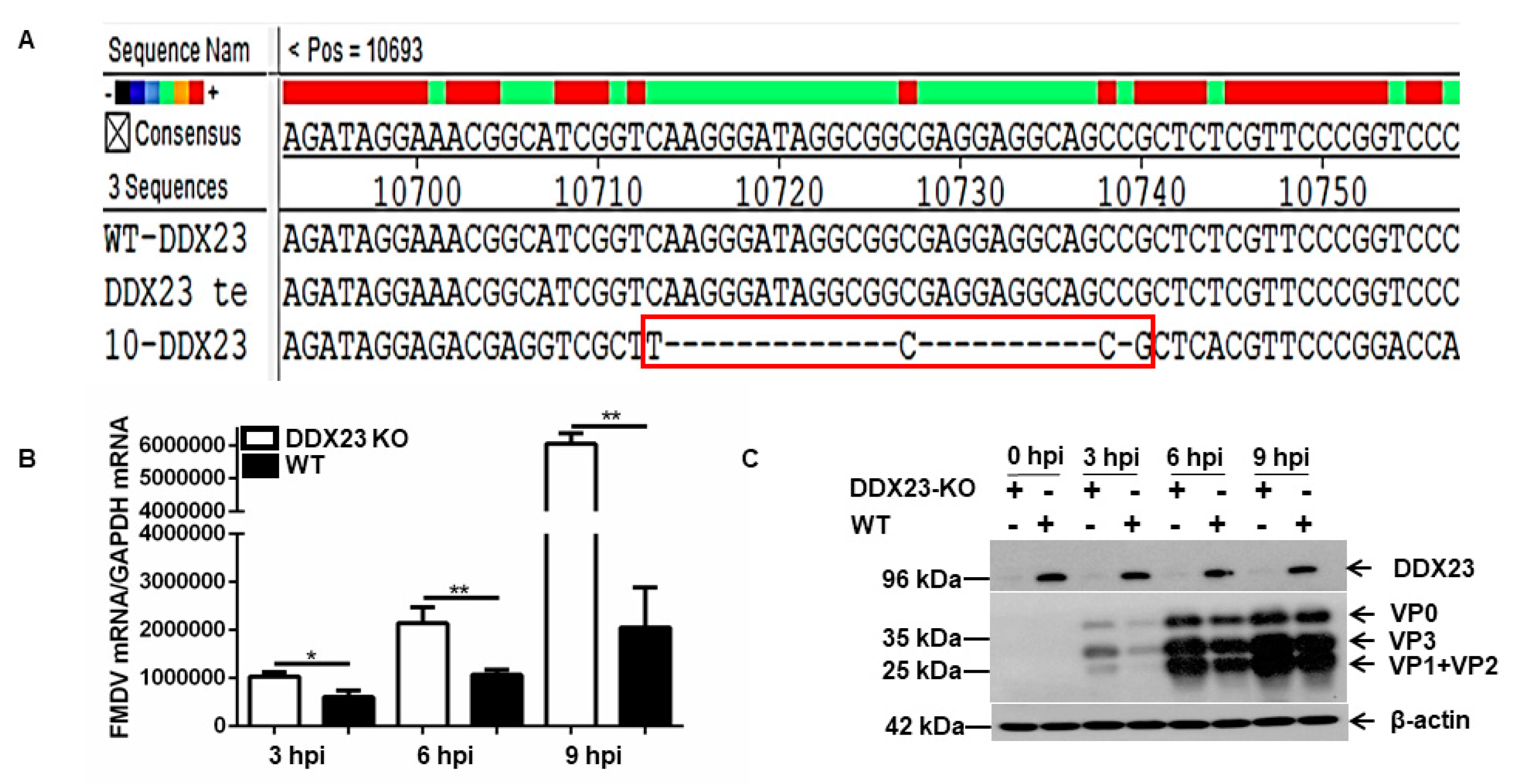
2.14. Establishment of DDX23-Knockout PK-15 Cell Line Using the Clustered Regularly Interspaced Short Palindromic Repeats ( CRISPR)/Cas9 System
2.15. Statistical Analysis
3. Results
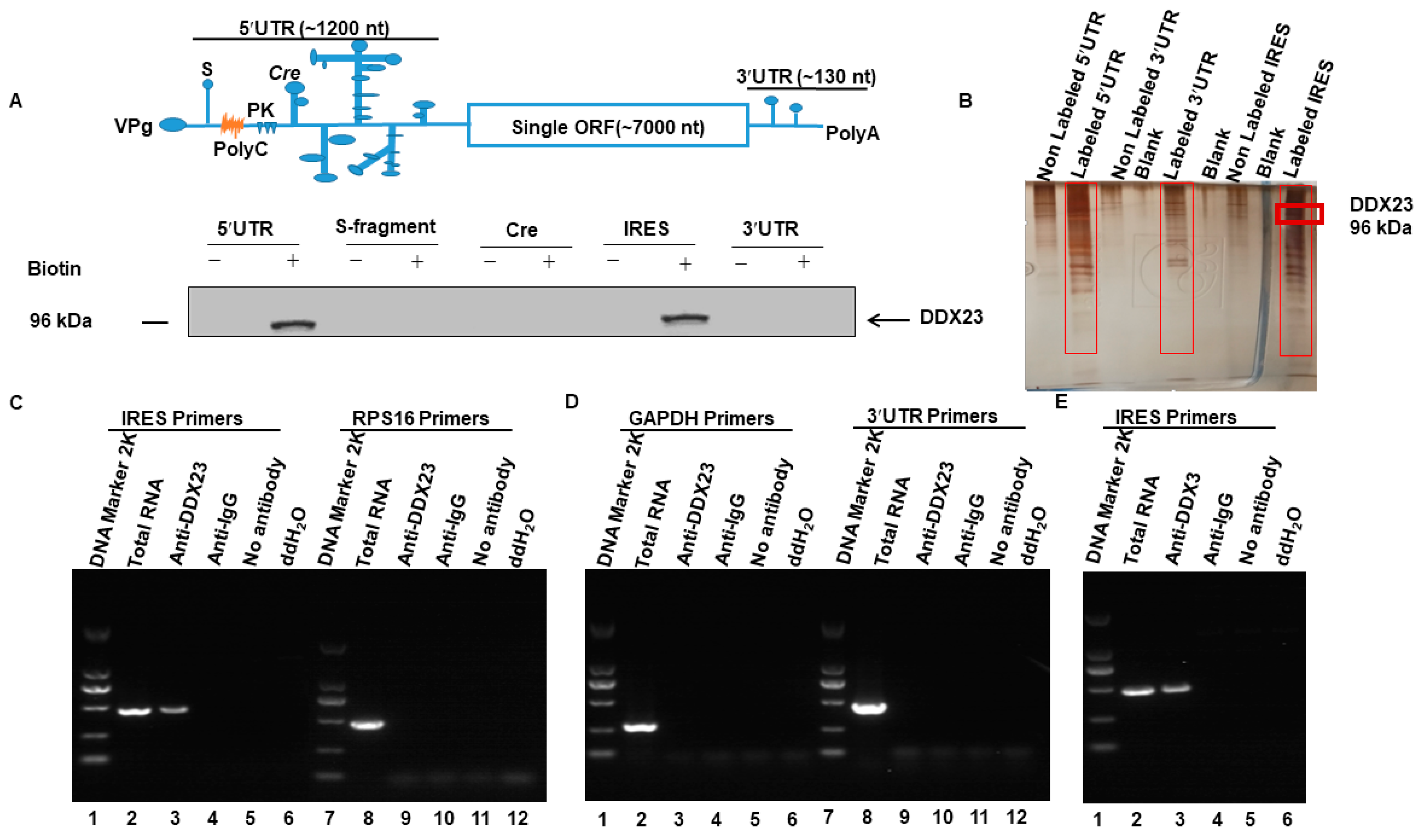
3.1. DDX23 Interacts with the FMDV IRES
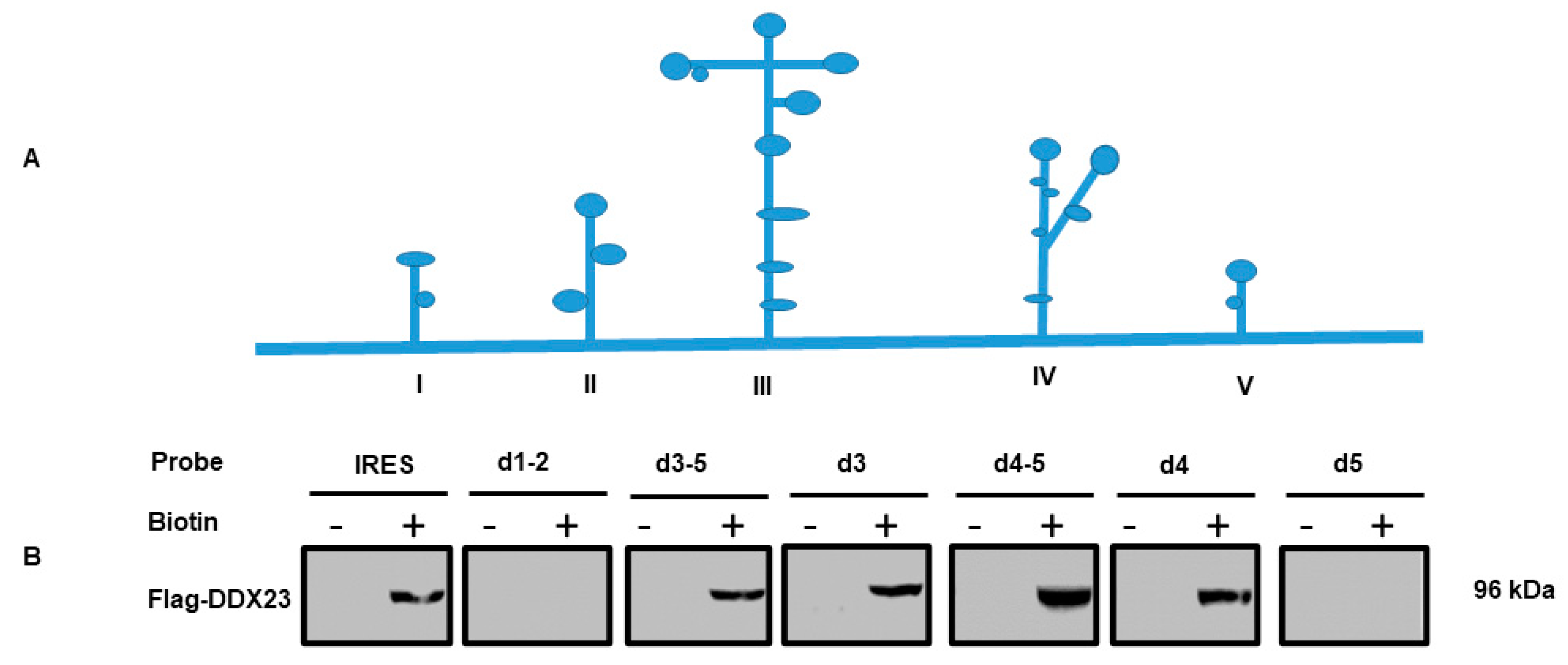
3.2. DDX23 Interact with the Specific Regions of the FMDV IRES
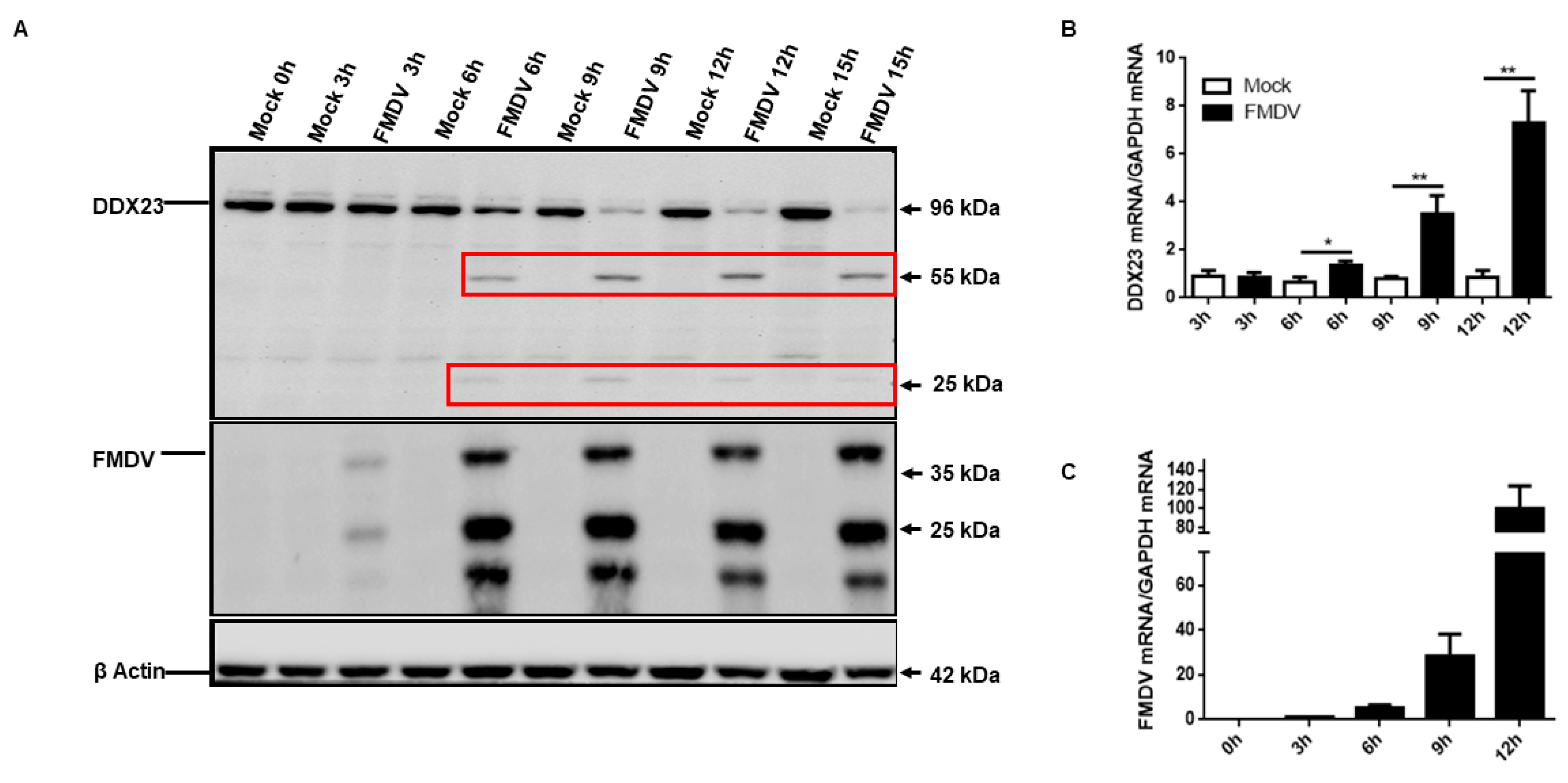
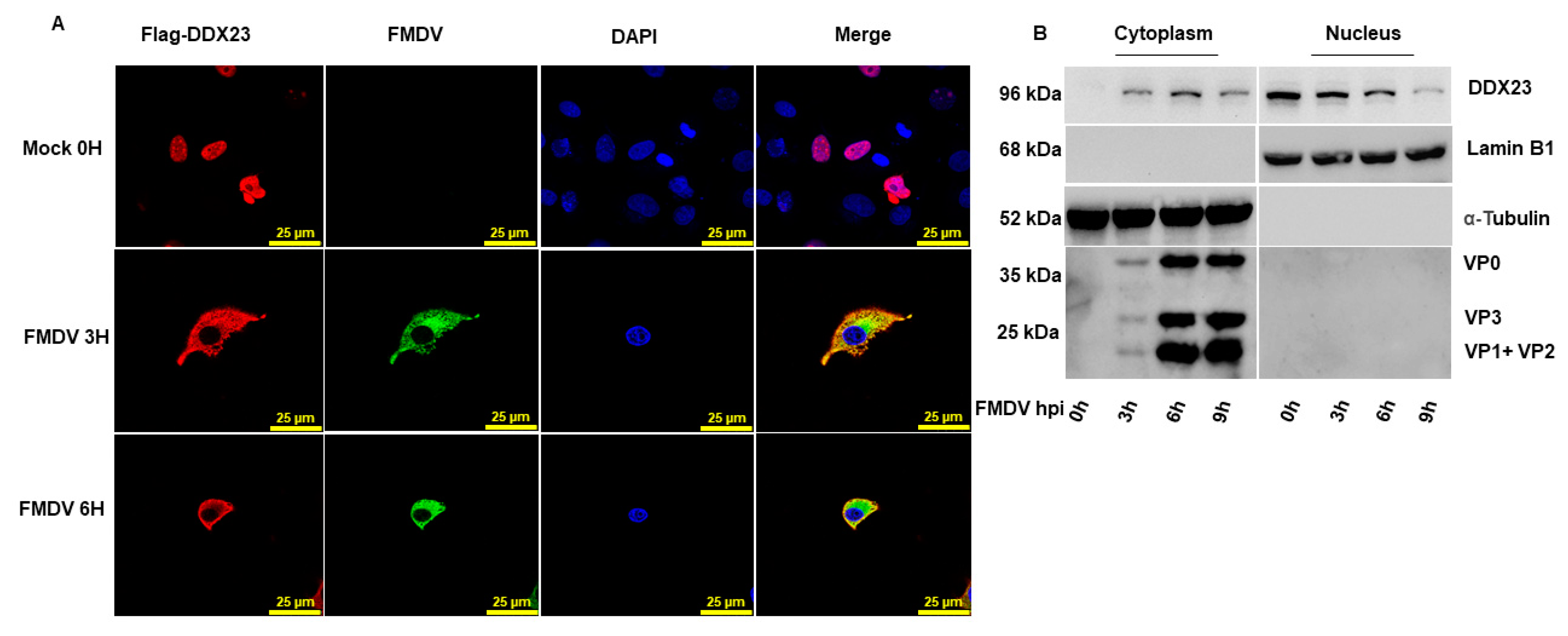
3.3. FMDV Infection Reduces DDX23 Protein Expression
3.4. DDX23 Inhibits FMDV Replication
3.5. DDX23 Suppresses IRES-Dependent Translation and Viral Replication
3.6. DDX23 Translocates during FMDV Infection
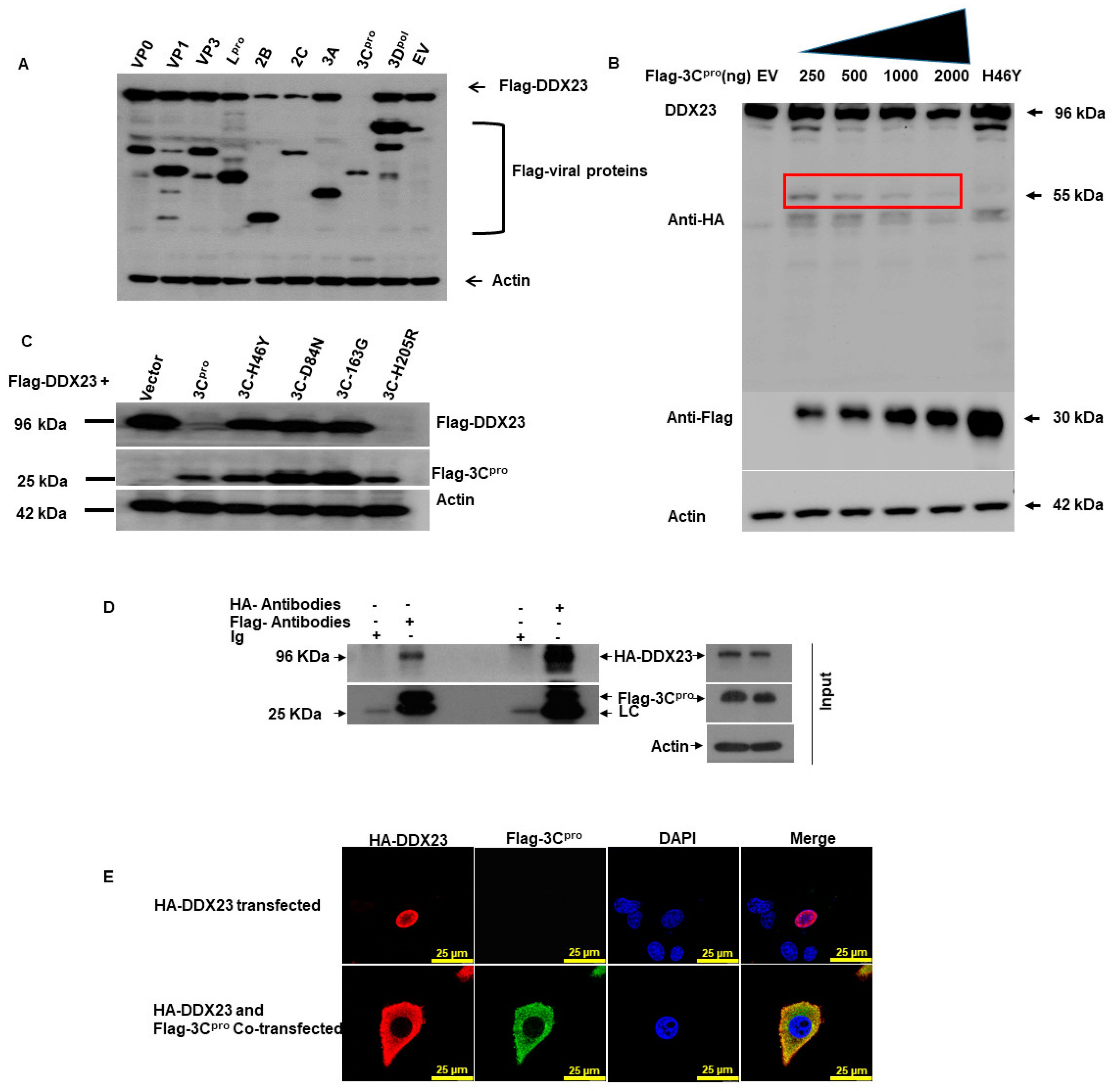
3.7. FMDV 3Cpro Interacts with DDX23, and Its Proteinase Activity Reduced DDX23 Expression
3.8. Involvement of the Lysosomal Pathway in FMDV-Dependent Reduction of DDX23
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R.; et al. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, R.K.; Leung, F.C. Evolutionary trend of foot-and-mouth disease virus in Hong Kong. Vet. Microbiol. 2012, 159, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Valdazo-Gonzalez, B.; Timina, A.; Scherbakov, A.; Abdul-Hamid, N.F.; Knowles, N.J.; King, D.P. Multiple introductions of serotype O foot-and-mouth disease viruses into East Asia in 2010-2011. Vet. Res. 2013, 44, 76. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.N.; Nguyen, T.; Kim, S.M.; Park, J.H.; Do, H.T.; Ngo, H.T.; Mai, D.T.; Lee, S.Y.; Nguyen, C.V.; Yoon, S.H.; et al. Direct typing and molecular evolutionary analysis of field samples of foot-and-mouth disease virus collected in Viet Nam between 2006 and 2007. Vet. Microbiol. 2011, 147, 244–252. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, S.Q.; Guo, H.C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Nunez, S.; Gismondi, M.I.; Konig, G.; Berinstein, A.; Taboga, O.; Rieder, E.; Martinez-Salas, E.; Carrillo, E. Enhanced IRES activity by the 3’UTR element determines the virulence of FMDV isolates. Virology 2014, 448, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Plank, T.D.; Kieft, J.S. The structures of nonprotein-coding RNAs that drive internal ribosome entry site function. Wiley Interdiscip. Rev. RNA 2012, 3, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Lozano, G.; Martinez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 2015, 12, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Lozano, G.; Fernandez, N.; Martinez-Salas, E. Magnesium-dependent folding of a picornavirus IRES element modulates RNA conformation and eIF4G interaction. FEBS J. 2014, 281, 3685–3700. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Miragall, O.; Martinez-Salas, E. Structural organization of a viral IRES depends on the integrity of the GNRA motif. RNA 2003, 9, 1333–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafasla, P.; Morgner, N.; Poyry, T.A.; Curry, S.; Robinson, C.V.; Jackson, R.J. Polypyrimidine tract binding protein stabilizes the encephalomyocarditis virus IRES structure via binding multiple sites in a unique orientation. Mol. Cell 2009, 34, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Quinto, S.L.; Lafuente, E.; Martinez-Salas, E. IRES interaction with translation initiation factors: Functional characterization of novel RNA contacts with eIF3, eIF4B, and eIF4GII. RNA 2001, 7, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monie, T.P.; Perrin, A.J.; Birtley, J.R.; Sweeney, T.R.; Karakasiliotis, I.; Chaudhry, Y.; Roberts, L.O.; Matthews, S.; Goodfellow, I.G.; Curry, S. Structural