Polymerization Reactions and Modifications of Polymers by Ionizing Radiation

 ,
, 
Abstract
:1. Introduction
1.1. Fundamental of Radiation Effects on Polymers
1.2. The Complexity of the Chemical Structures of New Polymeric Materials Used in Advanced Technology
2. Fundamental and Technological Aspects of Radiation-Induced Polymerization
2.1. Specificities of Radiation-Initiated Polymerization
2.1.1. Instantaneous Impact of Radiation Treatment
2.1.2. Spatial Control of Radiation-Induced Effects
2.1.3. Random Energy Deposition
2.1.4. Decoupling of Primary Initiation Steps from Thermal Activation
2.2. Basic Aspects
2.2.1. Free Radical Polymerization
2.2.2. Ionic Polymerization
2.2.3. Cationic Polymerization
2.2.4. Anionic Polymerization
2.2.5. Controlled Free Radical Polymerization
2.3. Radiation-Induced Cross-Linking Polymerization
2.3.1. General Description
2.3.2. Initiation Mechanisms
2.3.3. Gelation and Vitrification during Network Formation
3. Graft Copolymerization Induced by Ionizing Radiation
3.1. Radiation-Induced Grafting of Solid Polymers
3.2. Radiation-Induced Grafting Processing
3.3. Parameters Affecting the RIG
3.3.1. Irradiation Dose and Dose Rate
3.3.2. Polymer Substrate Chemistry
3.3.3. Monomer Concentration
3.3.4. Solvent
3.3.5. Grafting Temperature
3.3.6. Presence of Inhibitor of Homopolymerization
3.4. Grafting Front Mechanism
3.5. Radiation-Induced Grafting of Semi-Crystalline Polymers
3.6. Nature and Trapping of Radicals
3.7. Ion-Track Grafting
3.8. RIG in Ion Track-Etched Polymer Membranes
4. Radiation Synthesis of Polymer-Based Nanogels
4.1. Nanogels
4.2. General Synthetic Approaches
4.3. Synthesis of Nanogels by Radiation-Induced Intramolecular Crosslinking of Polymers
4.3.1. Radiation Chemistry of Polymers in Aqueous Solution
4.3.2. Synthesis of Nanogels by Intramolecular Crosslinking
4.3.3. Controlling the Physicochemical Properties of Radiation-Synthesized Nanogels
4.3.4. Controlling the Chemistry of Radiation-Synthesized Nanogels
4.4. Biomedical Applications of Radiation Engineered Nanogels
5. Radiation Chemistry of Natural Polymers
5.1. Radiation Chemistry of Polysaccharides
5.2. Sold State Irradiation
5.3. Aqueous Irradiation
5.4. The Effect of Oxygen
5.5. Radiation Crosslinking of Polysaccharides
5.6. Radical Lifetime in Polysaccharides
5.7. Radiation Chemistry of Lignin:
5.8. Radiation Chemistry of Natural Rubber
5.9. Radiation Chemistry of Peptides and Proteins
5.10. Radiation Chemistry of RNA and DNA
6. Summary
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Charlesby, A. Atomic Radiation and Polymers; Pergamon Press: Oxford, UK, 1960. [Google Scholar]
- Chapiro, A. Radiation Chemistry of Polymeric Systems; Interscience: New York, NY, USA, 1962. [Google Scholar]
- Dole, M. The Radiation Chemistry of Macromolecules; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- Schnabel, W. Polymer Degradation. Principles and Practical Applications; Hanser: Muenchen, Germany, 1981. [Google Scholar]
- Rosiak, J.M. Radiation Effects of Polymers. In ACS Symposium Series; ACS Symposium, Series; Clough, R.L., Shalaby, S.W., Eds.; American Chemical Society: Washington, DC, USA, 1991; Volume 475. [Google Scholar]
- Singh, A.; Silverman, J. (Eds.) Radiation Processing of Polymers; Hanser: Muenchen, Germany, 1992. [Google Scholar]
- Ivanov, V.S. Radiation Chemistry of Polymers; VSP: Utrecht, The Netherlands, 1992. [Google Scholar]
- Coqueret, X. Obtaining High-Performance Polymeric materials by Radiation. In Radiation Chemistry: From Basics to Applications in Material and Life Sciences; Spotheim-Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Les Ulis, France, 2008; pp. 131–150. [Google Scholar]
- Drobny, J.G. Ionizing Radiation and Polymers: Principles, Technology, and Applications; Wiliam Andrew: Norwich, NY, USA, 2013. [Google Scholar]
- Coqueret, X.; Sabharwal, S.; Khairul Zaman, H.M.D.; Czechowska-Biskup, R.; Wach, R.A.; Rosiak, J.M.; Ulanski, P.; Gulrez, S.K.H.; Al-Assaf, S. Introdcution to the radiation chemistry of polymers. In The Radiation Chemistry of Polysaccharides; Al-Assaf, S., Coqueret, X., Khairul Zaman, H.M.D., Sen, M., Ulanski, P., Eds.; International Atomic Energy Agency: Vienna, Austria, 2016. [Google Scholar]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Ferry, M.N.; Ngono-Ravache, Y.; Aymes-Chodur, C.; Clochard, M.C.; Coqueret, X.; Cortella, L.; Pellizzi, E.; Rouif, S.; Esnouf, S. Ionizing radiation effects in polymers. In Reference Module in Materials Science and Materials Engineering; Hashmi, S., Ed.; Elsevier: Oxford, UK, 2016; pp. 131–149. [Google Scholar]
- Wojnarovits, L. Radiation chemistry. In Handbook of Nuclear Chemistry; Vértes, A.N.S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Boston, MA, USA, 2011; pp. 1263–1331. [Google Scholar]
- Chapiro, A. Radiation-Induced Polymerization. Radiat. Phys. Chem 1979, 14, 101–116. [Google Scholar] [CrossRef]
- Hopwood, F.L.; Phillips, J.T. Polymerization of liquids by irradiation with neutrons and other rays. Nature 1939, 143, 640. [Google Scholar] [CrossRef]
- Chapiro, A. Polymerisation par les rayons-gamma.2. C.R. Acad. Sci. Chim. 1949, 229, 827–829. [Google Scholar]
- Zivic, N.; Kuroishi, P.K.; Dumur, F.; Gigmes, D.; Dove, A.P.; Sardon, H. Recent advances and challenges in the design of organic photoacid and photobase generators for polymerizations. Angew.Chem. Int. Ed. 2019, 58, 10410–10422. [Google Scholar] [CrossRef]
- Zandi Shafagh, R.; Vastesson, A.; Guo, W.; van der Wijngaart, W.; Haraldsson, T. E-beam nanostructuring and direct click biofunctionalization of thiol-ene resist. ACS Nano 2018, 12, 9940–9946. [Google Scholar] [CrossRef]
- Sarapas, J.M.; Tew, G.N. Thiol-ene step-growth as a versatile route to functional polymers. Angew. Chem. Int. Ed. Engl. 2016, 55, 15860–15863. [Google Scholar] [CrossRef]
- Kinoshita, S. Basics and applications of uv/eb curing technology. J. Photopolym. Sci. Technol. 2006, 19, 93–98. [Google Scholar] [CrossRef]
- Wojnarovits, L.; TakACS, E.; Dobo, J.; Foldiak, G. Pulse-radiolysis studies on the polymerization of 1,6-hexanediol diacrylate in cyclohexane solvent. Radiat. Phys. Chem. 1992, 39, 59–63. [Google Scholar] [CrossRef]
- Silverman, J.; Weiss, D.E. The role of pulse frequency and acrylic acid in the radiation-induced bulk polymerization of 2-ethylhexyl acrylate. Radiat. Phys. Chem. 2003, 67, 347–352. [Google Scholar] [CrossRef]
- Defoort, B.; Larnac, G.; Coqueret, X. Electron-beam initiated polymerization of acrylate compositions 4: Effects of pulsed irradiation parameters on curing kinetics. Radiat. Phys. Chem. 2001, 62, 47–53. [Google Scholar] [CrossRef]
- Feng, H.X.; Al-Sheikhly, M.; Silverman, J.; Weiss, D.E.; Neta, P. Polymerization of neat 2-ethylhexyl acrylate induced by a pulsed electron beam. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 196–203. [Google Scholar] [CrossRef]
- Berger, K. Energy curing: Advancing the graphic arts through innovation and performance. RadTech Rep. 2001, 15, 44–46. [Google Scholar]
- Sakaguchi, S.; Sakurai, T.; Ma, J.; Sugimoto, M.; Yamaki, T.; Chiba, A.; Saito, Y.; Seki, S. Conjugated nanowire sensors via high-energy single-particle-induced linear polymerization of 9,9 ‘-spirobi[9h-fluorene] derivatives. J. Phys. Chem. B 2018, 122, 8614–8623. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Kamiya, K.; Sakurai, T.; Seki, S. Interactions of single particle with organic matters: A facile bottom-up approach to low dimensional nanostructures. Quantum Beam Sci. 2020, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Beziers, D.; Perilleux, P.; Grenie, Y. Composite structures obtained by ionization curing. Radiat. Phys. Chem. 1996, 48, 171–177. [Google Scholar] [CrossRef]
- Vinje, J.; Beckwith, K.S.; Sikorski, P. Electron beam lithography fabrication of su-8 polymer structures for cell studies. J. Microelectromech. Syst. 2020, 29, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Furtak-Wrona, K.; Kozik-Ostrowka, P.; Jadwiszczak, K.; Maigret, J.E.; Aguie-Beghin, V.; Coqueret, X. Polyurethane acrylate networks including cellulose nanocrystals: A comparison between uv and eb- curing. Radiat. Phys. Chem. 2018, 142, 94–99. [Google Scholar] [CrossRef]
- Herman, J.A.; Roberge, P. X-ray induced polymerization of vinyl iodide in solution. J. Polym. Sci. 1962, 62, S116–S118. [Google Scholar] [CrossRef]
- Herman, J.A.; Roberge, P.C. Radiolysis of vinyl iodide.3. Formation of polymer in carbon tetrachloride solutions. Trans. Faraday Soc. 1969, 65, 1315–1324. [Google Scholar] [CrossRef]
- Roberge, P.C.; Herman, J.A. Radiolysis of vinyl iodide.2. Solutions of vinyl iodide in carbon tetrachloride. Trans. Faraday Soc. 1969, 65, 1303–1314. [Google Scholar] [CrossRef]
- Anderson, W.S. Radiation-induced cationic polymerization of butadiene. J. Phys. Chem. 1959, 63, 765–766. [Google Scholar] [CrossRef]
- Verdurmen, E.M.; German, A.L.; Sudol, E.D.; Gilbert, R.G. Particle growth in butadiene emulsion polymerization. 2. Gamma-radiolysis. Macromol. Chem. Phys. 1994, 195, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Z.P.; Ying, S.K. Study of gamma-ray radiation-induced polymerization of butadiene in ethanol. Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1991, 37, 263–266. [Google Scholar]
- Chatani, Y.; Nakatani, S.; Tadokoro, H. Structural evidence of radiation-induced thiourea canal polymerization of 2,3-disubstituted-1,3-butadienes. Acta Cryst. A 1972, 28, S123. [Google Scholar] [CrossRef]
- Miyata, M.; Takemoto, K. Radiation-induced polymerization of 2,3-disubstituted butadienes in deoxycholic-acid inclusion compounds. J. Polym. Sci. Part C Polym. Symp. 1975, 13, 221–223. [Google Scholar] [CrossRef]
- Cataldo, F.; Ragni, P.; Ursini, O.; Angelini, G. Synthesis of highly crystalline poly(dimethylbutadiene) (PDMB) by radiation-induced inclusion polymerization: A comparison with pdmbs synthesized by bulk and emulsion polymerization. Radiat. Phys. Chem. 2008, 77, 941–948. [Google Scholar] [CrossRef]
- Cataldo, F.; Ursini, O.; Ragni, P.; Rosati, A. Radiation-induced polymerization of 2,3-dimethyl-1,3-butadiene clathrate in deoxycholic acid. J. Radioanal. Nucl. Chem. 2009, 280, 99–106. [Google Scholar] [CrossRef]
- Yoshii, F.; Abe, T.; Kobayashi, Y. Radiation-induced in-source polymerization of acrylonitrile in urea canal complex—Radiation-induced polymerization of vinyl monomer in urea canal complex.4. Kobunshi Ronbunshu 1975, 32, 477–483. [Google Scholar]
- Zou, J.-T.; Wang, Y.-S.; Pang, W.-M.; Shi, L.; Lu, F. Radiation-induced inclusion polymerization of acrylonitrile in urea canals: Toward synthesis of completely isotactic polyacrylonitrile with controlled molecular weight. Macromolecules 2013, 46, 1765–1771. [Google Scholar] [CrossRef]
- Stannett, V.T. Radiation-induced ionic polymerization and grafting of vinyl monomers. Br. Polym. J. 1981, 13, 93–98. [Google Scholar] [CrossRef]
- Charlesby, A. Solid-state polymerization induced by radiation. Rep. Prog. Phys. 1965, 28, 463–518. [Google Scholar] [CrossRef]
- Mah, S.; Yamamoto, Y.; Hayashi, K. Effects of triphenylsulfonium hexafluorophosphate in the radiation-induced cationic polymerization of styrene. J. Polym. Sci. Part A Polym. Chem. 1982, 20, 1709–1716. [Google Scholar] [CrossRef]
- Mah, S.; Yamamoto, Y.; Hayashi, K. Effects of diphenyliodonium hexafluorophosphate in the radiation-induced cationic polymerization of styrene—Formation and decay processes of styrene dimer radical cation. J. Polym. Sci. Part A Polym. Chem. 1982, 20, 2151–2158. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Teramoto, M.; Ma, X.H.; Hayashi, K. Radiation-induced cationic polymerization by aryl onium salts. Abstr. Pap. Am. Chem. Soc. 1989, 198, 43. [Google Scholar]
- Henley, E.J.; Ng, C.C. Gamma-radiation-induced solution polymerization of ethylene. J. Polym. Sci. 1959, 36, 511–517. [Google Scholar] [CrossRef]
- Colombo, P.; Chapman, R.N.; Steinberg, M.; Fontana, J.; Kukacka, L.E. High-pressure ethylene polymerization reactions with cobalt-60 gamma radiation. Trans. Am. Nuc. Soc. 1964, 7, 313–3214. [Google Scholar]
- Wiley, R.H.; Parrish, C.F.; Lipscomb, N.T.; Guillet, J.E. Kinetics of gamma-radiation-induced polymerization of ethylene in alkyl chlorides. J. Polym. Sci. Part A Polym. Chem. 1964, 2, 2503–2511. [Google Scholar] [CrossRef]
- Hagiwara, M.; Mitsui, H.; Machi, S.; Kagiya, T. Liquid carbon dioxide as a solvent for radiation polymerization of ethylene. J. Polym. Sci. Part A Polym. Chem. 1968, 6, 603–608. [Google Scholar] [CrossRef]
- Takehisa, M.; Machi, S.; Watanabe, H.; Ueno, T.; Takahashi, S.; Tsuchiya, R.; Otaguro, K.; Motoda, I.; Takasaka, Y.; Miyanaga, K.; et al. Radiation-induced polymerization of ethylene in a pilot-plant.1. Bulk process. J. Appl. Polym. Sci. 1979, 24, 853–864. [Google Scholar] [CrossRef]
- Takehisa, M.; Watanabe, H.; Kurihara, H.; Takasaka, Y.; Maruyama, Y.; Miyanaga, K.; Suwa, T.; Nakajima, H.; Yamaguchi, K.; Tohei, M.; et al. Radiation-induced polymerization of ethylene in a pilot-plant. 2. Development of wet-wall process. J. Appl. Polym. Sci. 1979, 24, 865–882. [Google Scholar] [CrossRef]
- Takehisa, M.; Watanabe, H.; Kurihara, H.; Yamaguchi, K.; Nakajima, H.; Yagi, T.; Watanabe, T.; Sugo, T.; Suwa, T.; Maruyama, S.; et al. Radiation-induced polymerization of ethylene in pilot-plant. 3. Heavy-phase recycling process. J. Appl. Polym. Sci. 1979, 24, 1831–1844. [Google Scholar] [CrossRef]
- Watanabe, H.; Machi, S.; Kurihara, H.; Wada, T.; Yamaguchi, K.; Watanabe, T.; Takehisa, M. Radiation-induced polymerization of ethylene in pilot-plant. 4. Kinetic-analysis. J. Appl. Polym. Sci. 1980, 25, 277–285. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Watanabe, H.; Sugo, T.; Watanabe, T.; Takehisa, M.; Machi, S. Radiation-induced polymerization of ethylene in pilot-plant. 5. Molecular-weight distribution of polyethylene. J. Appl. Polym. Sci. 1980, 25, 1633–1638. [Google Scholar] [CrossRef]
- Machi, S.; Tamura, T.; Hagiwara, M.; Gotoda, M.; Kagiya, T. Short-chain branching in gamma-radiation-induced polymerization of ethylene. J. Polym. Sci. Part A Polym. Chem. 1966, 4, 283–291. [Google Scholar] [CrossRef]
- Wiley, R.H.; Lipscomb, M.T. Gamma-radiation-induced polymerization of styrene. J. Polym. Sci. 1960, 45, 271–274. [Google Scholar] [CrossRef]
- Ueno, K.; Hayashi, K.; Okamura, S. Radiation induced polymerization of styrene in a dry system. J. Polym. Sci. Part B Polym. Lett. 1965, 3, 363–368. [Google Scholar] [CrossRef]
- Ueno, K.; Williams, F.; Hayashi, K.; Okamura, S. Radiation-induced polymerization by free ions.1. Polymerization of styrene under anhydrous conditions. Trans. Faraday Soc. 1967, 63, 1478–1488. [Google Scholar] [CrossRef]
- Stannett, V.; Meyer, J.A.; Szwarc, M.; Bahstetter, F.C. Radiation polymerization of isobutylene and styrene. Appl. Radiat. Isot. 1964, 15, 747–757. [Google Scholar] [CrossRef]
- Gotoh, T.; Yamamoto, M.; Nishijima, Y. Radiation-induced cationic dimerization and polymerization of styrene in methylene-chloride solution.1. J. Polym. Sci. Part A Polym. Chem. 1981, 19, 1047–1060. [Google Scholar] [CrossRef]
- Chapiro, A. Cationic polymerizations initiated by high-energy radiation. Makromol. Chem. 1974, 175, 1181–1197. [Google Scholar] [CrossRef]
- Chen, B.C.; Stamm, R.F. Polymerization induced by ionizing radiation at low temperatures. I. Evidence for the simultaneous existence of ionic and free-radical mechanisms in the polymerization of styrene and 2,4-dimethylstyrene. J. Polym. Sci. 1962, 58, 369–388. [Google Scholar] [CrossRef]
- Guarise, G.B.; Palma, G.; Siviero, E.; Talamini, G. Polymerization induced by gamma radiation of styrene under pressure. Polymer 1970, 11, 613–625. [Google Scholar] [CrossRef]
- Tabata, Y.; Sobue, H.; Oda, E. Radiation induced ionic polymerization of butadiene. J. Phys. Chem. 1961, 65, 1645–1647. [Google Scholar] [CrossRef]
- Ishigure, K.; Oneill, T.; Stahel, E.P.; Stannett, V. Radiation-induced polymerization and copolymerization of butadiene in emulsion. J. Macromol. Sci. Chem. 1974, 8, 353–372. [Google Scholar] [CrossRef]
- Radzinskii, S.A.; Sheinker, A.P.; Abkin, A.D. Radiation-induced polymerization of butadiene. Vysok. Soedin. Seriya A 1977, 19, 2066–2072. [Google Scholar]
- Verdurmen, E.M.; Verstegen, J.M.; German, A.L. Particle growth in butadiene emulsion polymerization—4. The promoting effect of mercaptans. Macromol. Chem. Phys. 1994, 195, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Challa, R.R.; Drew, J.H.; Stahel, E.P.; Stannett, V. Radiation-induced emulsion polymerization of vinyl-acetate in a pilot-plant reactor.2. Kinetics. J. Appl. Polym. Sci. 1986, 31, 27–38. [Google Scholar] [CrossRef]
- Verdurmen, E.M.; Geurts, J.M.; German, A.L. Particle growth in butadiene emulsion polymerization, 1. The use of Fremy salt as aqueous radical scavenger. Macromol. Chem. Phys. 1994, 195, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Verdurmen, E.M.; Geurts, J.M.; German, A.L. Particle growth in butadiene emulsion polymerization, 3. Radical adsorption and desorption rate coefficients. Macromol. Chem. Phys. 1994, 195, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Sundardi, F. Emulsion polymerization of vinyl-acetate initiated by intermittent gamma-radiation. J. Appl. Polym. Sci. 1979, 24, 1031–1038. [Google Scholar] [CrossRef]
- Xu, X.L.; Zhang, Z.C.; Zhang, M.W. Microemulsion polymerization of butyl acrylate initiated by gamma rays. J. Appl. Polym. Sci. 1996, 62, 1179–1183. [Google Scholar] [CrossRef]
- Yu, H.; Peng, J.; Zhai, M.; Li, J.; Wei, G.; Qiao, J. Radiation-induced copolymerization of styrene/n-butyl acrylate in the presence of ultra-fine powdered styrene-butadiene rubber. Radiat. Phys. Chem. 2007, 76, 1736–1740. [Google Scholar] [CrossRef]
- Okada, T.; Ishigaki, I.; Suwa, T.; Machi, S. Synthesis of cationic flocculant by radiation-induced co-polymerization of methyl-chloride salt of n,n-dimethylaminoethyl methacrylate with acrylamide in aqueous-solution. J. Appl. Polym. Sci. 1979, 24, 1713–1721. [Google Scholar] [CrossRef]
- Tabata, Y.; Ito, W.; Oshima, K. Radiation-induced polymerization of tetrafluoroethylene. J. Macromol. Sci. Part A Chem. 1970, 4, 789–799. [Google Scholar] [CrossRef]
- Suwa, T.; Watanabe, T.; Okamoto, J.; Machi, S. Emulsifier-free emulsion polymerization of tetrafluoroethylene by radiation.5. Effect of reaction conditions on the stability of polytetrafluoroethylene latex. J. Polym. Sci. Part A Polym. Chem. 1979, 17, 503–516. [Google Scholar] [CrossRef]
- Brown, D.W.; Lowry, R.E. Radiation-induced co-polymerization of tetrafluoroethylene and styrene at high-pressure. J. Polym. Sci. Part A Polym. Chem. 1979, 17, 759–768. [Google Scholar] [CrossRef]
- Lowry, R.E.; Brown, D.W.; Wall, L.A. Radiation-induced polymerization of hexafluoropropylene at high temperature and pressure. J. Polym. Sci. Part A Polym. Chem. 1966, 4, 2229–2240. [Google Scholar] [CrossRef]
- Matsuda, O.; Watanabe, T.; Tabata, Y.; Machi, S. Radiation-induced co-polymerization of methyl trifluoroacrylate with ethylene. J. Polym. Sci. Part A Polym. Chem. 1979, 17, 1789–1793. [Google Scholar] [CrossRef]
- Matsuda, O.; Watanabe, T.; Tabata, Y.; Machi, S. Radiation-induced co-polymerization of methyl trifluoroacrylate with propylene. J. Polym. Sci. Part A Polym. Chem. 1979, 17, 1795–1800. [Google Scholar] [CrossRef]
- Allcock, H.R. X-ray-induced polymerization of diphenylvinylphosphine oxide. J. Polym. Sci. Part A Polym. Chem. 1964, 2, 4087–4095. [Google Scholar] [CrossRef]
- Wiley, R.H.; Gensheimer, D.E. Gamma-radiation-induced polymerization of ethenesulfonamide. J. Polym. Sci. 1960, 42, 119–123. [Google Scholar] [CrossRef]
- Kumar, V.; Bhardwaj, Y.K.; Sabharwal, S.; Mohan, H. The role of radiolytically generated species in radiation-induced polymerization of vinylbenzyltrimethylammonium chloride (VBT) in aqueous solution: Steady-state and pulse radiolysis study. J. Radiat. Res. 2003, 44, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, R.S.; Ellis, R.J.; Wilkinson, S.A. The electron-beam curing of dialkyltin diacrylates. Polymer 1992, 33, 1836–1842. [Google Scholar] [CrossRef]
- Batten, R.J.; Davidson, R.S.; Ellis, R.J.; Wilkinson, S.A. New surface-coating materials—Electron-beam curing of some silicon-containing acrylates. Polymer 1992, 33, 3037–3043. [Google Scholar] [CrossRef]
- Tyagi, M.; Seshadri, G.; Sabharwal, S.; Niyogi, U.K.; Khandal, R.K. Studies on development of polymeric materials using gamma irradiation for contact and intra-ocular lenses. Int. J. Polym. Sci. 2009. [Google Scholar] [CrossRef]
- Lipscomb, N.T.; Purcell, T.O. Intensity dependence of gamma-radiation-induced polymerization of solid N-(1,1-dimethyl-3-oxobutyl)-acrylamide (diacetone acrylamide). J. Polym. Sci. Part B Polym. Lett. 1971, 9, 831–837. [Google Scholar] [CrossRef]
- Hardy, G.; Varga, J.; Nagy, G.; Cser, F.; Ero, J. Gamma-radiation-initiated polymerization of N-vinylsuccinimide in liquid and solid states. J. Polym. Sci. Part C Polym. Symp. 1967, 16, 2583–2596. [Google Scholar] [CrossRef]
- Hardy, G.; Nagy, G.; Varga, J.; Nemesheg, G. Investigations in field of radiation-induced solid-state polymerization.31. Eur. Polym. J. 1973, 9, 399–410. [Google Scholar] [CrossRef]
- Hayakawa, K.; Yamakita, H.; Kawase, K. Gamma-radiation-induced and ultraviolet-radiation-induced solid-state polymerization of maleimide in a few binary-systems. J. Polym. Sci. Part A Polym. Chem. 1972, 10, 1363–1375. [Google Scholar] [CrossRef]
- Allayarov, S.R.; Barkalov, I.M.; Kiryukhin, D.P. Radiation-induced polymerization of trifluorochloroethylene in perfluoroalkane glass. High Energy Chem. 1987, 21, 112–115. [Google Scholar]
- Allayarov, S.R.; Kiryukhin, D.P.; Barkalov, I.M. Radiation-induced polymerization of vinyl-chloride in glassy matrix of perfluoroalkanes. Vysok. Soedin. Seriya A 1983, 25, 1655–1659. [Google Scholar]
- Usanmaz, A.; Ozdemir, T.; Polat, O. Solid state polymerization of N-vinylcaprolactam via gamma irradiation and characterization. J. Macromol. Sci. Chem. 2009, 46, 597–606. [Google Scholar] [CrossRef]
- Wegner, G. Topochemical polymerization of monomers with conjugated triple bonds. Angew. Chem. Int. Ed. 1971, 10, 355–363. [Google Scholar]
- Tabata, Y.; Shibano, H.; Hara, K.; Sobue, H. Radiation-induced cationic polymerization of ethylene. J. Polym. Sci. Part A Polym. Chem. 1963, 1, 1049–1053. [Google Scholar] [CrossRef]
- Ueno, K.; Yamaoka, H.; Hayashi, K.; Okamura, S. Studies on radiation-induced ionic polymerization.2. Effect of solvent on polymerization of isobutene at low temperature. Appl. Radiat. Isot. 1966, 17, 595–602. [Google Scholar] [CrossRef]
- Marek, M.T.L.; Pecka, J. Process For Radiation Polymerization and Copolymerization of Monomers with Olefinic Double Bonds Using Halide Catalysts. U.S. Patent US3997417A, 14 December 1976. [Google Scholar]
- Cataldo, F.; Ragni, P.; Ursini, O. Radiation-induced polymerization of beta(-)pinene: A further insight. J. Radioanal. Nucl. Chem. 2007, 272, 29–36. [Google Scholar] [CrossRef]
- Goineau, A.M.; Kohler, J.; Stannett, V. Radiation-induced polymerization of a series of vinyl ethers. J. Macromol. Sci. Chem. 1977, 11, 99–114. [Google Scholar] [CrossRef]
- Parrish, C.F.; Harmer, D.E. Solid-state polymerization of 1,2,3,4-diepoxybutane initiated by cobalt-60 gamma-radiation. J. Polym. Sci. Part A Polym. Chem. 1967, 5, 1015–1020. [Google Scholar] [CrossRef]
- Nishii, M.; Hayashi, K.; Okamura, S. Radiation-induced polymerization of pentoxane in solid state. J. Polym. Sci. Part B Polym. Lett. 1969, 7, 891–895. [Google Scholar] [CrossRef]
- Ishigaki, I.; Ito, A.; Iwai, T.; Hayashi, K. Radiation-induced postpolymerization of trioxane in solid-state.5. Studies on copolymer composition. J. Polym. Sci. Part A Polym. Chem. 1972, 10, 1883–1893. [Google Scholar] [CrossRef]
- Naylor, D.M.; Stannett, V.T.; Deffieux, A.; Sigwalt, P. The radiation-induced polymerization of dimethylcyclosiloxanes in the liquid-state.3. Copolymerization of D3 with D4 and D4 with D5 reactivities and interpretation. Polymer 1994, 35, 1764–1768. [Google Scholar] [CrossRef]
- Crivello, J.V.; Fan, M.X.; Bi, D.S. The electron beam-induced cationic polymerization of epoxy-resins. J. Appl. Polym. Sci. 1992, 44, 9–16. [Google Scholar] [CrossRef]
- Crivello, J.V. Advanced curing technologies using photo- and electron beam induced cationic polymerization. Radiat. Phys. Chem. 2002, 63, 21–27. [Google Scholar] [CrossRef]
- Tsuji, K.; Yamaoka, H.; Hayashi, K.; Kamiyama, H.; Yoshida, H. ESR study on radiation-induced polymerization of nitroethylene. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 629–631. [Google Scholar] [CrossRef]
- Yamaoka, H.; Williams, F.; Hayashi, K. Radiation-induced polymerization of nitroethylene. Trans. Faraday Soc. 1967, 63, 376–381. [Google Scholar] [CrossRef]
- Ogasawara, M.; Arai, S.; Imamura, M. Radiation-induced polymerization of nitroethylene as studied by low-temperature pulse-radiolysis. Abstr. Pap. Am. Chem. Soc. 1979, 176. [Google Scholar] [CrossRef]
- DinhNgoc, B.; Schnabel, W. Primary reactions during free anionic-polymerization of beta-nitrostyrene. Z. Nat. A 1978, 33, 253–256. [Google Scholar] [CrossRef]
- Bai, R.K.; You, Y.Z.; Pan, C.Y. Co-60 gamma-irradiation-initiated “living” free-radical polymerization in the presence of dibenzyl trithiocarbonate. Macromol. Rapid Commun. 2001, 22, 315–319. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Pyun, J.; Matyjaszewski, K. Synthesis of nanocomposite organic/inorganic hybrid materials using controlled/”living” radical polymerization. Chem. Mater. 2001, 13, 3436–3448. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Dai, J.; Du, F.; Zheng, H.; Bai, R. Real-time and in situ investigation of “living”/controlled photopolymerization in the presence of a trithiocarbonate. Macromolecules 2013, 46, 2576–2582. [Google Scholar] [CrossRef]