insights into the transcriptional and translational roles of Ebp1. EMBO J. 2007, 26, 3936–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.T.; Kung, Y.A.; Li, M.L.; Brewer, G.; Lee, K.M.; Liu, S.T.; Shih, S.R. Additive Promotion of Viral Internal Ribosome Entry Site-Mediated Translation by Far Upstream Element-Binding Protein 1 and an Enterovirus 71-Induced Cleavage Product. PLoS Pathog. 2016, 12, e1005959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolbert, M.; Morgan, C.E.; Pollum, M.; Crespo-Hernandez, C.E.; Li, M.L.; Brewer, G.; Tolbert, B.S. HnRNP A1 Alters the Structure of a Conserved Enterovirus IRES Domain to Stimulate Viral Translation. J. Mol. Biol. 2017, 429, 2841–2858. [Google Scholar] [CrossRef]
- Galan, A.; Lozano, G.; Pineiro, D.; Martinez-Salas, E. G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 2017, 284, 3202–3217. [Google Scholar] [CrossRef] [Green Version]
- Wurth, L.; Papasaikas, P.; Olmeda, D.; Bley, N.; Calvo, G.T.; Guerrero, S.; Cerezo-Wallis, D.; Martinez-Useros, J.; Garcia-Fernandez, M.; Huttelmaier, S.; et al. UNR/CSDE1 Drives a Post-transcriptional Program to Promote Melanoma Invasion and Metastasis. Cancer Cell 2016, 30, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Chamorro, J.; Francisco-Velilla, R.; Ramajo, J.; Martinez-Salas, E. Rab1b and ARF5 are novel RNA-binding proteins involved in FMDV IRES-driven RNA localization. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, A.; Lopez de Quinto, S.; Ramajo, J.; Fernandez, N.; Martinez-Salas, E. A novel role for Gemin5 in mRNA translation. Nucleic Acids Res. 2009, 37, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.K.; Lawrence, P.; Kloc, A.; Schafer, E.; Rieder, E. Analysis of the interaction