- Barsbay, M.; Güven, O. A short review of radiation-induced raft-mediated graft copolymerization: A powerful combination for modifying the surface properties of polymers in a controlled manner. Radiat. Phys. Chem. 2009, 78, 1054–1059. [Google Scholar] [CrossRef]
- Nasef, M.M.; Gursel, S.A.; Karabelli, D.; Güven, O. Radiation-grafted materials for energy conversion and energy storage applications. Prog. Polym. Sci. 2016, 63, 1–41. [Google Scholar] [CrossRef]
- Bongiovanni, R.; Sangermano, M. UV-curing science and technology. In Encyclopedia of Polymer Science and Technology; Wiley Online Library: Hoboken, NJ, USA, 2014; pp. 1–20. [Google Scholar] [CrossRef]
- Wicks, Z.W.; Jones, F.N.; Pappas, S.P.; Wicks, D.A. Organic Coatings: Science and Technology, 4th ed.; John Wiley & Sons: New York, NY, USA, 2017; p. 512. [Google Scholar]
- Cortella, L.; Albino, C.; Tran, Q.-K.; Froment, K. 50 years of French experience in using gamma rays as a tool for cultural heritage remedial conservation. Radiat. Phys. Chem. 2020, 171, 8726. [Google Scholar] [CrossRef]
- Takács, E.; Wojnárovits, L. Pulse radiolysis studies on the polymerization of acrylates and methacrylates. Radiat. Phys. Chem. 1996, 47, 441–444. [Google Scholar] [CrossRef]
- Takács, E.; Wojnárovits, L. Kinetic study on the radiation-induced polymerization of HDDA and HDDMA. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1997, 131, 295–299. [Google Scholar] [CrossRef]
- Knolle, W.; Mehnert, R. On the mechanism of the electron-initiated curing of acrylates. Radiat. Phys. Chem. 1995, 46, 963–974. [Google Scholar] [CrossRef]
- Kozicki, M.; Kujawa, P.; Rosiak, J.M. Pulse radiolysis study of diacrylate macromonomer in aqueous solution. Radiat. Phys. Chem. 2002, 65, 133–139. [Google Scholar] [CrossRef]
- TakACS, E.; Dajka, K.; Wojnarovits, L.; Emmi, S.S. Protonation kinetics of acrylate radical anions. Phys. Chem. Chem. Phys. 2000, 2, 1431–1433. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Lalevée, J. Photoinitiators for Polymer Synthesis: Scope, Reactivity, and Efficiency; John Wiley & Sons: Weinheim, Germany, 2012. [Google Scholar]
- Emmi, S.S.; Corda, U.; Fuochi, P.; Lavalle, M.; Alessi, S.; Spadaro, G. Pulse radiolysis and theoretical investigation on the initial mechanism of the e-beam polymerization of epoxy resins. The results obtained on (phenoxymethyl)oxirane. Radiat. Phys. Chem. 2007, 76, 1251–1256. [Google Scholar] [CrossRef]
- Davidson, R.S.; Wilkinson, S.A. Electron-beam-induced polymerization of epoxides. J. Photochem. Photobiol. A 1991, 58, 123–134. [Google Scholar] [CrossRef]
- Schalek, R.L.; Defoort, B.; Drzal, L.T. A TEM investigation of the network structure of electron beam cured epoxy polymers. Microsc. Today 2018, 10, 24–25. [Google Scholar] [CrossRef] [Green Version]
- Defoort, B.; Defoort, D.; Coqueret, X. Electron-beam initiated polymerization of acrylate compositions, 2 - simulation of thermal effects in thin films. Macromol. Theory Simul. 2000, 9, 725–734. [Google Scholar] [CrossRef]
- Decker, C.; Moussa, K. Photopolymerization of multifunctional monomers.4. Acrylates with carbamate or oxazolidone structure. Eur. Polym. J. 1991, 27, 403–411. [Google Scholar] [CrossRef]
- Vandeberg, J.; Krongauz, V.V. Methods for curing optical fiber coatings and inks by low power electron beam radiation. U.S. Patent WO 98/41484, 24 September 1998. [Google Scholar]
- Taylor, D.H.; Cahil, V. Advances in Energy Cure Inkjet; Uv+Eb Technol: Boston, MA, USA, 2017; pp. 12–14. [Google Scholar]
- Du, Z.; Janke, C.J.; Li, J.; Wood, D.L., III. High-speed electron beam curing of thick electrode for high energy density li-ion batteries. Green Energy Environ. 2019, 4, 375–381. [Google Scholar] [CrossRef]
- Defoort, B.; Lopitaux, G.; Dupillier, J.M.; Larnac, G.; Coqueret, X. Electron-beam initiated polymerization of acrylate compositions, 6—Influence of processing parameters on the curing kinetics of an epoxy acrylate blend. Macromol. Chem. Phys. 2001, 202, 3149–3156. [Google Scholar] [CrossRef]
- Chuda, K.; Smolinski, W.; Defoort, B.; Rudz, W.; Gawdzik, B.; Rayss, J.; Coqueret, X. Effects of vitrification on the isothermal polymerization of acrylate blends under radiation. Polimery 2004, 49, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Al-Sheikhly, M.; McLaughlin, W. On the mechanisms of radiation-induced curing of epoxy-fiber composites. Radiat. Phys. Chem. 1996, 48, 201–206. [Google Scholar] [CrossRef]
- Krzeminski, M.; Molinari, M.; Troyon, M.; Coqueret, X. Characterization by atomic force microscopy of the nanoheterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules 2010, 43, 8121–8127. [Google Scholar] [CrossRef]
- McHugh, J.; Fideu, P.; Herrmann, A.; Stark, W. Determination and review of specific heat capacity measurements during isothermal cure of an epoxy using TM-DSC and standard DSC techniques. Polym. Test. 2010, 29, 59–765. [Google Scholar] [CrossRef]
- Coqueret, X.; Krzeminski, M.; Ponsaud, P.; Defoort, B. Recent advances in electron-beam curing of carbon fiber-reinforced composites. Radiat. Phys. Chem. 2009, 78, 557–561. [Google Scholar] [CrossRef]
- Defoort, B.; Coqueret, X.; Larnac, G.; Dupillier, J.M. Electron-beam curing of acrylate resins for composites: Modeling reaction kinetics. In Proceedings of the 45th International SAMPE Symposium and Exhibition, books 1 and 2. Long Beach, CA, USA, 21–25 May 2000; SAMPE International Business Office: Diamond Bar, CA, USA, 2000; Volume 45, pp. 2223–2234. [Google Scholar]
- Defoort, B.; Boursereau, F.; Dupillier, J.M.; Larnac, G.; Lopitaux, G.; Coqueret, X. Investigations to improve the properties of eb cured composites: A status report. In Affordable Materials Technology-Platform to Global Value and Performance; Books 1 and 2; In Proceedings of the 47th International SAMPE Symposium and Exhibition; SAMPE International Business Office: Diamond Bar, CA, USA, 2002; Volume 47, pp. 607–617. [Google Scholar]
- Martin, A.; Pietras-Ozga, D.; Ponsaud, P.; Kowandy, C.; Barczak, M.; Defoort, B.; Coqueret, X. Radiation-curing of acrylate composites including carbon fibres: A customized surface modification for improving. Mechanical performances. Radiat. Phys. Chem. 2014, 105, 63–68. [Google Scholar] [CrossRef]
- Manning, P.; Chapiro, A. Radiation chemistry of polymeric systems. In High Polymers; John Wiley & Sons: New York, NY, USA, London, UK, 1962; Volume XV, pp. 712. E7 18s. net. Endeavour 1963, 22, 100. [Google Scholar] [CrossRef]
- Nasef, M.M.; Güven, O. Radiation-grafted copolymers for separation and purification purposes: Status, challenges and future directions. Prog. Polym. Sci. 2012, 37, 1597–1656. [Google Scholar] [CrossRef]
- Hassan, M.I.U.; Taimur, S.; Yasin, T. Upcycling of polypropylene waste by surface modification using radiation-induced grafting. Appl. Surf. Sci. 2017, 422, 720–730. [Google Scholar] [CrossRef]
- Gao, Q.; Hua, J.; Li, R.; Xing, Z.; Pang, L.; Zhang, M.; Xu, L.; Wu, G. Radiation-induced graft polymerization for the preparation of highly efficient UHMWPE fibrous adsorbent for Cr(VI) removal. Rad. Phys. Chem. 2017, 130, 92–102. [Google Scholar] [CrossRef]
- Kavakli, C.; Barsbay, M.; Tilki, S.; Güven, O.; Kavakli, P.A. Activation of polyethylene/propylene non-woven fabric by radiation-induced grafting for the removal of Cr(VI) from aqueous solutions. Water Air Soil Pollut. 2016, 227, 473. [Google Scholar] [CrossRef]
- Kavakli, P.A.; Kavakli, C.; Seko, N.; Tamada, M.; Güven, O. Radiation induced emulsion graft polymerization of 4-vinylpyridine onto PE/PP non woven fabric for As(V) adsorption. Rad. Phys. Chem. 2016, 127, 13–20. [Google Scholar] [CrossRef]
- Gamma-radiation grafting on chitosan polymer substrate: a) Zhuang, S.; Yin, Y.; Wang, J. Removal of cobalt ions from aqueous solution using chitosan grafted with maleic acid by gamma radiation. Nucl. Eng. Techn. 2018, 50, 211–215. [Google Scholar] [CrossRef] b) Crini, G. Recent Developments in Polysaccharide-Based Materials Used as Adsorbents in Wastewater Treatment. Progr. Polym. Sci. 2005, 30, 38–70. [Google Scholar] [CrossRef] c) Sokker, H.H.; Abdel Ghaffar, A.M.; Gad, Y.H.; Aly, A.S. Synthesis and characterization of hydrogels based on grafted chitosan for the controlled drug release. Carbohydr. Polym. 2009, 75, 222–229. [Google Scholar] d) Casimiro, M.H.; Ferreira, L.M.; Leal, J.P.; Pereira, C.C.L.; Monteiro, B. Ionizing Radiation for Preparation and Functionalization of Membranes and Their Biomedical and Environmental Applications. Membranes 2019, 9, 163. [Google Scholar] [CrossRef] e) Cai, H.; Zhang, Z.P.; Sun, P.C.; He, B.L.; Zhu, X.X. Synthesis and characterization of thermo- and pH- sensitive hydrogels based on Chitosan-grafted N-isopropylacrylamide via γ-radiation. Radiat. Phys. Chem. 2005, 74, 26–30. [Google Scholar] [CrossRef]
- Saito, K.; Fujiwara, K.; Sugo, T. Commercial Products by Radiation-Induced Graft Polymerization. In Innovative Polymeric Adsorbents; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Chen, G.; Wang, Y.; Weng, H.; Wu, Z.; He, K.; Zhang, P.; Guo, Z.; Lin, M. Selective separation of Pd(II) on pyridine-functionalized graphene oxide prepared by radiation-induced simultaneous grafting polymerization and reduction. Acs App. Mater. Interf. 2019, 11, 24560–24570. [Google Scholar] [CrossRef]
- Ueki, Y.; Seko, N. Synthesis of fibrous metal adsorbent with a piperazinyl-dithiocarbamate group by radiation-induced grafting and its performance. ACS Omega 2020, 5, 2947–2959. [Google Scholar] [CrossRef]
- Shin, I.H.; Hong, S.; Lima, S.J.; Son, Y.-S.S.; Kim, T.-H. Surface modification of PVDF membrane by radiation-induced graft polymerization for novel membrane bioreactor. J. Indust. Eng. Chem. 2017, 46, 103–110. [Google Scholar] [CrossRef]
- Omichi, M.; Yamashita, S.; Okura, Y.; Ikutomo, R.; Ueki, Y.; Seko, N.; Kakuchi, R. Surface engineering of fluoropolymer films via the attachment of crown ether derivatives based on the combination of radiation-induced graft polymerization and the Kabacknik-fields reaction. Polymers 2019, 11, 1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fastow, E.; Cook, S.; Dean, P.; Ott, P.; Wilson, J.; Yoon, H.; Dietz, T.; Bateman, F.; Al-Sheikhly, M. Single-step synthesis of atmospheric CO2 sorbents through radiation-induced graft polymerization on commercial-grade fabrics. Rad. Res. 2019, 192, 219–230. [Google Scholar] [CrossRef]
- Ponce-Gonzalez, J.; Ouachan, I.; Varcoe, J.R.; Whelligan, D.K. Radiation-induced grafting of a butyl-spacer styrene monomer onto ETFE: The synthesis of the most alkali stable radiation-grafted anion-exchange membranes to date. J. Mater. Chem. A 2018, 6, 823–827. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, K.; Hiroki, A.; Yu, H.-C.; Zhao, Y.; Shishitani, H.; Yamaguchi, S.; Tanaka, H.; Maekawa, Y. Alkaline durable 2-methylimidazolium containing anion-conducting electrolyte membranes synthesized by radiation-induced grafting for direct hydrazine hydrate fuel cells. J. Memb. Sci. 2019, 574, 403–410. [Google Scholar] [CrossRef]
- Sawada, S.-I.; Hasegawa, S.; Zhao, Y.; Maekawa, Y. Block-type proton exchange membranes prepared by a combination of radiation-induced grafting and atom-transfer radical polymerization. J. Memb. Sci. 2017, 532, 105–114. [Google Scholar] [CrossRef]
- Sadeghi, S.; Sanli, L.I.; Güler, E.; Gürsel, S.A. Enhancing proton conductivity via sub-micron structures in proton conducting membranes originating from sulfonate PVDF powder by radiation-induced grafting. Solid State Ion. 2018, 314, 66–73. [Google Scholar] [CrossRef]
- Ren, Y.; Qin, Y.; Liu, X.; Hut, T.; Jiang, L.; Tian, T. Flame-retardant polyacrylonitrile fabric prepared by ultraviolet-induced grafting with glycidyl methacrylate followed by ammoniation and phosphorylation. J. Appl. Polym. Sci. 2018, 135, 46752. [Google Scholar] [CrossRef]
- Zheng, X.; Ding, X.; Guan, J.; Gu, Y.; Su, Z.; Zhao, Y.; Tu, Y.; Li, X.; Li, Y.; Li, J. Ionic liquid-grafted polyamide 6 by radiation-induced grafting: New strategy to prepare covalently bonded ion-containing polymers and their application as functional fibers. ACS App. Mater. Interfaces 2019, 11, 5462–5475. [Google Scholar] [CrossRef]
- Ma, J.; Peng, J.; Zhai, M. Chapter Seven—Radiation technology for applications in Renewable Energy technology. In Radiation Technology for Advanced Materials; from basic to modern applications; Academic Press: Waltham, MA, USA, 2019; pp. 207–247. [Google Scholar] [CrossRef]
- Chapiro, A. Chemical modifications in irradiated polymers. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1988, 32, 111–114. [Google Scholar] [CrossRef]
- Khelifa, F.; Erskov, S.; Habibi, Y.; Snyders, R.; Dubois, P. Free-radical-induced grafting from plasma polymer surfaces. Chem. Rev. 2016, 116, 3975–4005. [Google Scholar] [CrossRef]
- Kim, B.; Weaver, A.; Chumakov, M.; Pazos, I.M.; Poster, D.L.; Gaskell, K.; Han, D.H.; Scherer, G.; Yandrasits, M.A.; Lee, B.C.; et al. Mechanisms and characterization of the pulsed electron-induced grafting of styrene onto poly(tetrafluoroethylene-co-hexafluoropropylene) to prepare a polymer electrolyte membrane. Radiat. Res. 2018, 190, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Jain, R.; Anjum, N.; Singh, H. Preirradiation grafting of acrylonitrile onto polypropylene monofilament for biomedical applications: I. Influence of synthesis conditions. Rad. Phys. Chem. 2006, 75, 161–167. [Google Scholar] [CrossRef]
- Binh, D.; Huy, H.T. The effect of concentration of acrylic acid, dose rates and temperature on preirradiated graft of natural rubber-based thermoplastic elastomer. Rad. Phys. Chem. 1998, 53, 177–180. [Google Scholar] [CrossRef]
- Gupta, B.; Büchi, F.N.; Scherer, G.G. Cation exchange membranes by pre-irradiation grafting of styrene into FEP films. I. Influence of synthesis conditions. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1931–1938. [Google Scholar] [CrossRef]
- Ping, X.; Wang, M.; Ge, X. Radiation induced graft copolymerization of n-butyl acrylate onto poly(ethylene terephthalate) (PET) films and thermal properties of the obtained graft copolymer. Rad. Phys. Chem. 2011, 80, 632–637. [Google Scholar] [CrossRef]
- Makhlouf, C.; Marais, S.; Roudesli, S. Graft copolymerization of acrylic acid onto polyamide fibers. Appl. Surf. Sci. 2007, 253, 5521–5528. [Google Scholar] [CrossRef]
- Nasr, H.I.; Haggag, K.M.; El Kharadly, E.A. Polyamides with improved moisture regain via γ-rays. Rad. Phys. Chem. 1980, 16, 447–449. [Google Scholar] [CrossRef]
- Chumakov, M.K.; Shahamat, L.; Weaver, A.; LeBlanc, J.; Chaychian, M.; Silverman, J.; Benjamin Richter, K.; Weiss, D.; Al-Sheikhly, M. Electron beam induced grafting of N-isopropylacrylamide to a poly(ethylene-terephthalate) membrane for rapid cell sheet detachment. Rad. Phys. Chem. 2011, 80, 182–189. [Google Scholar] [CrossRef]
- Zhitariuk, N.I.; Shtanko, N.I. Influence of some factors on radiation grafting of styrene on poly(ethylene terephthalate) nuclear membranes. Eur. Polym. J. 1990, 26, 847–851. [Google Scholar] [CrossRef]
- Walo, M.; Przybytniak, G.; Kavaklı, P.A.; Güven, O. Radiation-induced graft polymerization of N-vinylpyrrolidone onto segmented polyurethane based on isophorone diisocyanate. Rad. Phys. Chem. 2013, 84, 85–90. [Google Scholar] [CrossRef]
- Alves, P.; Coelho, J.F.J.; Haack, J.; Rota, A.; Bruinink, A.; Gil, M.H. Surface modification and characterization of thermoplastic polyurethane. Eur. Polym. J. 2009, 45, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Gubler, L.; Gürsel, S.A.; Scherer, G.G. Radiation Grafted Membranes for Polymer Electrolyte Fuel Cells. Fuel Cells 2005, 5, 317–335. [Google Scholar] [CrossRef]
- Qiu, J.; Ni, J.; Zhai, M.; Peng, J.; Zhou, H.; Li, J.; Wei, G. Radiation grafting of styrene and maleic anhydride onto PTFE membranes and sequent sulfonation for applications of vanadium redox battery. Rad. Phys. Chem. 2007, 76, 1703–1707. [Google Scholar] [CrossRef]
- Nasef, M.M.; Sithambaranathan, P.; Ahmad, A.; Abouzari-lotf, E. Intensifying radiation induced grafting of 4-vinylpyridine/glycidyl methacrylate mixtures onto poly(ethylene-co-tetrafluoroethylene) films using ultrasound. Rad. Phys. Chem. 2017, 134, 56–61. [Google Scholar] [CrossRef]
- Estrada-Villegas, G.M.; Bucio, E. Comparative study of grafting a polyampholyte in a fluoropolymer membrane by gamma radiation in one or two-steps. Rad. Phys. Chem. 2013, 92, 61–65. [Google Scholar] [CrossRef]
- Ferreira, H.P.; Parra, D.F.; Lugao, A.B. Radiation-induced grafting of styrene into poly(vinylidene fluoride) film by simultaneous method with two different solvents. Rad. Phys. Chem. 2012, 81, 1341–1344. [Google Scholar] [CrossRef]
- Clochard, M.-C.; Bègue, J.; Lafon, A.; Caldemaison, D.; Bittencourt, C.; Pireaux, J.-J.; Betz, N. Tailoring bulk and surface grafting of poly(acrylic acid) in electron-irradiated PVDF. Polymer 2004, 45, 8683–8694. [Google Scholar] [CrossRef] [Green Version]
- Madrid, J.F.; Abad, L.V. Modification of microcrystalline cellulose by gamma radiation-induced grafting. Rad. Phys. Chem. 2015, 115, 143–147. [Google Scholar] [CrossRef]
- Wojnárovits, L.; Földváry, C.M.; Takács, E. Radiation-induced grafting of cellulose for adsorption of hazardous water pollutants: A review. Rad. Phys. Chem. 2010, 79, 848–862. [Google Scholar] [CrossRef]
- Przybytniak, G.; Kornacka, E.M.; Mirkowski, K.; Walo, M.; Zimek, Z. Functionalization of polymer surfaces by radiation-induced grafting. Nukleonika 2008, 53, 89–95, ISSN: 0029-5922. [Google Scholar]
- Clough, R. High-energy radiation and polymers: A review of commercial processes and emerging applications. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 2001, 185, 8–33. [Google Scholar] [CrossRef]
- Makuuchi, K.; Cheng, S. Radiation Processing of Polymer Materials and its Industrial Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 9781118162798. [Google Scholar]
- Lin, Z.; Xu, T.; Zhang, L. Radiation-induced grafting of N-isopropylacrylamide onto the brominated poly(2,6-dimethyl-1,4-phenylene oxide) membranes. Rad. Phys. Chem. 2006, 75, 532–540. [Google Scholar] [CrossRef]
- Phadnis, S.; Patri, M.; Hande, V.R.; Deb, P.C. Proton exchange membranes by grafting of styrene-acrylic acid onto FEP by preirradiation technique. I. Effect of synthesis conditions. J. Appl. Polym. Sci. 2003, 90, 2572–2577. [Google Scholar] [CrossRef]
- Ellinghorst, G.; Fuehrer, J.; Vierkotten, D. Radiation initiated grafting on fluoro polymers for membrane preparation. Rad. Phys. Chem. 1981, 18, 889–897. [Google Scholar] [CrossRef]
- Nasef, M.M.; Saidi, H.; Dessouki, A.M.; EI-Nesr, E.M. Radiation-induced grafting of styrene onto poly(tetrafluoroethylene) (PTFE) films. I. Effect of grafting conditions and properties of the grafted films. Polymer. Int. 2000, 49, 399–406. [Google Scholar] [CrossRef]
- Dargaville, T.R.; George, G.A.; Hill, D.J.T.; Whittaker, A.K. High energy radiation grafting of fluoropolymers. Prog. Polym. Sci. 2003, 28, 1355–1376. [Google Scholar] [CrossRef]
- Oshima, A.; Seguchi, T.; Tabata, Y. ESR study on free radicals trapped in crosslinked polytetrafluoroethylene(PTFE)—II radical formation and reactivity. Rad. Phys. Chem. 1999, 55, 61–71. [Google Scholar] [CrossRef]
- Hill, D.J.T.; Mohajerani, S.; Pomery, P.J.; Whittaker, A.K. An ESR study of the radiation chemistry of poly (tetrafluoroethylene-co-hexafluoropropylene) at 77 and 300 K. Rad. Phys. Chem. 2000, 59, 295–302. [Google Scholar] [CrossRef]
- Dargaville, T.R.; Hill, D.J.T.; Whittaker, A. An ESR study of irradiated poly(tetrafluoroethylene-co-perfluoropropyl vinyl ether) (PFA). Rad. Phys. Chem. 2001, 62, 25–31. [Google Scholar] [CrossRef]
- Seguchi, T.; Makuuchi, K.; Suwa, T.; Tamura, N.; Abe, T.; Takehisa, M. Electron Spin Resonance studies on irradiated poly (vinylidene fluoride). Nippon Kagaku Kaishi 1974, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Aymes-Chodur, C.; Esnouf, S.; Le Moël, A. ESR studies in γ-irradiated and PS-radiation-grafted poly(vinylidene fluoride). J. Polym. Sci. Part B Polym. Phys. 2001, 39, 1437–1448. [Google Scholar] [CrossRef]
- Adem, E.; Burillo, G.; Muñoz, E.; Rickards, J.; Cota, L.; Avalos-Borja, M. Electron and proton irradiation of poly(vinylidene fluoride): Characterization by electron paramagnetic resonance. Polym. Degrad. Stab. 2003, 81, 75–79. [Google Scholar] [CrossRef]
- Betz, N.; Petersohn, E.; Le Moël, A. Swift heavy ions effects in fluoropolymers: Radicals and crosslinking. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 1996, 116, 207–211. [Google Scholar] [CrossRef]
- Komaki, Y.; Ishikawa, N.; Morishita, N.; Takamura, S. Radicals in heavy ion-irradiated polyvinylidene fluoride. Rad. Meas. 1996, 26, 123–129. [Google Scholar] [CrossRef]
- Goslar, J.; Hilczer, B.; Smogór, H. Studies of fast electron irradiated ferroelectric poly(vinylidene fluoride). Acta Phys. Pol. A 2005, 108, 89–94. [Google Scholar] [CrossRef]
- Suryanarayana, D.; Kevan, L. Peroxy spin probe studies of motion in poly(vinylidene fluoride). J. Am. Chem. Soc. 1982, 104, 6251–6254. [Google Scholar] [CrossRef]
- Goslar, J.; Hilczer, B.; Smogór, H. Radiation-induced modification of P(VDF/TrFE) copolymers studied by ESR and vibrational spectroscopy. Appl. Magn. Reson. 2008, 34, 37–45. [Google Scholar] [CrossRef]
- Le Bouëdec, A.; Betz, N.; Esnouf, S.; Le Moël, A. Swift heavy ion irradiation effects in α poly(vinylidene fluoride): Spatial distribution of defects within the latent track. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 1999, 151, 89–96. [Google Scholar] [CrossRef]
- Aymes-Chodur, C.; Betz, N.; Porte-Durrieu, M.-C.; Baquey, C.; Le Moël, A. A FTIR and SEM study of PS radiation grafted fluoropolymers: Influence of the nature of the ionizing radiation on the film structure. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 1999, 151, 377–385. [Google Scholar] [CrossRef]
- Gebel, G.; Ottomani, E.; Allegraud, J.J.; Betz, N.; Le Moël, A. Structural study of polystyrene grafted in irradiated polyvinylidene fluoride thin films. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 1995, 105, 145–149. [Google Scholar] [CrossRef]
- Betz, N. Ion track grafting. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 1995, 105, 55–62. [Google Scholar] [CrossRef]
- Clochard, M.-C.; Berthelot, T.; Baudin, C.; Betz, N.; Balanzat, E.; Gébel, G.; Morin, A. Ion track grafting: A way of producing low-cost and highly proton conductive membranes for fuel cell applications. J. Power Sources 2010, 195, 223–231. [Google Scholar] [CrossRef]
- Yamaki, T.; Nuryanthi, N.; Kitamura, A.; Koshikawa, H.; Sawada, S.; Voss, K.-O.; Severin, D.; Trautmann, C. Fluoropolymer-based nanostructured membranes created by swift-heavy-ion irradiation and their energy and environmental applications. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 2018, 435, 162–168. [Google Scholar] [CrossRef]
- Melilli, G.; Lairez, D.; Gorse, D.; Garcia-Caurel, E.; Peinado, A.; Cavani, O.; Boizot, B.; Clochard, M.-C. Conservation of the piezoelectric response of PVDF films under irradiation. Rad. Phys. Chem. 2018, 142, 54–59. [Google Scholar] [CrossRef]
- Apel, P.Y. Fabrication of functional micro- and nanoporous materials from polymers modified by swift heavy ions. Rad. Phys. Chem. 2019, 159, 25–34. [Google Scholar] [CrossRef]
- Zdorovets, M.; Yerszhanov, A.; Korolkov, I.; Güven, O.; Dosmagambetova, S.S.; Shlimas, D.I.; Zhatkanbayeva, Z.K.; Zhidkov, I.S.; Kharkin, P.V.; Gluchshenko, V.N.; et al. Liquid low-level radioactive waste treatment by using hydrophobized track-etched membranes. Prog. Nucl. Energy 2020, 118, 103128. [Google Scholar] [CrossRef]
- Korolkov, I.; Yerzshanov, A.; Zodorevets, M.; Gorin, Y.G.; Güven, O.; Dosmagambetova, S.S.; Khlebnikov, N.A.; Serkov, K.V.; Krasnopyorova, M.V.; Milts, O.S.; et al. Modification of PET track-etched membranes for membrane distillation of low-level liquid radioactive waste and salt solutions. Sep. Pur. Technol. 2019, 227, 115694. [Google Scholar] [CrossRef]
- Korolkov, I.; Mashentseva, A.; Güven, O.; Gorin, Y.G.; Kozlovskiy, A.L.; Zdorovets, M.V.; Zhidkov, I.S.; Cholach, S.O. Electron/gamma radiation-induced synthesis and catalytic activity of gold nanoparticles supported on Track-etched PET membranes. Mat. Chem. Phys. 2018, 217, 31–39. [Google Scholar] [CrossRef]
- Mazzei, R.; Bermúdez, G.G.; Betz, N.; Cabanillas, E. Swift heavy ion induced graft polymerization in track etched membranes’submicroscopic pores. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 2004, 226, 575–584. [Google Scholar] [CrossRef]
- Cuscito, O.; Clochard, M.-C.; Esnouf, S.; Betz, N.; Lairez, D. Nanoporous β-PVDF membranes with selectively functionalized pores. Nucl. Instr. Meth. Phys. Res. Sect. B Beam Interact. Mater. At. 2007, 265, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Barsbay, M.; Güven, O.; Bessbousse, H.; Wade, T.L.; Beuneu, F.; Clochard, M.-C. Nanopore size tuning of polymeric membranes using the RAFT-mediated radical polymerization. J. Memb. Sci. 2013, 445, 135–145. [Google Scholar] [CrossRef]
- Bessbousse, H.; Nandhakumar, I.; Decker, M.; Barsbay, M.; Cuscito, O.; Lairez, D.; Clochard, M.-C.; Wade, T.L. Functionalized nanoporous track-etched β-PVDF membrane electrodes for lead(ii) determination by square wave anodic stripping voltammetry. Anal. Methods. 2011, 3, 1351–1359. [Google Scholar] [CrossRef]