between host factor Sam68 and viral elements during foot-and-mouth disease virus infections. Virol. J. 2015, 12, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Yang, D.; Sun, C.; Wang, H.; Zhao, B.; Zhou, G.; Yu, L. hnRNP K Is a Novel Internal Ribosomal Entry Site-Transacting Factor That Negatively Regulates Foot-and-Mouth Disease Virus Translation and Replication and Is Antagonized by Viral 3C Protease. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Chen, C.J.; Shih, S.R. Regulation Mechanisms of Viral IRES-Driven Translation. Trends Microbiol. 2017, 25, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Petersen, A.; Meyer, K.; Beck, E. Functional involvement of polypyrimidine tract-binding protein in translation initiation complexes with the internal ribosome entry site of foot-and-mouth disease virus. J. Virol. 1997, 71, 8330–8339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. The DEAD-box protein family of RNA helicases. Gene 2006, 367, 17–37. [Google Scholar] [CrossRef]
- Ranji, A.; Boris-Lawrie, K. RNA helicases: Emerging roles in viral replication and the host innate response. RNA Biol. 2010, 7, 775–787. [Google Scholar] [CrossRef] [Green Version]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef]
- Konishi, T.; Uodome, N.; Sugimoto, A. The Caenorhabditis elegans DDX-23, a homolog of yeast splicing factor PRP28, is required for the sperm-oocyte switch and differentiation of various cell types. Dev. Dyn. 2008, 237, 2367–2377. [Google Scholar] [CrossRef]
- Ruan, J.; Cao, Y.; Ling, T.; Li, P.; Wu, S.; Peng, D.; Wang, Y.; Jia, X.; Chen, S.; Xu, A.; et al. DDX23, an Evolutionary Conserved dsRNA Sensor, Participates in Innate Antiviral Responses by Pairing With TRIF or MAVS. Front. Immunol. 2019, 10, 2202. [Google Scholar] [CrossRef]