- Pinaeva, U.; Lairez, D.; Oral, O.; Faber, A.; Clochard, M.-C.; Wade, T.L.; Moreau, P.; Ghestem, J.-P.; Vivier, M.; Ammor, S.; et al. Early warning sensors for monitoring mercury in water. J. Hazard. Mater. 2019, 376, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Pinaeva, U.; Dietz, T.C.; Al Sheikhly, M.; Balanzat, E.; Castellino, M.; Wade, T.L.; Clochard, M.C. Bis[2-(methacryloyloxy)ethyl] phosphate radiografted into track-etched PVDF for uranium (VI) determination by means of cathodic stripping voltammetry. React. Funct. Polym. 2019, 142, 77–86. [Google Scholar] [CrossRef]
- Langer, R. Drug delivery and targeting. Nature 1998, 392 (Suppl. 6679), 5–10. [Google Scholar]
- Funke, W.; Okay, O.; Joos-Müller, B. Microgels—Intramolecularly crosslinked macromolecules with a globular structure. Adv. Polym.Sci. 1998, 136, 139–234. [Google Scholar]
- Oh, J.K.; Drumright, R.; Siegwart, D.J.; Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar] [CrossRef]
- Dispenza, C.; Grimaldi, N.; Sabatino, M.A.; Soroka, I.L.; Jonsson, M. Radiation-Engineered Functional Nanoparticles in Aqueous Systems. J. Nanosci. Nanotechnol. 2015, 15, 3445–3467. [Google Scholar] [CrossRef]
- Dispenza, C.; Spadaro, G.; Jonsson, M. Radiation Engineering of Multifunctional Nanogels. Top.Curr. Chem. 2016, 374. [Google Scholar] [CrossRef]
- Sabatino, M.A.; Ditta, L.A.; Conigliaro, A.; Dispenza, C. A multifuctional nanoplatform for drug targeted delivery based on radiation-engineered nanogels. Radiat. Phys. Chem. 2020, 169, 108059. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as pharmaceutical carriers: Finite networks of infinite capabilities. Angew. Chem. Int. Ed. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, L.; Banik, B.; Alexis, F. Stimulus responsive nanogels for drug delivery. Soft Matter 2011, 7, 5908–5916. [Google Scholar] [CrossRef]
- Chacko, R.T.; Ventura, J.; Zhuang, J.; Thayumanavan, S. Polymer nanogels: A versatile nanoscopic drug delivery platform. Adv. Drug Deliv. Rev. 2012, 64, 836–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef]
- Karg, M.; Pich, A.; Hellweg, T.; Hoare, T.; Lyon, L.A.; Crassous, J.J.; Suzuki, D.; Gumerov, R.A.; Schneider, S.; Potemkin, I.I.; et al. Nanogels and microgels: From model colloids to applications, recent developments, and future trends. Langmuir 2019, 35, 6231–6255. [Google Scholar] [CrossRef]
- Pich, A.; Richtering, W. Polymer Nanogels and Microgels. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Moeller, M., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2012; Volume 6, pp. 309–350. ISBN 978-0-08-087862-1. [Google Scholar]
- Vashist, A.; Kaushik, A.K.; Ahmad, S.; Nair, M. (Eds.) Nanogels for Biomedical Applications; Royal Society of Chemistry: London, UK, 2018; ISBN 978-1782628620. [Google Scholar]
- Kröger, A.P.P.; Paulusse, J.M.J. Single-chain polymer nanoparticles in controlled drug delivery and targeted imaging. J. Control. Release 2018, 286, 326–347. [Google Scholar] [CrossRef]
- Ekkelenkamp, A.E.; Elzes, M.R.; Engbersen, J.F.J.; Paulusse, J.M.J. Responsive crosslinked polymer nanogels for imaging and therapeutics delivery. J. Mater. Chem. B 2018, 6, 210–235. [Google Scholar] [CrossRef]
- Graham, N.B.; Cameron, A. Nanogels and microgels: The new polymeric materials playground. Pure Appl. Chem. 1998, 70, 1271–1275. [Google Scholar] [CrossRef] [Green Version]
- Kadlubowski, S. Radiation-induced synthesis of nanogels based on poly(N-vinyl-2-pyrrolidone)—A review. Radiat. Phys. Chem. 2014, 102, 29–39. [Google Scholar] [CrossRef]
- Al-Assaf, S.; Phillips, G.O.; Deeble, D.J.; Parsons, B.; Starnes, H.; von Sonntag, C. The enhanced stability of the cross-linked hylan structure to the hydroxyl (OH) radicals compared with the uncross-linked Hyaluran. Radiat. Phys. Chem. 1995, 46, 207–217. [Google Scholar] [CrossRef]
- Ulanski, P.; Janik, I.; Kadlubowski, S.; Kozicki, M.; Kujawa, P.; Pietrzak, M.; Stasica, P.; Rosiak, J.M. Polymeric biomaterials synthesized by radiation techniques—Current studies at IARC, Poland. Polym. Adv. Technol. 2002, 13, 951–959. [Google Scholar] [CrossRef]
- Ulanski, P.; Rosiak, J.M. Polymeric Nano/Microgels. In Encyclopedia of Nanoscience and Nanotechnology; Nalwa, H.S., Ed.; American Scientific Publishers: Stevenson Ranch, CA, USA, 2004; pp. 845–871. [Google Scholar]
- Kadlubowski, S.; Grobelny, J.; Olejniczak, W.; Cichomski, M.; Ulanski, P. Pulses of fast electrons as a tool to synthesize poly(acrylic acid) nanogels. Intramolecular cross-linking of linear polymer chains in additive-free aqueous solution. Macromolecules 2003, 36, 2484–2492. [Google Scholar] [CrossRef]
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Mangione, M.R.; Walo, M.; Murugan, E.; Jonsson, M. On the origin of functionalization in one-pot radiation synthesis of nanogels from aqueous polymer solutions. RSC Adv. 2016, 6, 2582–2591. [Google Scholar] [CrossRef]
- Ditta, L.A.; Dahlgren, B.; Sabatino, M.A.; Dispenza, C.; Jonsson, M. The role of molecular oxygen in the formation of radiation-engineered multifunctional nanogels. Europ. Polym. J. 2019, 114, 164–175. [Google Scholar] [CrossRef]
- Staudinger, H.; Heuer, W.; Husemann, E.; Rabinovitch, I.J. The insoluble polystyrene. Trans. Faraday Soc. 1936, 32, 323–332. [Google Scholar] [CrossRef]
- Staudinger, H.; Huseman, E. Highly polymerized compounds. CXVI. The limiting swelling capability of polystyrene. Berichte Dtsch. Chem. Gesellschaft 1935, 68, 1618–1634. [Google Scholar] [CrossRef]
- Krzeminski, M.; Molinari, M.; Troyon, M.; Coqueret, X. Calorimetric characterization of the heterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules 2010, 43, 3757–3763. [Google Scholar] [CrossRef]
- Kowandy, C.; Ranoux, G.; Walo, M.; Vissouvanadin, B.; Teyssedre, G.; Laurent, C.; Berquand, A.; Molinari, M.; Coqueret, X. Microstructure aspects of radiation-cured networks: Cationically polymerized aromatic epoxy resins. Radiat. Phys. Chem. 2018, 143, 20–26. [Google Scholar] [CrossRef]
- Cao, Z.; Ziener, U. Synthesis of nanostructured materials in inverse miniemulsions and their applications. Nanoscale 2013, 5, 10093–10107. [Google Scholar] [CrossRef]
- Puig, J.E.; Rabelero, M. Semicontinuous microemulsion polymerization. Curr. Opin. Colloid Interface Sci. 2016, 25, 83–88. [Google Scholar] [CrossRef]
- Hayashi, K.; Kijima, T.; Okamura, S.; Egusa, S.; Makuuchi, K. Formation of fine particle emulsions by high-dose-rate polymerization. J. Polym. Sci. Polym. Lett. Ed. 1982, 20, 643–645. [Google Scholar] [CrossRef]
- Pusch, J.; Van Herk, A.M. Emulsion polymerization of novel transparent latices by pulsed electron beam initiation. Macromolecules 2005, 38, 6939–6945. [Google Scholar] [CrossRef]
- Landfester, K.; Eisenblaetter, J.; Rothe, R. Preparation of polymerizable miniemulsions by ultrasonication. JCT Res. 2004, 1, 65–68. [Google Scholar] [CrossRef]
- Teo, B.M.; Suh, S.K.; Hatton, T.A.; Ashokkumar, M.; Grieser, F. Sonochemical synthesis of magnetic Janus nanoparticles. Langmuir 2011, 27, 30–33. [Google Scholar] [CrossRef]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional polymers by atom transfer radical polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Radical addition–fragmentation chemistry in polymer synthesis. Polymer (Guildf) 2008, 49, 1079–1131. [Google Scholar] [CrossRef] [Green Version]
- Barsbay, M.; Güven, O.; Davis, T.P.; Barner-Kowollik, C.; Barner, L. RAFT-mediated polymerization and grafting of sodium 4-styrenesulfonate from cellulose initiated via γ-radiation. Polymer 2009, 50, 973–982. [Google Scholar] [CrossRef]
- Kiraç, F.; Güven, O. Gamma radiation induced synthesis of poly(N-isopropylacrylamide) mediated by Reversible Addition–Fragmentation Chain Transfer (RAFT) process. Radiat. Phys. Chem. 2015, 112, 76–82. [Google Scholar] [CrossRef]
- Barsbay, M.; Güven, O. Nanostructuring of polymers by controlling of ionizing radiation-induced free radical polymerization, copolymerization, grafting and crosslinking by RAFT mechanism. Radiat. Phys. Chem. 2020, 169, 107816. [Google Scholar] [CrossRef]
- Brasch, U.; Burchard, W. Preparation and solution properties of microhydrogels from poly(vinyl alcohol). Macromol. Chem. Phys. 1996, 197, 223–235. [Google Scholar] [CrossRef]
- Ulański, P.; Janik, I.; Rosiak, J.M. Radiation formation of polymeric nanogels. Radiat. Phys. Chem. 1998, 52, 289–294. [Google Scholar] [CrossRef]
- Matusiak, M.; Kadlubowski, S.; Rosiak, J.M. Nanogels synthesized by radiation-induced intramolecular crosslinking of water-soluble polymers. Radiat.Phys.Chem. 2018, 169, 108099. [Google Scholar] [CrossRef]
- Von Sonntag, C. The Chemical Basis of Radiation Biology; Taylor and Francis: London, UK, 1987. [Google Scholar]
- Spotheim-Maurizot, M.; Mostafavi, M.; Douki, T.; Belloni, J. (Eds.) Radiation Chemistry—From Basics to Applications in Material and Life Sciences; EDP Sciences: Les Ulis, France, 2008. [Google Scholar]
- The Radiation Chemistry of Polysaccharides; Al-Assaf, S.; Coqueret, X.; Zaman, K.; Sen, M.; Ulanski, P. (Eds.) International Atomic Energy Agency: Vienna, Austria, 2016. [Google Scholar]
- Abs, M.; Jongen, Y.; Poncelet, E.; Bol, J.-L. The IBA rhodotron TT1000: A very high power E-beam accelerator. Radiat. Phys. Chem. 2004, 71, 287–290. [Google Scholar] [CrossRef]
- Ziaie, F.; Tahami, S.M.R. Mass throughput rate calculation for X-ray facilities. Nukleonika 2005, 50, 121–124. [Google Scholar]
- Cleland, M.R.; Stichelbaut, F. Radiation processing with high-energy X-rays. Radiat. Phys. Chem. 2013, 84, 91–99. [Google Scholar] [CrossRef]
- Şen, M.; Toprak, D.; Güven, O. The effect of environmental humidity on radiation-induced degradation of carrageenans. Carbohydr. Polym. 2014, 114, 546–552. [Google Scholar] [CrossRef]
- Wach, R.A.; Mitomo, H.; Yoshii, F.; Kume, T. Hydrogel of radiation-induced cross-linked hydroxypropylcellulose. Macromol. Mater. Eng. 2002, 287, 285–295. [Google Scholar] [CrossRef]
- Al-Assaf, S.; Gulrez, S.K.H.; Czechowska-Biskup, R.; Wach, R.A.; Rosiak, J.M.; Ulanski, P. Radiation Modification of Polysaccharides. In The Radiation Chemistry of Polysaccharides; Al-Assaf, S., Coqueret, X., Zaman, K., Sen, M., Ulanski, P., Eds.; International Atomic Energy Agency: Vienna, Austria, 2006; pp. 77–115. [Google Scholar]
- Buxton, G.V. Basic radiation chemistry of liquid water. In The Study of First Processes and Transient Species by Electron; Baxendale, H.J., Busi, F., Eds.; Springer: Dordrecht, The Netherlands, 1982; pp. 241–266. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/.O-) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.B.; Mallard, W.G.; Helman, W.P.; Buxton, G.V.; Huie, R.E.; Neta, P. NDRL-NIST Solution Kinetics Database; Ver. 2; NIST Standard Reference Data: Gaithersburg, MD, USA, 1994. [Google Scholar]
- Von Sonntag, C. Peroxyl radicals in aqueous media. In Oxygen Radicals in Biology and Medicin; Simic, M.G., Taylor, K.A., Ward, J.F., von Sonntag, C., Eds.; Plenum: New York, NY, USA, 1988; pp. 47–54. [Google Scholar]
- Al-Sheikhly, M.; Simic, M.G. Oxygen uptake in characterization of autoxidation and antioxidation processes. In Free Radicals: Methodology and Concepts; Rice-Evans, C., Halliwell, B., Eds.; Richelieu Press: London, UK, 1988; pp. 481–497. [Google Scholar]
- Dahlgren, B.; Sabatino, M.A.; Dispenza, C.; Jonsson, M. Numerical simulations of nanogel synthesis using pulsed electron beam. Macromol. Theory Simul. 2020, 29, 1900046. [Google Scholar] [CrossRef]
- Ulanski, P.; Bothe, E.; Hildenbrand, K.; Rosiak, J.M.; von Sonntag, C. Hydroxyl-radical-induced reactions of poly(acrylic acid): A pulse radiolysis, EPR and product study. Part, I. Deoxygenated aqueous solution. J. Chem. Soc.Perkin Trans. 2 1996, 13–22. [Google Scholar] [CrossRef]
- von Sonntag, C.; Bothe, E.; Ulanski, P.; Adhikary, A. Radical transfer reactions in polymers. Radiat.Phys.Chem. 1999, 55, 599–603. [Google Scholar] [CrossRef]
- Jeszka, J.K.; Kadlubowski, S.; Ulanski, P. Monte Carlo simulations of nanogels formation by intramolecular recombination of radicals on polymer chain. Dispersive kinetics controlled by chain dynamics. Macromolecules 2006, 39, 857–870. [Google Scholar] [CrossRef]
- Ulanski, P.; Bothe, E.; Hildenbrand, K.; von Sonntag, C. Free-radical-induced chain breakage and depolymerization of poly(methacrylic acid): Equilibrium polymerization in aqueous solution at room temperature. Chem.A Eur. J. 2000, 6, 3922–3934. [Google Scholar] [CrossRef]
- Ulanski, P.; Bothe, E.; Rosiak, J.M.; Von Sonntag, C. Radiolysis of the poly(acrylic acid) model 2,4-dimethylglutaric acid: A pulse radiolysis and product study. J. Chem. Soc. Perkin Trans. 2 1996, 1, 5–12. [Google Scholar] [CrossRef]
- Ulanski, P.; Bothe, E.; Rosiak, J.M.; von Sonntag, C. OH-Radical-induced crosslinking and strand breakage of poly(vinyl alcohol) in aqueous solution in the absence and presence of oxygen. A pulse radiolysis and product study. Die Makromol. Chemie 1994, 195, 1443–1461. [Google Scholar] [CrossRef]
- Rosiak, J.M. Hydrogel dressings HDR. In Radiation Effects on Polymers; ACS Symposium Series, 475; Clough, R.C., Shalaby, S.W., Eds.; American Chemical Society: Washington, DC, USA, 1991; pp. 271–299. [Google Scholar]
- Rosiak, J.M. Radiation formation of hydrogels for drug delivery. J. Control. Release 1994, 31, 9–19. [Google Scholar] [CrossRef]
- Behar, D.; Rabani, J. Pulse radiolysis of poly(styrenesulfonate) in aqueous solutions. J.Phys.Chem. 1988, 92, 5288–5292. [Google Scholar] [CrossRef]
- Ulanski, P.; Bothe, E.; Hildenbrand, K.; von Sonntag, C.; Rosiak, J.M. The influence of repulsive electrostatic forces on the lifetimes of poly(acrylic acid) radicals in aqueous solution. Nukleonika 1997, 42, 425–436. [Google Scholar]
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Bulone, D.; Bondì, M.L.; Casaletto, M.P.; Rigogliuso, S.; Adamo, G.; Ghersi, G. Minimalism in radiation synthesis of biomedical functional nanogels. Biomacromolecules 2012, 13, 1805–1817. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, M.A.; Bulone, D.; Veres, M.; Spinella, A.; Spadaro, G.; Dispenza, C. Structure of e-beam sculptured poly(N-vinylpyrrolidone) networks across different length-scales, from macro to nano. Polymer (Guildf) 2013, 54, 54–64. [Google Scholar] [CrossRef]
- An, J.-C.; Weaver, A.; Kim, B.; Barkatt, A.; Poster, D.; Vreeland, W.N.; Silverman, J.; Al-Sheikhly, M. Radiation-induced synthesis of poly(vinylpyrrolidone) nanogel. Polymer (Guildf) 2011, 52, 5746–5755. [Google Scholar] [CrossRef]
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Spadaro, G.; Bulone, D.; Bondì, M.L.; Adamo, G.; Rigogliuso, S. Large-scale radiation manufacturing of hierarchically assembled nanogels. Chem. Eng. Trans. 2012, 27, 229–234. [Google Scholar] [CrossRef]
- Sütekin, S.D.; Güven, O. Application of radiation for the synthesis of poly(N-vinyl pyrrolidone) nanogels with controlled sizes from aqueous solutions. Appl. Radiat. Isot. 2019, 145, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Adamo, G.; Grimaldi, N.; Campora, S.; Bulone, D.; Bondì, M.L.; Al-Sheikhly, M.; Sabatino, M.A.; Dispenza, C.; Ghersi, G. Multi-functional nanogels for tumor targeting and redox-sensitive drug and siRNA delivery. Molecules 2016, 21, 1594. [Google Scholar] [CrossRef]
- Ulanski, P.; Rosiak, J.M.; Zainuddin, A. Pulse radiolysis of poly(ethylene oxide) in aqueous solution. II. Decay of macroradicals. Radiat.Phys.Chem. 1995, 46, 917–920. [Google Scholar] [CrossRef]
- Ulański, P.; Kadłubowski, S.; Jeszka, J.K. Nanogel formation by intrachain radiation-induced cross-linking. Simulation and experiment. Mater. Sci. Pol. 2006, 24, 467–476. [Google Scholar]
- Dispenza, C.; Sabatino, M.A.; Grimaldi, N.; Dahlgren, B.; Al-Sheikhly, M.; Wishart, J.F.; Tsinas, Z.; Poster, D.L.; Jonsson, M. On the nature of macroradicals formed upon radiolysis of aqueous poly(N-vinylpyrrolidone) solutions. Radiat. Phys. Chem. 2020, 174, 108900. [Google Scholar] [CrossRef]
- Dahlgren, B.; Dispenza, C.; Jonsson, M. Numerical simulation of the kinetics of radical decay in single-pulse high-energy electron-irradiated polymer aqueous solutions. J. Phys. Chem. A 2019, 123, 5043–5050. [Google Scholar] [CrossRef]
- Plonka, A. Developments in dispersive kinetics. Prog.Reaction Kinet. 1991, 16, 157–333. [Google Scholar]
- Plonka, A. Dispersive Kinetics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; ISBN 0-7923-7128-3. [Google Scholar]
- Polanowski, P.; Pakula, T. Studies of mobility, interdiffusion, and self-diffusion in two-component mixtures using the dynamic lattice liquid model. J. Chem. Phys. 2003, 118, 11139–11146. [Google Scholar] [CrossRef]
- Polanowski, P.; Pakula, T. Simulation of polymer-polymer interdiffusion using the dynamic lattice liquid model. J. Chem. Phys. 2004, 120, 6306–6311. [Google Scholar] [CrossRef] [PubMed]
- Polanowski, P.; Jeszka, J.K.; Li, W.; Matyjaszewski, K. Effect of dilution on branching and gelation in living copolymerization of monomer and divinyl cross-linker: Modeling using dynamic lattice liquid model (DLL) and Flory-Stockmayer (FS) model. Polymer 2011, 52, 5092–5101. [Google Scholar] [CrossRef]
- Kozanecki, M.; Halagan, K.; Saramak, J.; Matyjaszewski, K. Diffusive properties of solvent molecules in the neighborhood of a polymer chain as seen by Monte-Carlo simulations. Soft Matter 2016, 12, 5519–5528. [Google Scholar] [CrossRef] [Green Version]
- Kiełbik, R.; Hałagan, K.; Zatorski, W.; Jung, J.; Ulański, J.; Napieralski, A.; Rudnicki, K.; Amrozik, P.; Jabłoński, G.; Stożek, D.; et al. ARUZ—Large-scale, massively parallel FPGA-based analyzer of real complex systems. Comput. Phys. Commun. 2018, 232, 22–34. [Google Scholar] [CrossRef]
- Ulanski, P.; Rosiak, J.M. The use of radiation technique in the synthesis of polymeric nanogels. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1999, 151, 356–360. [Google Scholar] [CrossRef]
- Kadlubowski, S.; Ulanski, P.; Rosiak, J.M. Synthesis of tailored nanogels by means of two-stage irradiation. Polymer 2012, 53, 1985–1991. [Google Scholar] [CrossRef]
- Dispenza, C.; Grimaldi, N.; Sabatino, M.-A.; Todaro, S.; Bulone, D.; Giacomazza, D.; Przybytniak, G.; Alessi, S.; Spadaro, G. Studies of network organization and dynamics of e-beam crosslinked PVPs: From macro to nano. Radiat. Phys. Chem. 2012, 81, 1349–1353. [Google Scholar] [CrossRef] [Green Version]
- Dispenza, C.; Rigogliuso, S.; Grimaldi, N.; Sabatino, M.A.; Bulone, D.; Bondì, M.L.; Ghersi, G. Structure and biological evaluation of amino-functionalized PVP nanogels for fast cellular internalization. React. Funct. Polym. 2013, 73, 1103–1113. [Google Scholar] [CrossRef]
- Grimaldi, N.; Sabatino, M.A.; Przybytniak, G.; Kaluska, I.; Bondì, M.L.; Bulone, D.; Alessi, S.; Spadaro, G.; Dispenza, C. High-energy radiation processing, a smart approach to obtain PVP-graft-AA nanogels. Radiat. Phys. Chem. 2014, 94, 76–79. [Google Scholar] [CrossRef]
- Dispenza, C.; Grimaldi, N.; Sabatino, M.A.; Adamo, G.; Rigogliuso, S.; Ghersi, G. Large-scale manufacturing of radiation sculptured therapeutic nanogels. In Proceedings of the Technical 2013 NSTI Nanotechnology Conference and Expo, NSTI-Nanotech, Washington, DC, USA, 12–14 June 2013; Volume 3, pp. 202–205. [Google Scholar]
- Adamo, G.; Grimaldi, N.; Campora, S.; Sabatino, M.A.; Dispenza, C.; Ghersi, G. Glutathione-sensitive nanogels for drug release. Chem. Eng. Trans. 2014, 38, 457–462. [Google Scholar] [CrossRef]
- Dispenza, C.; Adamo, G.; Sabatino, M.A.; Grimaldi, N.; Bulone, D.; Bondì, M.L.; Rigogliuso, S.; Ghersi, G. Oligonucleotides-decorated-poly(N -vinyl pyrrolidone) nanogels for gene delivery. J. Appl. Polym. Sci. 2014, 131, 39774. [Google Scholar] [CrossRef]
- Matusiak, M.; Kadlubowski, S.; Ulanski, P. Radiation-induced synthesis of poly(acrylic acid) nanogels. Radiat. Phys. Chem. 2018, 142, 125–129. [Google Scholar] [CrossRef]
- Michaeli, I.; Katchalsky, A. Potentiometric titration of polyelectrolyte gels. J. Polym. Sci. 1957, 23, 683–696. [Google Scholar] [CrossRef]
- Katchalsky, A.; Gillis, J. Theory of the Potentiometric Titration of Polymeric Acids; D.B. Ceten Uitgevers: Amsterdam, The Netherlands, 1950; pp. 277–295. [Google Scholar]
- Oosawa, F. Polyelectrolytes; Marcel Dekker: New York, NY, USA, 1971. [Google Scholar]
- Schmidt, T.; Janik, I.; Kadlubowski, S.; Ulanski, P.; Rosiak, J.M.; Reichelt, R.; Arndt, K.-F. Pulsed electron beam irradiation of dilute aqueous poly(vinyl methyl ether) solutions. Polymer (Guildf) 2005, 46, 9908–9918. [Google Scholar] [CrossRef]
- Reichelt, R.; Schmidt, T.; Kuckling, D.; Arndt, K.F. Structural characterization of temperature-sensitive hydrogels by field emission scanning electron microscopy (FESEM). Macromol.Symp. 2004, 210, 501–511. [Google Scholar] [CrossRef]
- Picone, P.; Ditta, L.A.; Sabatino, M.A.; Militello, V.; San Biagio, P.L.; Di Giacinto, M.L.; Cristaldi, L.; Nuzzo, D.; Dispenza, C.; Giacomazza, D.; et al. Ionizing radiation-engineered nanogels as insulin nanocarriers for the development of a new strategy for the treatment of Alzheimer’s disease. Biomaterials 2016, 80, 179–194. [Google Scholar] [CrossRef]
- Dispenza, C.; Sabatino, M.A.; Ajovalasit, A.; Ditta, L.A.; Ragusa, M.; Purrello, M.; Costa, V.; Conigliaro, A.; Alessandro, R. Nanogel-antimiR-31 conjugates affect colon cancer cells behaviour. RSC Adv. 2017, 7, 52039–52047. [Google Scholar] [CrossRef] [Green Version]
- Khutoryanskiy, V.V.; Staikos, G. Hydrogen-Bonded Interpolymer Complexes; World Scientific: Singapore, 2009; ISBN 978-981-270-785-7. [Google Scholar]
- Henke, A.; Kadłubowski, S.; Wolszczak, M.; Ulański, P.; Boyko, V.; Schmidt, T.; Arndt, K.-F.; Rosiak, J.M. The structure and aggregation of hydrogen-bonded interpolymer complexes of poly(acrylic acid) with poly(N-vinylpyrrolidone) in dilute aqueous solution. Macromol. Chem. Phys. 2011, 212, 2529–2540. [Google Scholar] [CrossRef]
- Nakamura, K.; Murray, R.J.; Joseph, J.I.; Peppas, N.A.; Morishita, M.; Lowman, A.M. Oral insulin delivery using P(MAA-g-EG) hydrogels: Effects of network morphology on insulin delivery characteristics. J. Control. Release 2004, 95, 589–599. [Google Scholar] [CrossRef]
- Bromberg, L.; Temchenko, M.; Hatton, T.A. Dually responsive microgels from polyether-modified poly(acrylic acid): Swelling and drug loading. Langmuir 2002, 18, 4944–4952. [Google Scholar] [CrossRef]
- Bromberg, L. Intelligent hydrogels for the oral delivery of chemotherapeutics. Expert Opin. Drug Deliv. 2005, 2, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Henke, A.; Kadlubowski, S.; Ulanski, P.; Rosiak, J.M.; Arndt, K.-F. Radiation-induced cross-linking of polyvinylpyrrolidone-poly(acrylic acid) complexes. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2005, 236, 391–398. [Google Scholar] [CrossRef]
- Nurkeeva, Z.S.; Khutoryanskiy, V.V.; Mun, G.A.; Bitekenova, A.B.; Kadlubowski, S.; Zhilina, Y.A.; Ulanski, P.; Rosiak, J.M. Interpolymer complexes of poly(acrylic acid) nanogels with some non-ionic polymers in aqueous solutions. Colloid Surf.A 2004, 236, 141–146. [Google Scholar] [CrossRef]
- Henke, A.; Ulanski, P.; Rosiak, J.M. Radiation Cross-linked Hydrogen-Bonding Interpolymer Complexes. In Hydrogen-Bonded Interpolymer Complexes. Formation, Structure and Applications; Khutoryanskiy, V.V., Staikos, G., Eds.; World Scientific: Singapore, 2009; pp. 259–300. ISBN 978-981-270-785-7. [Google Scholar]
- Ghaffarlou, M.; Sütekin, S.D.; Güven, O. Preparation of nanogels by radiation-induced cross-linking of interpolymer complexes of poly (acrylic acid) with poly (vinyl pyrrolidone) in aqueous medium. Radiat. Phys. Chem. 2018, 142, 130–136. [Google Scholar] [CrossRef]
- Rattanawongwiboon, T.; Ghaffarlou, M.; Sütekin, S.D.; Pasanphan, W.; Güven, O. Preparation of multifunctional poly(acrylic acid)-poly(ethylene oxide) nanogels from their interpolymer complexes by radiation-induced intramolecular crosslinking. Colloid Polym. Sci. 2018, 296, 1599–1608. [Google Scholar] [CrossRef]
- El-Hag Ali, A.; Shawky, H.A.; Abd El Rehim, H.A.; Hegazy, E.A. Synthesis and characterization of PVP/AAc copolymer hydrogel and its applications in the removal of heavy metals from aqueous solution. Eur.Polym.J. 2003, 39, 2337–2344. [Google Scholar] [CrossRef]
- Abd El-Rehim, H.A.; Hegazy, E.A.; Khalil, F.H.; Hamed, N.A. Radiation preparation of drug carriers based polyacrylic acid (PAAc) using poly(vinyl pyrrolidone) (PVP) as a template polymer. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2007, 254, 105–112. [Google Scholar] [CrossRef]
- Abd El-Rehim, H.A.; Hegazy, E.S.A.; Hamed, A.A.; Swilem, A.E. Controlling the size and swellability of stimuli-responsive polyvinylpyrrolidone-poly(acrylic acid) nanogels synthesized by gamma radiation-induced template polymerization. Eur. Polym. J. 2013, 49, 601–612. [Google Scholar] [CrossRef]
- Abd El-Rehim, H.A.; Swilem, A.E.; Klingner, A.; Hegazy, E.S.A.; Hamed, A.A. Developing the potential ophthalmic applications of pilocarpine entrapped into polyvinylpyrrolidone-poly(acrylic acid) nanogel dispersions prepared by γ radiation. Biomacromolecules 2013, 14, 688–698. [Google Scholar] [CrossRef]
- Neamtu, I.; Rusu, A.G.; Diaconu, A.; Nita, L.E.; Chiriac, A.P. Basic concepts and recent advances in nanogels as carriers for medical applications. Drug Deliv. 2017, 24, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Rangaraj, N.; Laxmikeshav, K.; Sampathi, S. Nanogels as drug carriers—Introduction, chemical aspects, release mechanisms and potential applications. Int. J. Pharm. 2020, 581, 119268. [Google Scholar] [CrossRef] [PubMed]