- Krishnan, V.; Zeichner, S.L. Alterations in the expression of DEAD-box and other RNA binding proteins during HIV-1 replication. Retrovirology 2004, 1, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Q.; Zhang, S.; Lin, Q.; Qu, R.L.; Li, Y.F.; Zhang, F.H.; Xu, A.L. Immune response- and viral control-related pathways in the progression of chronic hepatitis B. Microb. Pathog. 2017, 105, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Murillo, A.; Vera-Estrella, R.; Barkla, B.J.; Mendez, E.; Arias, C.F. Identification of Host Cell Factors Associated with Astrovirus Replication in Caco-2 Cells. J. Virol. 2015, 89, 10359–10370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtley, J.R.; Knox, S.R.; Jaulent, A.M.; Brick, P.; Leatherbarrow, R.J.; Curry, S. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 2005, 280, 11520–11527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Grubman, M.J.; Zellner, M.; Bablanian, G.; Mason, P.W.; Piccone, M.E. Identification of the active-site residues of the 3C proteinase of foot-and-mouth disease virus. Virology 1995, 213, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.M.; Grigera, P.R.; Bergmann, I.E.; Zibert, A.; Multhaup, G.; Beck, E. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 1990, 64, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, P.; Schafer, E.A.; Rieder, E. The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 2012, 425, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ross-Smith, N.; Proud, C.G.; Belsham, G.J. Cleavage of translation initiation factor 4AI (eIF4AI) but not eIF4AII by foot-and-mouth disease virus 3C protease: Identification of the eIF4AI cleavage site. FEBS Lett. 2001, 507, 1–5. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, S.; Roque-Rosell, N.; Sweeney, T.R.; Zunszain, P.A.; Leatherbarrow, R.J. Structural analysis of foot-and-mouth disease virus 3C protease: A viable target for antiviral drugs? Biochem. Soc. Trans. 2007, 35, 594–598. [Google Scholar] [CrossRef]
- Zhi, X.; Zhang, Y.; Sun, S.; Zhang, Z.; Dong, H.; Luo, X.; Wei, Y.; Lu, Z.; Dou, Y.; Wu, R.; et al. NLRP3 inflammasome activation by Foot-and-mouth disease virus infection mainly induced by viral RNA and non-structural protein 2B. RNA Biol. 2020, 17, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Su, W.C.; Jeng, K.S.; Chang, T.H.; Lai, M.M. Attenuation of 40S ribosomal subunit abundance differentially affects host and HCV translation and suppresses HCV replication. PLoS Pathog. 2012, 8, e1002766. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Sun, S.; Li, P.; Liu, Q.; Zhang, Z.; Dong, H.; Sun, M.; Wu, W.; Wang, X.; Guo, H. Ribosomal Protein L13 Promotes IRES-Driven Translation of Foot-and-Mouth Disease Virus in a Helicase DDX3-Dependent Manner. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Lee, J.T.Y.; Tsang, W.H.; Chow, K.L. Simple Modifications to Standard TRIzol® Protocol Allow High-Yield RNA Extraction from Cells on Resorbable Materials. J. Biomater. Nanobiotechnol. 2011, 2, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Andreev, D.E.; Fernandez-Miragall, O.; Ramajo, J.; Dmitriev, S.E.; Terenin, I.M.; Martinez-Salas, E.; Shatsky, I.N. Differential factor requirement to assemble translation initiation complexes at the alternative start codons of foot-and-mouth disease virus RNA. RNA 2007, 13, 1366–1374. [Google Scholar] [CrossRef] [Green Version]
- Komar, A.A.; Hatzoglou, M. Exploring Internal Ribosome Entry Sites as Therapeutic Targets. Front. Oncol. 2015, 5, 233. [Google Scholar] [CrossRef] [Green Version]