- Manickam, P.; Pierre, M.; Jayant, R.D.; Nair, M.; Bhansali, S. Future of Nanogels for Sensing Applications in Nanogels for Biomedical Applications. In Nanogels for Biomedical Applications; Chapter 13; Vashist, A., Kaushik, A.K., Ahmad, S., Nair, M., Eds.; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Chiriac, A.P.; Ghilan, A.; Neamtu, I.; Nita, L.E.; Rusu, A.G.; Chiriac, V.M. Advancement in the biomedical applications of the (nano)gel structures based on particular polysaccharides. Macromol. Biosci. 2019, 19, 1900187. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Cui, X.; Caranasos, T.G.; Hensley, M.T.; Vandergriff, A.C.; Hartanto, Y.; Shen, D.; Zhang, H.; Zhang, J.; Cheng, K. Heart Repair Using Nanogel-Encapsulated Human Cardiac Stem Cells in Mice and Pigs with Myocardial Infarction. ACS Nano 2017, 11, 9738–9749. [Google Scholar] [CrossRef]
- Schmidt, S.; Zeiser, M.; Hellweg, T.; Duschl, C.; Fery, A.; Möhwald, H. Adhesion and Mechanical Properties of PNIPAM Microgel Films and Their Potential Use as Switchable Cell Culture Substrates. Adv. Funct. Mater. 2010, 20, 3235–3243. [Google Scholar] [CrossRef]
- Sanzari, I.; Buratti, E.; Huang, R.; Tusan, C.G.; Dinelli, F.; Evans, N.D.; Bertoldo, M. Poly(N-isopropylacrylamide) based thin microgel films for use in cell culture applications. Sci. Rep. 2020, 10, 6126. [Google Scholar] [CrossRef]
- Ferrer, M.C.C.; Dastgheyb, S.; Hickok, N.J.; Eckmann, D.M.; Composto, R.J. Designing nanogel carriers for antibacterial applications. Acta Biomater. 2014, 10, 2105–2111. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, C.A.; Correa, D.S.; Zucolotto, V. Polycaprolactone nanofiber mats decorated with photoresponsive nanogels and silver nanoparticles: Slow release for antibacterial control. Mater. Sci. Eng. C 2020, 107, 110334. [Google Scholar] [CrossRef]
- Zhu, J.; Li, F.; Wang, X. Hyaluronic acid and polyethylene glycol hybrid hydrogel encapsulating nanogel with hemostasis and sustainable antibacterial property for wound healing. ACS Appl. Mater. Interfaces 2018, 10, 13304–13316. [Google Scholar] [CrossRef]
- Lin, X.; Guan, X.; Wu, Y.; Zhuang, S.; Wu, Y.; Du, L.; Zhao, J.; Rong, J.; Zhao, J.; Tu, M. An alginate/poly(N-isopropylacrylamide)-based composite hydrogel dressing with stepwise delivery of drug and growth factor for wound repair. Mater. Sci. Eng. C 2020, 115, 111123. [Google Scholar] [CrossRef]
- Liu, Z.; Qiao, J.; Nagy, T.; Xiong, M.P. ROS-triggered degradable iron-chelating nanogels: Safely improving iron elimination in vivo. J Control. Release 2018, 283, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Gattu, S.; Crihfield, C.L.; Holland, L.A. Microscale measurements of Michaelis–Menten constants of neuraminidase with nanogel capillary electrophoresis for the determination of the sialic acid linkage. Anal. Chem 2017, 89, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Luo, F.; Peng, H.; Zhang, S.; Xie, R.; Ju, X.; Wang, W.; Faraj, Y.; Chu, L. A novel smart membrane with ion-recognizable nanogels as gates on interconnected pores for simple and rapid detection of trace lead(II) ions in water. J. Membr. Sci. 2019, 575, 28–37. [Google Scholar] [CrossRef]
- Shi, H.; Liu, Y.; Qu, R.; Li, Y.; Ma, R.; An, Y.; Shi, L. A facile one-pot method to prepare peroxidase-like nanogel artificial enzymes for highly efficient and controllable catalysis. Colloids Surf. B Biointerfaces 2019, 174, 352–359. [Google Scholar] [CrossRef]
- Molina, M.; Asadian-Birjand, M.; Balach, J.; Bergueiro, J.; Miceli, E.; Calderón, M. Stimuli-responsive nanogel composites and their application in nanomedicine. Chem. Soc. Rev. 2015, 44, 6161. [Google Scholar] [CrossRef] [Green Version]
- Eslami, P.; Rossi, F.; Fedeli, S. Hybrid Nanogels: Stealth and biocompatible structures for drug delivery applications. Pharmaceutics 2019, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Das, R.P.; Gandhi, V.V.; Singh, B.G.; Kunwar, A. Passive and active drug targeting: Role of nanocarriers in rational design of anticancer formulations. Curr. Pharm. Des. 2019, 25, 3034. [Google Scholar] [CrossRef]
- Swain, S.; Sahu, P.K.; Beg, S.; Babu, S.M. Nanoparticles for Cancer Targeting: Current and Future Directions. Curr. Drug Deliv. 2016, 13, 1290. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Lehner, R.; Wang, X.; Marsch, S.; Hunziker, P. Intelligent nanomaterials for medicine: Carrier platforms and targeting strategies in the context of clinical application. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 742–757. [Google Scholar] [CrossRef]
- Zilkowski, I.; Theodorou, I.; Albrecht, K.; Ducongé, F.; Groll, J. Subtle changes in network composition impact the biodistribution and tumor accumulation of nanogels. Chem. Commun. 2018, 54, 11777–11780. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Vashist, A.; Shah, P.; Tiwari, S.; Jayant, R.D.; Nair, M. Scale-up and current clinical trials for nanogels in therapeutics Chapter 14. In Nanogels for Biomedical Applications; Vashist, A., Kaushik, A.K., Ahmad, S., Nair, M., Eds.; Royal Society of Chemistry: London, UK, 2017. [Google Scholar] [CrossRef]
- Yoshii, F.; Zhao, L.; Wach, R.A.; Nagasawa, N.; Mitomo, H.; Kume, T. Hydrogels of polysaccharide derivatives crosslinked with irradiation at paste-like condition. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2003, 208, 320–324. [Google Scholar] [CrossRef]
- Al-Assaf, S. Polysaccharides: Origin, Source and Properties. In The Radiation Chemistry of Polysaccharides; Al-Assaf, S., Coqueret, X., Zaman, K., Sen, M., Ulanski, P., Eds.; International Atomic Energy Agency: Vienna, Austria, 2016. [Google Scholar]
- LaVerne, J.A.; Driscoll, M.S.; Al-Sheikhly, M. Radiation stability of lignocellulosic material components. Radiat. Phys. Chem. 2020, 171, 108716. [Google Scholar] [CrossRef]
- Linhardt, R.J. Polysaccharides I: Structure, Characterization and Use. Advances in Polymer Science, 186. J. Am. Chem. Soc. 2006, 128, 6268. [Google Scholar] [CrossRef]
- Klemm, D. Polysaccharides II; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Liu, J.; Willför, S.; Xu, C. A review of bioactive plant polysaccharides: Biological activities, functionalization, and biomedical applications. Bioact. Carbohydr. Diet. Fibre. 2015, 5, 31–61. [Google Scholar] [CrossRef]
- Ye, J.; Pei, Y. Degradation of carboxymethyl cellulose (CMC) in solid state by electron beams. He Jishu/Nucl. Tech. 2012, 35, 512–514. [Google Scholar]
- Stepanik, T.M.; Rajagopal, S.; Ewing, D.; Whitehouse, R. Electron-processing technology: A promising application for the viscose industry. Radiat. Phys. Chem. 1998, 52, 505–509. [Google Scholar] [CrossRef]
- Saeman, J.F.; Millett, M.A.; Lawton, E.J. Effect of high-energy cathode rays on cellulose. Ind. Eng. Chem. 1952, 44, 2848–2852. [Google Scholar] [CrossRef]
- El-Ashhab, F.; Sheha, L.; Abdalkhalek, M.; Khalaf, H.A. The influence of gamma irradiation on the intrinsic properties of cellulose acetate polymers. J. Assoc. Arab Univ. Basic Appl. Sci. 2013, 14, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Tissot, C.; Grdanovska, S.; Barkatt, A.; Silverman, J.; Al-Sheikhly, M. On the mechanisms of the radiation-induced degradation of cellulosic substances. Radiat. Phys. Chem. 2013, 84, 185–190. [Google Scholar] [CrossRef]
- Brenner, W.; Rugg, B.; Arnon, J.; Cleland, M.; Rogers, C. Radiation pretreatments for optimizing the sugar yield in the acid hydrolysis of waste cellulose. Radiat. Phys. Chem. 1979, 14, 299–308. [Google Scholar] [CrossRef]
- Postek, M.T.; Poster, D.L.; Vládar, A.E.; Driscoll, M.S.; LaVerne, J.A.; Tsinas, Z.; Al-Sheikhly, M.I. Ionizing radiation processing and its potential in advancing biorefining and nanocellulose composite materials manufacturing. Radiat. Phys. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Bergey, N.S.; Salamanca-Cardona, L.; Stipanovic, A.; Driscoll, M. Electron beam pretreatment of switchgrass to enhance enzymatic hydrolysis to produce sugars for biofuels. Carbohydr. Polym. 2014, 100, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, M.; Stipanovic, A.; Winter, W.; Cheng, K.; Manning, M.; Spiese, J.; Cleland, M.R. Electron beam irradiation of cellulose. Radiat. Phys. Chem. 2009, 78. [Google Scholar] [CrossRef]
- Driscoll, M.S.; Stipanovic, A.J.; Cheng, K.; Barber, V.A.; Manning, M.; Smith, J.L.; Sundar, S. Ionizing radiation and a wood-based biorefinery. Radiat. Phys. Chem. 2014, 94. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, J.I.; Kim, J.H.; Lee, K.W.; Chung, Y.J.; Shin, M.H.; Lee, J.W. Investigation on radiation degradation of carboxymethylcellulose by ionizing irradiation. Appl. Radiat. Isot. 2009, 67, 1513–1515. [Google Scholar] [CrossRef]
- Fei, B.; Wach, R.A.; Mitomo, H.; Yoshii, F.; Kume, T. Hydrogel of biodegradable cellulose derivatives. I. Radiation-induced crosslinking of CMC. J. Appl. Polym. Sci. 2000, 78, 278–283. [Google Scholar] [CrossRef]
- Wach, R.A.; Mitomo, H.; Nagasawa, N.; Yoshii, F. Radiation crosslinking of methylcellulose and hydroxyethylcellulose in concentrated aqueous solutions. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2003, 211, 533–544. [Google Scholar] [CrossRef]
- Wach, R.A.; Mitomo, H.; Yoshii, F.; Kume, T. Hydrogel of Biodegradable Cellulose Derivatives Radiation-Induced Crosslinking of HPC, Japan. 2002. Available online: http://jolisf.tokai-sc.jaea.go.jp/pdf/conf/JAERI-Conf-2002-003.pdf (accessed on 22 September 2020).
- Blouin, F.A.; Arthur, J.C. The effects of gamma radiation on cotton: Part I: Some of the properties of purified cotton irradiated in oxygen and nitrogen atmospheres. Text. Res. J. 1958, 28, 198–204. [Google Scholar] [CrossRef]
- Shin, S.; Sung, Y.-J. Improving enzymatic saccharification of hybrid poplar by electron beam irradiation pretreatment. J. Biobased Mater. Bioenergy 2010, 4, 23–26. [Google Scholar] [CrossRef]
- Takacs, E.; Wojnárovits, L.; Borsa, J.; Földváry, C.; Hargittai, P.; Zöld, O. Effect of γ-irradiation on cotton-cellulose. Radiat. Phys. Chem. 1999, 55, 663–666. [Google Scholar] [CrossRef]
- Gondhalekar, S.C.; Pawar, P.J.; Dhumal, S.S. Use of electron beam irradiation for improving reactivity of dissolving pulp in viscose process. J. Radioanal. Nucl. Chem. 2019, 322, 67–72. [Google Scholar] [CrossRef]
- Ulański, P.; Rosiak, J. Preliminary studies on radiation-induced changes in chitosan. Int. J. Radiat. Appl. Instrum. Part C. Radiat. Phys. Chem. 1992, 39, 53–57. [Google Scholar] [CrossRef]
- Baroudi, A.; García-Payo, C.; Khayet, M. Structural, mechanical, and transport properties of electron beam-irradiated chitosan membranes at different doses. Polymers 2018, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Czechowska-Biskup, R.; Rokita, B.; Ulanski, P.; Rosiak, J.M. Radiation-induced and sonochemical degradation of chitosan as a way to increase its fat-binding capacity. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2005, 236, 383–390. [Google Scholar] [CrossRef]
- Rahman, M.; Rashid, T. Impact of high energy irradiation on chitin and chitosan: A short review. Crustac. Struct. Ecol. Life Cycle 2013, 37–70. [Google Scholar]
- Chmielewski, A.G.; Migdal, W.; Swietoslawski, J.; Swietoslawski, J.; Jakubaszek, U.; Tarnowski, T. Chemical-radiation degradation of natural oligoamino-polysaccharides for agricultural application. Radiat. Phys. Chem. 2007, 76, 1840–1842. [Google Scholar] [CrossRef]
- Chmielewski, A.G. Chitosan and radiation chemistry. Radiat. Phys. Chem. 2010, 79, 272–275. [Google Scholar] [CrossRef]
- Vasiliev, M.; Vasilieva, T.; Hein, A.M. Hybrid plasma chemical reactors for bio-polymers processing. J. Phys. D. Appl. Phys. 2019, 52, 335202. [Google Scholar] [CrossRef]
- Abagian, G.V.; Krutova, I.N.; Putilova, I.N.; Butiagin, P.I. Study of free radicals in gamma-irradiated starch by the electron paramagnetic resonance technic. Biofizika 1967, 12, 820–823. [Google Scholar] [PubMed]
- Braşoveanu, M.; Nemţanu, M.R. Pasting properties modeling and comparative analysis of starch exposed to ionizing radiation. Radiat. Phys. Chem. 2020, 168, 108492. [Google Scholar] [CrossRef]
- Fernando, I.P.S.; Kim, D.; Nah, J.-W.; Jeon, Y.-J. Advances in functionalizing fucoidans and alginates (bio)polymers by structural modifications: A. review. Chem. Eng. J. 2019, 355, 33–48. [Google Scholar] [CrossRef]
- Nagasawa, N.; Mitomo, H.; Yoshii, F.; Kume, T. Radiation-induced degradation of sodium alginate. Polym. Degrad. Stab. 2000, 69, 279–285. [Google Scholar] [CrossRef]
- Munarin, F.; Bozzini, S.; Visai, L.; Tanzi, M.C.; Petrini, P. Sterilization treatments on polysaccharides: Effects and side effects on pectin. Food Hydrocoll. 2013, 31, 74–84. [Google Scholar] [CrossRef]
- Purwanto, Z.I.; Broek, L.; Schols, H.A.; Pilnik, W.; Voragen, A. Degradation of low molecular weight fragments of pectin and alginates by gamma-irradiation. Acta Aliment. 1998, 27, 29–42. [Google Scholar]