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belsham, G.J. Divergent picornavirus IRES elements. Virus Res. 2009, 139, 183–192. [Google Scholar] [CrossRef]
- Pichon, X.; Wilson, L.A.; Stoneley, M.; Bastide, A.; King, H.A.; Somers, J.; Willis, A.E. RNA binding protein/RNA element interactions and the control of translation. Curr. Protein Pept. Sci. 2012, 13, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luz, N.; Beck, E. Interaction of a cellular 57-kilodalton protein with the internal translation initiation site of foot-and-mouth disease virus. J. Virol. 1991, 65, 6486–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, M.E.; Seamons, R.A.; Belsham, G.J. A selection system for functional internal ribosome entry site (IRES) elements: Analysis of the requirement for a conserved GNRA tetraloop in the encephalomyocarditis virus IRES. RNA 1999, 5, 1167–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.E.; Lieberman, P.M.; Berk, A.J.; Dasgupta, A. Direct cleavage of human TATA-binding protein by poliovirus protease 3C in vivo and in vitro. Mol. Cell. Biol. 1993, 13, 1232–1237. [Google Scholar] [CrossRef] [Green Version]
- Gustin, K.E.; Sarnow, P. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 2001, 20, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wu, F.; Han, J.; Qi, F.; Ni, T.; Qian, F. Exploitation of nuclear protein SFPQ by the encephalomyocarditis virus to facilitate its replication. Biochem. Biophys. Res. Commun. 2019, 510, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wang, G.; Yang, F.; Cao, W.; Mao, R.; Du, X.; Zhang, X.; Li, C.; Li, D.; Zhang, K.; et al. Foot-and-Mouth Disease Virus Viroporin 2B Antagonizes RIG-I-Mediated Antiviral Effects by Inhibition of Its Protein Expression. J. Virol. 2016, 90, 11106–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhu, Z.; Du, X.; Cao, W.; Yang, F.; Zhang, X.; Feng, H.; Li, D.; Zhang, K.; Liu, X.; et al. Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication. Virology 2017, 509, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Han, S.; Yan, D.; Gao, Y.; Wei, Y.; Liu, X.; Liao, Y.; Guo, H.; Sun, S. Foot-and-mouth disease virus infection suppresses autophagy and NF-small ka, CyrillicB antiviral responses via degradation of ATG5-ATG12 by 3C(pro). Cell Death Dis. 2017, 8, e2561. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, H.; Zeng, Q.; Zheng, H.; Xue, Q.; Cai, X. The DEAD-Box RNA Helicase DDX1 Interacts with the Viral Protein 3D and Inhibits Foot-and-Mouth Disease Virus Replication. Virol. Sin. 2019, 34, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.L. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah, S.W.; Han, S.; Wu, J.; Zhang, Y.; Bai, M.; Jin, Y.; Zhi, X.; Guan, J.; Sun, S.; Guo, H. The DDX23 Negatively Regulates Translation and Replication of Foot-and-Mouth Disease Virus and Is Degraded by 3C Proteinase. Viruses 2020, 12, 1348. https://doi.org/10.3390/v12121348
Abdullah SW, Han S, Wu J, Zhang Y, Bai M, Jin Y, Zhi X, Guan J, Sun S, Guo H. The DDX23 Negatively Regulates Translation and Replication of Foot-and-Mouth Disease Virus and Is Degraded by 3C Proteinase. Viruses. 2020; 12(12):1348. https://doi.org/10.3390/v12121348
Chicago/Turabian StyleAbdullah, Sahibzada Waheed, Shichong Han, Jin’en Wu, Yun Zhang, Manyuan Bai, Ye Jin, Xiaoying Zhi, Junyong Guan, Shiqi Sun, and Huichen Guo. 2020. "The DDX23 Negatively Regulates Translation and Replication of Foot-and-Mouth Disease Virus and Is Degraded by 3C Proteinase" Viruses 12, no. 12: 1348. https://doi.org/10.3390/v12121348