- Bachman, S.; Zegota, A.; Zegota, H. Radiation Sterilization of Dextran Irradiated in Dry State. Probl. Tech. W Med. 1973, 331–337. [Google Scholar]
- Flynn, J.H.; Wall, L.A.; Morrow, W.L. Irradiation of Dextran and its Aqueous Solutions with Cobalt-60 Gamma Rays. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1967, 71, 25–31. [Google Scholar] [CrossRef]
- Korotchenko, K.; Sharpatyĭ, V. The role of adsorbed water in polysaccharide radiation destruction. Radiats. Biol. Radioecol. 2000, 40, 133–137. [Google Scholar]
- Dzaugis, M.E.; Spivack, A.J.; D’Hondt, S. A quantitative model of water radiolysis and chemical production rates near radionuclide-containing solids. Radiat. Phys. Chem. 2015, 115, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Spinks, J.W.T.; Woods, R.J. An Introduction to Radiation Chemistry; John Wiley and Sons Inc: Hoboken, NJ, USA, 1990. [Google Scholar]
- Fekete, T.; Borsa, J.; Takács, E.; Wojnárovits, L. Synthesis of carboxymethylcellulose/starch superabsorbent hydrogels by gamma-irradiation. Chem. Cent. J. 2017, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozalewska, W.; Czechowska-Biskup, R.; Olejnik, A.K.; Wach, R.A.; Ulański, P.; Rosiak, J.M. Chitosan-containing hydrogel wound dressings prepared by radiation technique. Radiat. Phys. Chem. 2017, 134, 1–7. [Google Scholar] [CrossRef]
- Fan, L.; Yang, H.; Yang, J.; Peng, M.; Hu, J. Preparation and characterization of chitosan/gelatin/PVA hydrogel for wound dressings. Carbohydr. Polym. 2016, 146, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Uddin, I.; Islam, J.M.M.; Haque, A.; Zubair, A.; Barua, R.; Rahaman, S.; Rahman, L.; Khan, M.A. Significant influence of gamma-radiation-treated chitosan and Alginate on increased productivity as well as improved taste and flavor of pineapple. Int. J. Fruit Sci. 2020, 1–15. [Google Scholar] [CrossRef]
- Ahmad, B.; Jahan, A.; Sadiq, Y.; Shabbir, A.; Jaleel, H.; Khan, M.M.A. Radiation-mediated molecular weight reduction and structural modification in carrageenan potentiates improved photosynthesis and secondary metabolism in peppermint (Mentha piperita L.). Int. J. Biol. Macromol. 2019, 124, 1069–1079. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Xu, L.; Wei, S.; Zhai, M. Radiation cross-linked collagen/dextran dermal scaffolds: Effects of dextran on cross-linking and degradation. J. Biomater. Sci. Polym. Ed. 2015, 26, 162–180. [Google Scholar] [CrossRef]
- Zegota, H. Some quantitative aspects of hydroxyl radical induced reactions in γ-irradiated aqueous solutions of pectins. Food Hydrocoll. 2002, 16, 353–361. [Google Scholar] [CrossRef]
- Tenchurin, T.; Pavlovsky, M.; Shepelev, A.; Mamagulashvilli, V.; Gomzyak, V.; Sedush, N.; Krasheninnikov, S.; Puchkov, A.; Malakhov, S.; Sharikov, R.; et al. Modification of non-woven materials based on sodium alginate for tissue-engineering. J. Phys. Conf. Ser. 2019, 1347, 12072. [Google Scholar] [CrossRef]
- Kovalev, G.; Bugaenko, L. On the crosslinking of cellulose under exposure to radiation. High Energy Chem. 2003, 37, 209–215. [Google Scholar] [CrossRef]
- Yang, J.; Medronho, B.; Lindman, B.; Norgren, M. Simple one pot preparation of chemical hydrogels from cellulose dissolved in cold LiOH/urea. Polymers 2020, 12, 373. [Google Scholar] [CrossRef] [Green Version]
- Abou Taleb, M.F.; Alkahtani, A.; Mohamed, S.K. Radiation synthesis and characterization of sodium alginate/chitosan/hydroxyapatite nanocomposite hydrogels: A drug delivery system for liver cancer. Polym. Bull. 2015, 72, 725–742. [Google Scholar] [CrossRef]
- Katayama, T.; Nakauma, M.; Todoriki, S.; Phillips, G.; Tada, M. Radiation-induced polymerization of gum arabic (Acacia senegal) in aqueous solution. Food Hydrocoll. 2006, 983–989. [Google Scholar] [CrossRef]
- Al-Assaf, S.; Phillips, G.O.; Williams, P.A.; du Plessis, T.A. Application of ionizing radiations to produce new polysaccharides and proteins with enhanced functionality. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2007, 265, 37–43. [Google Scholar] [CrossRef]
- Al-Assaf, S.; Phillips, G.O.; Williams, P.A. Controlling the molecular structure of food hydrocolloids. Food Hydrocoll. 2006, 20, 369–377. [Google Scholar] [CrossRef]
- Hayrabolulu, H.; Şen, M.; Çelik, G.; Kavaklı, P.A. Synthesis of carboxylated locust bean gum hydrogels by ionizing radiation. Radiat. Phys. Chem. 2014, 94, 240–244. [Google Scholar] [CrossRef]
- Gulrez, S.; Al-Assaf, S. Phillips, G. Hydrogels: Methods of Preparation, Characterisation and Applications. In Progress in Molecular and Environmental Bioengineering; IntechOpen: Rijeka, Croatia, 2011. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Nagai, S. Studies on the radiation-induced coloration mechanism of the cellulose triacetate film dosimeter. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1991, 42, 1215–1221. [Google Scholar] [CrossRef]
- Blouin, F.A.; Ott, V.J.; Mares, T.; Arthur, J.C. The effects of gamma radiation on the chemical properties of methyl cellulose. Text. Res. J. 1964, 34, 153–158. [Google Scholar] [CrossRef]
- Hegazy, E.-S.; Abdel-Rehim, H.; Diaa, D.A.; Elbarbary, A. Controlling of degradation effects in radiation processing of polymers. In Controlling of Degradation Effects in Radiation Processing of Polymers; International Atomic Energy Agency: Vienna, Austria, 2009; pp. 67–84. [Google Scholar]
- Drábková, K.; Ďurovič, M.; Kučerová, I. Influence of gamma radiation on properties of paper and textile fibres during disinfection. Radiat. Phys. Chem. 2018, 152, 75–80. [Google Scholar] [CrossRef]
- Rosiak, J.; Ulański, P.; Kucharska, M.; Dutkiewicz, J.; Judkiewicz, L. Radiation sterilization of chitosan sealant for vascular prostheses. J. Radioanal. Nucl. Chem. 1992, 159, 87–96. [Google Scholar] [CrossRef]
- Piroonpan, T.; Katemake, P.; Panritdam, E.; Pasanphan, W. Alternative chitosan-based EPR dosimeter applicable for a relatively wide range of gamma radiation doses. Radiat. Phys. Chem. 2017, 141, 57–65. [Google Scholar] [CrossRef]
- Pearl, I.W. The Chemistry of Lignin; Marcel Dekker: New York, NY, USA, 1967. [Google Scholar]
- Lanzalunga, O.; Bietti, M. Photo- and radiation chemical induced degradation of lignin model compounds. J. Photochem. Photobiol. B Biol. 2000, 56, 85–108. [Google Scholar] [CrossRef]
- Skvortsov, S.V. Radiation degradation of lignin. Chem. Nat. Compd. 1990, 26, 1–9. [Google Scholar] [CrossRef]
- Ponomarev, A.V.; Ershov, B.G. Radiation-thermal decomposition of lignin: Products and the mechanism of their formation (Review). High Energy Chem. 2018, 52, 58–70. [Google Scholar] [CrossRef]
- Rajeswara Rao, N.; Venkatappa Rao, T.; Ramana Reddy, S.V.S.; Sanjeeva Rao, B. The effect of gamma irradiation on physical, thermal and antioxidant properties of kraft lignin. J. Radiat. Res. Appl. Sci. 2015, 8, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Chen, L.; Chen, J.; Su, X.; Liu, Y.; Wang, K.; Qin, W.; Qi, H.; Deng, M. The effect of 60Co γ-irradiation on the structure and thermostability of alkaline lignin and its irradiation derived degradation products. Waste Biomass Valorization. 2019, 10, 3025–3035. [Google Scholar] [CrossRef]
- Glazer, A.W.; Nikaido, H. Microbial Biotechnology: Fundamentals of Applied Microbiology; W.H. Freeman: San Francisco, CA, USA, 1995. [Google Scholar]
- Newton, E.B. Method of Vulcanizing Rubber. U.S. Patent US1906402A, 2 May 1933. [Google Scholar]
- Haque, M.D.E.; Makuuchi, K.; Mitomo, H.; Yoshii, F.; Ikeda, K. A new trend in radiation vulcanization of natural rubber latex with a low energy electron beam. Polym. J. 2005, 37, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.D.; Alliger, G. Rubber—Application of radiation to tire manufacture. Radiat. Phys. Chem. 1979, 14, 39–53. [Google Scholar] [CrossRef]
- Chirinos, H.; Yoshii, F.; Makuuchi, K.; Lugao, A. Radiation vulcanization of natural rubber latex using 250 keV electron beam machine. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At 2003, 208, 256–259. [Google Scholar] [CrossRef]
- Makuuchi, K.; Yoshii, F.; Gunewardena, J.A.G.S.G. Radiation vulcanization of NR latex with low energy electron beams. Radiat. Phys. Chem. 1995, 46, 979–982. [Google Scholar] [CrossRef]
- Moustafa, A.B.; Mounir, R.; El Miligy, A.A.; Mohamed, M.A. Effect of gamma irradiation on the properties of natural rubber/styrene butadiene rubber blends. Arab. J. Chem. 2016, 9, S124–S129. [Google Scholar] [CrossRef] [Green Version]
- Essawy, H.; El-Nashar, D. The use of montmorillonite as a reinforcing and compatibilizing filler for NBR/SBR rubber blend. Polym. Test. 2004, 23, 803–807. [Google Scholar] [CrossRef]
- Kim, H.J.; Hamed, G.R. On the reason that passenger tire sidewalls are based on blends of natural rubber and cis-polybutadiene. Rubber Chem. Technol. 2000, 73, 743–752. [Google Scholar] [CrossRef]
- Stephen, R.; Jose, S.; Joseph, K.; Thomas, S.; Oommen, Z. Thermal stability and ageing properties of sulphur and gamma radiation vulcanized natural rubber (NR) and carboxylated styrene butadiene rubber (XSBR) latices and their blends. Polym. Degrad. Stab. 2006, 91, 1717–1725. [Google Scholar] [CrossRef]
- Aoshuang, Y.; Zhengtao, G.; Li, L.; Ying, Z.; Peng, Z. The mechanical properties of radiation-vulcanized NR/BR blending system. Radiat. Phys. Chem. 2002, 63, 497–500. [Google Scholar] [CrossRef]
- Manaila, E.; Craciun, G.; Stelescu, M.-D.; Ighigeanu, D.; Ficai, M. Radiation vulcanization of natural rubber with polyfunctional monomers. Polym. Bull. 2014, 71, 57–82. [Google Scholar] [CrossRef]
- Bhowmick, A.K.; Vijayabaskar, V. Electron beam curing of elastomers. Rubber Chem. Technol. 2006, 79, 402–428. [Google Scholar] [CrossRef]
- Henning, S. The Use of Coagents in the Radical Cure of Elastomers. In Proceedings of the 56th IWCS Conf.—Proc. Int. Wire Cable Symp. Inc., 11 November 2007; IWCS: Annandale, VA, USA, 2007. [Google Scholar]
- Haines, J.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Kempner, E.S. Effects of high-energy electrons and gamma rays directly on protein molecules. J. Pharm. Sci. 2001, 90, 1637–1646. [Google Scholar] [CrossRef]
- Wang, W.; Yu, Z.; Su, W. Ion irradiation induced direct damage to proteins and their components. arXiv 2008, arXiv:0807.0084. [Google Scholar]
- Garrison, W.M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chem. Rev. 1987, 87, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Chapelier, A.; Desmadril, M.; Houée-Levin, C. Gamma radiation effects on α-lactalbumin: Structural modifications. Can. J. Physiol. Pharmacol. 2001, 79, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Weik, M.; Bergès, J.; Raves, M.; Gros, P.; McSweeney, S.; Silman, I.; Sussman, J.; Houée-Levin, C.; Ravelli, R. Evidence for the formation of disulfide radicals in protein crystals upon X-ray irradiation. J. Synchrotron Radiat. 2002, 9, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Weik, M.; Ravelli, R.; Kryger, G.; McSweeney, S.; Raves, M.; Harel, M.; Gros, P.; Silman, I.; Kroon, J.; Sussman, J. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biaglow, J.E.; Varnes, M.E.; Epp, E.R.; Clark, E.P.; Tuttle, S.W.; Held, K.D. Role of glutathione in the aerobic radiation response. Int. J. Radiat. Oncol. 1989, 16, 1311–1314. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Saha, A.; Mandal, P.C. Radiation-induced inactivation of flavocytochrome b2 in dilute aqueous solution. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 1995, 1250, 1–8. [Google Scholar] [CrossRef]
- Durchschlag, H.; Fochler, C.; Feser, B.; Hausmann, S.; Seroneit, T.; Swientek, M.; Swoboda, E.; Winklmair, A.; Wlček, C.; Zipper, P. Effects of X- and UV-irradiation on proteins. Radiat. Phys. Chem. 1996, 47, 501–505. [Google Scholar] [CrossRef]
- Durchschlag, H.; Feser, B.; Fochler, C.; Seroneit, T.; Swoboda, E.; Wlček, C.; Zipper, P. Radiation damage and modification of radiation action of X-rays and UV-light on enzymes. In Trends in Colloid and Interface Science VII; Laggner, P., Glatter, O., Eds.; Steinkopff: Darmstadt, Germany, 1992; pp. 223–224. [Google Scholar]
- Davies, K.J. Protein damage and degradation by oxygen radicals. I. general aspects. J. Biol. Chem. 1987, 262, 9895–9901. [Google Scholar]
- Barata-Vallejo, S.; Ferreri, C.; Postigo, A.; Chatgilialoglu, C. Radiation chemical studies of methionine in aqueous solution: Understanding the role of molecular oxygen. Chem. Res. Toxicol. 2010, 23, 258–263. [Google Scholar] [CrossRef]









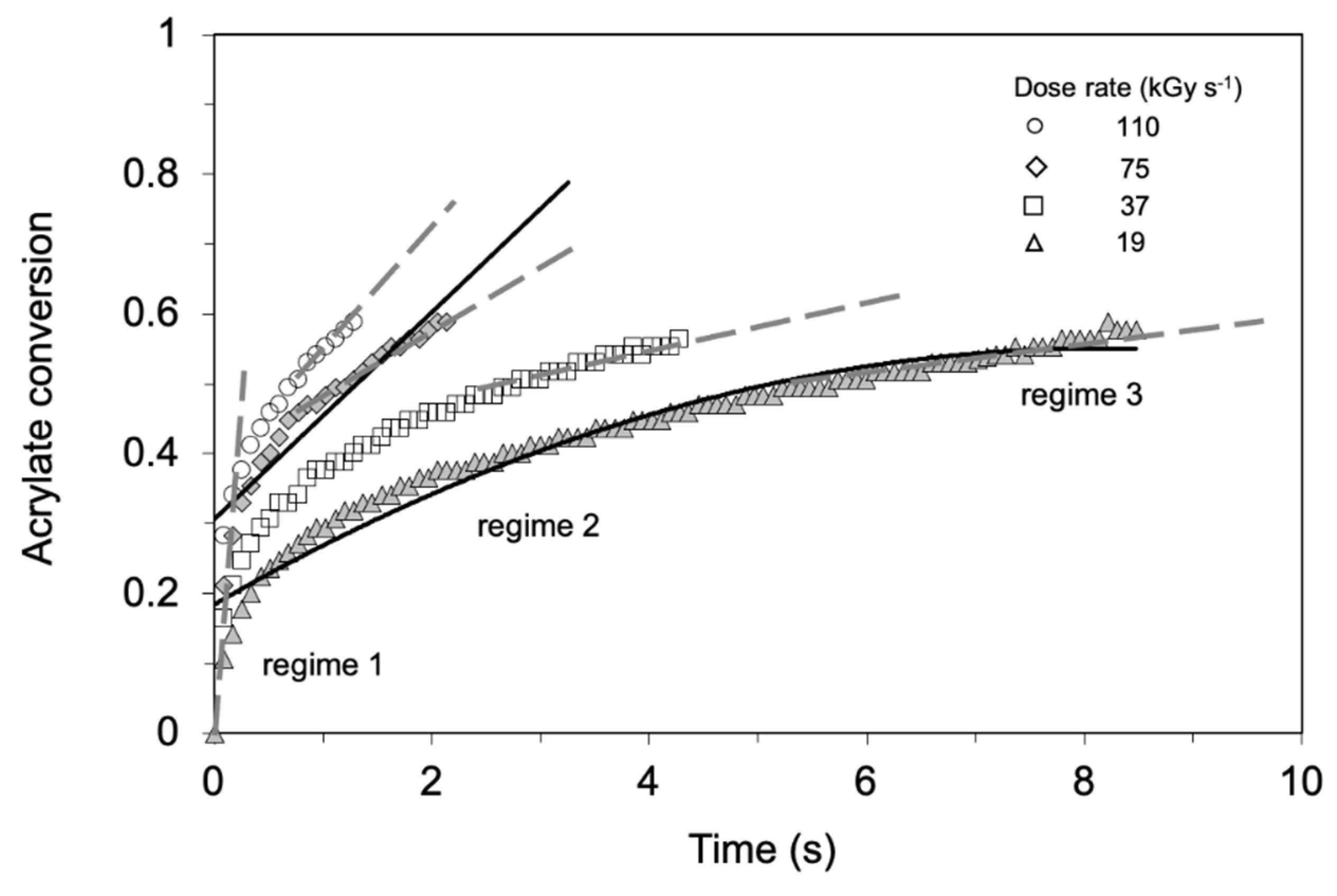
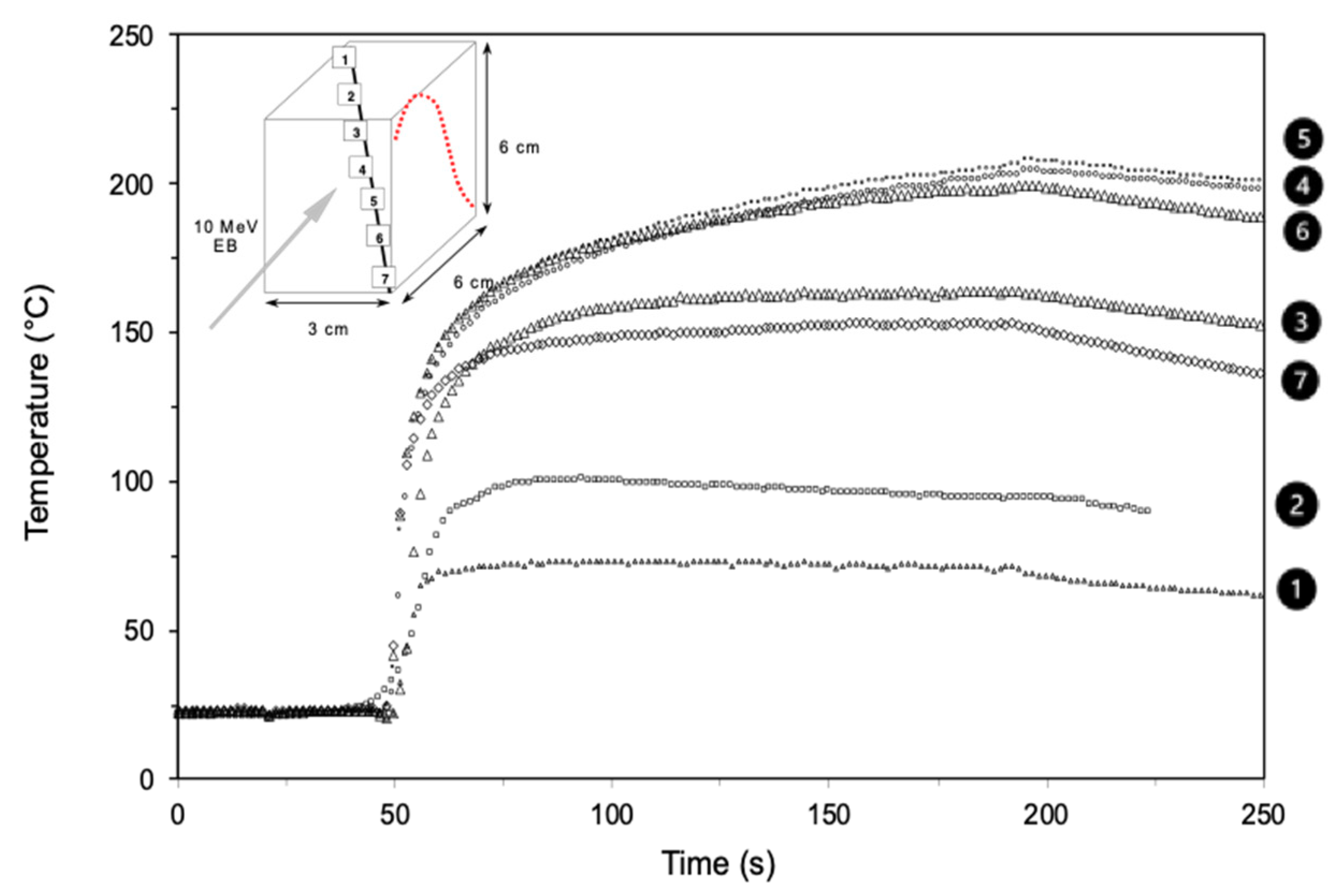






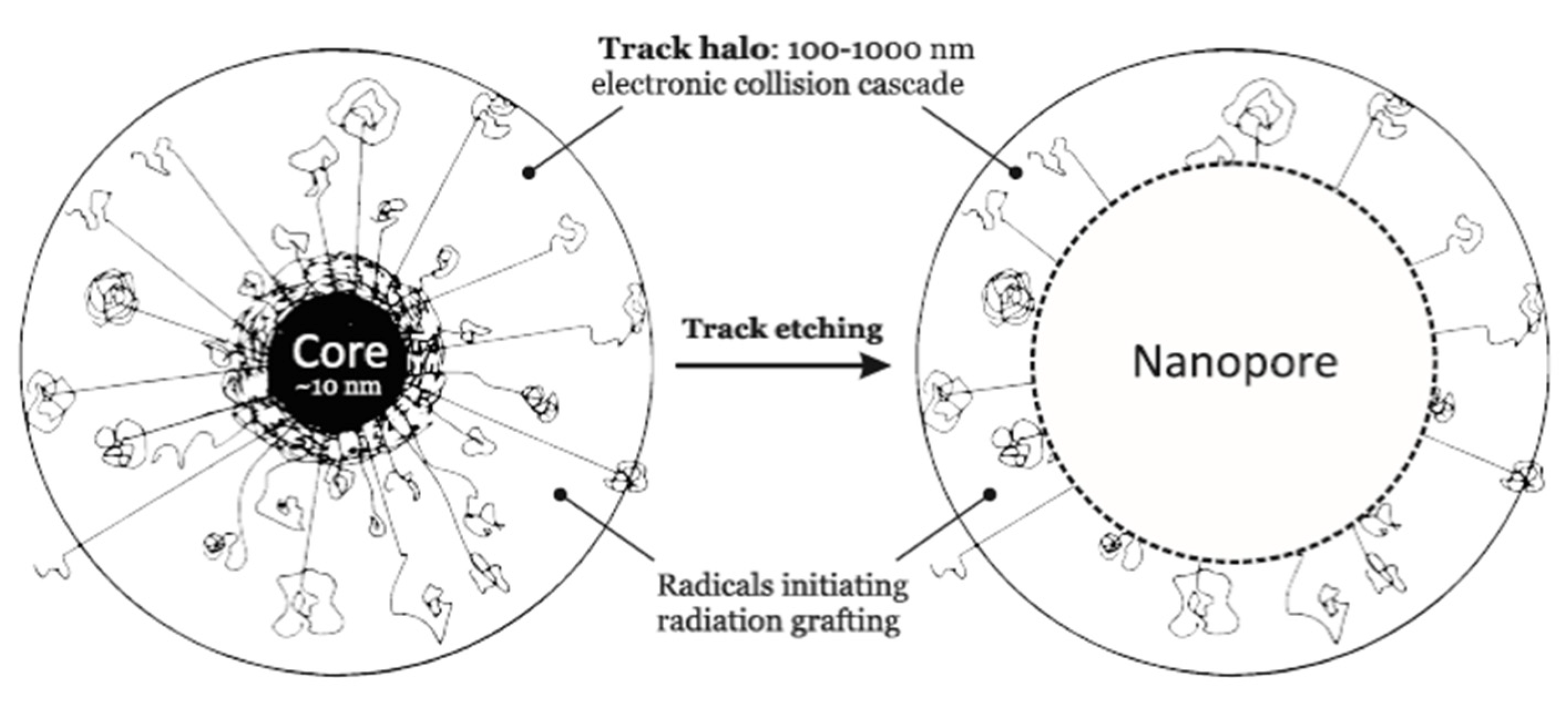

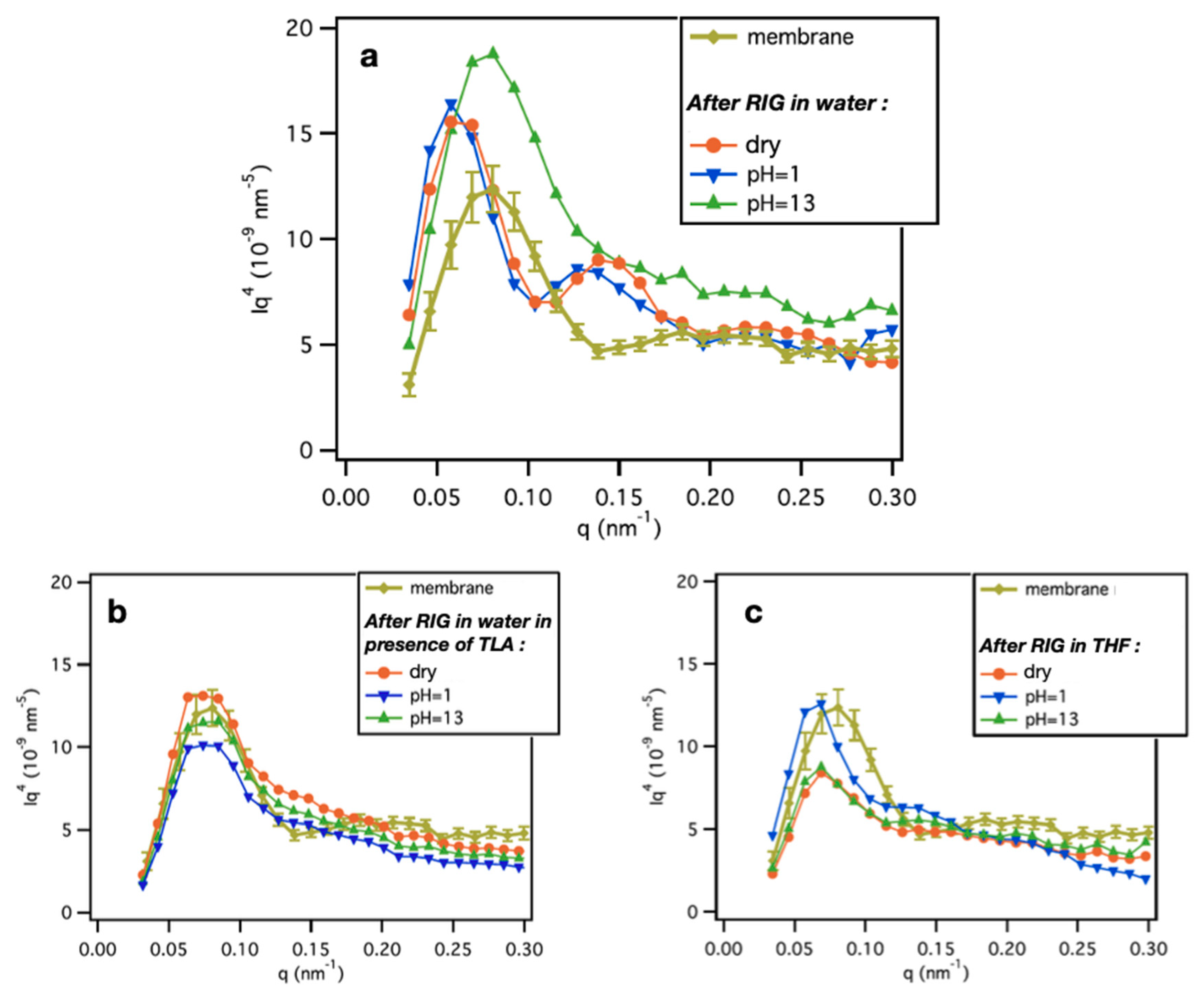

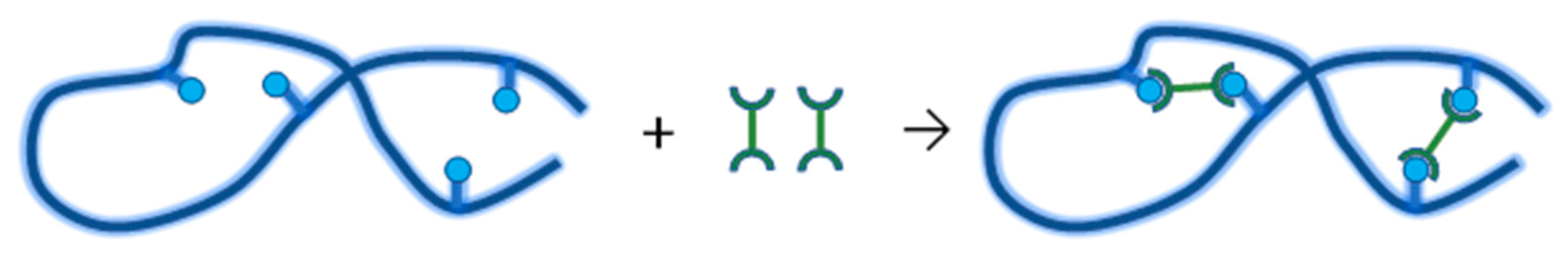







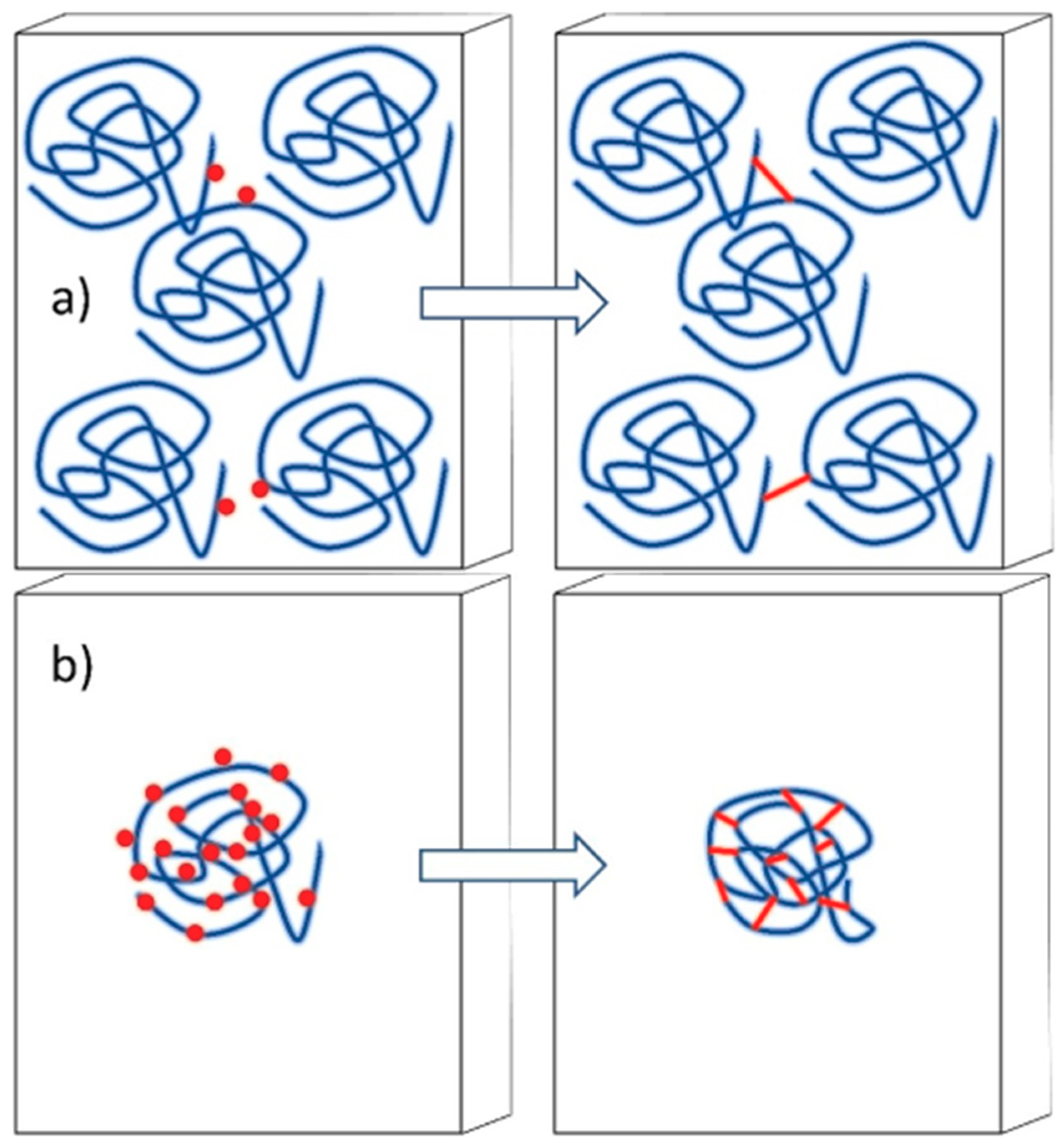

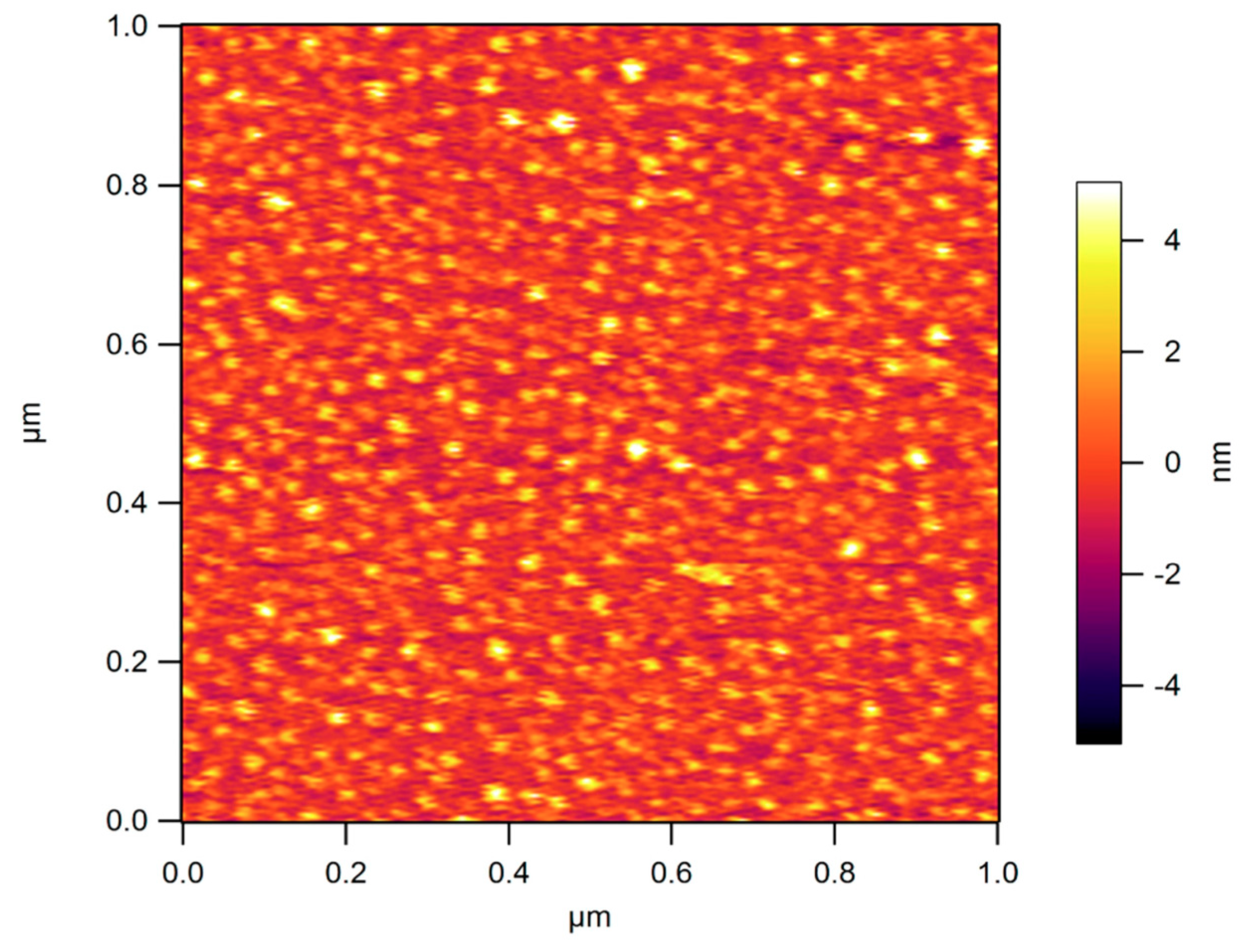







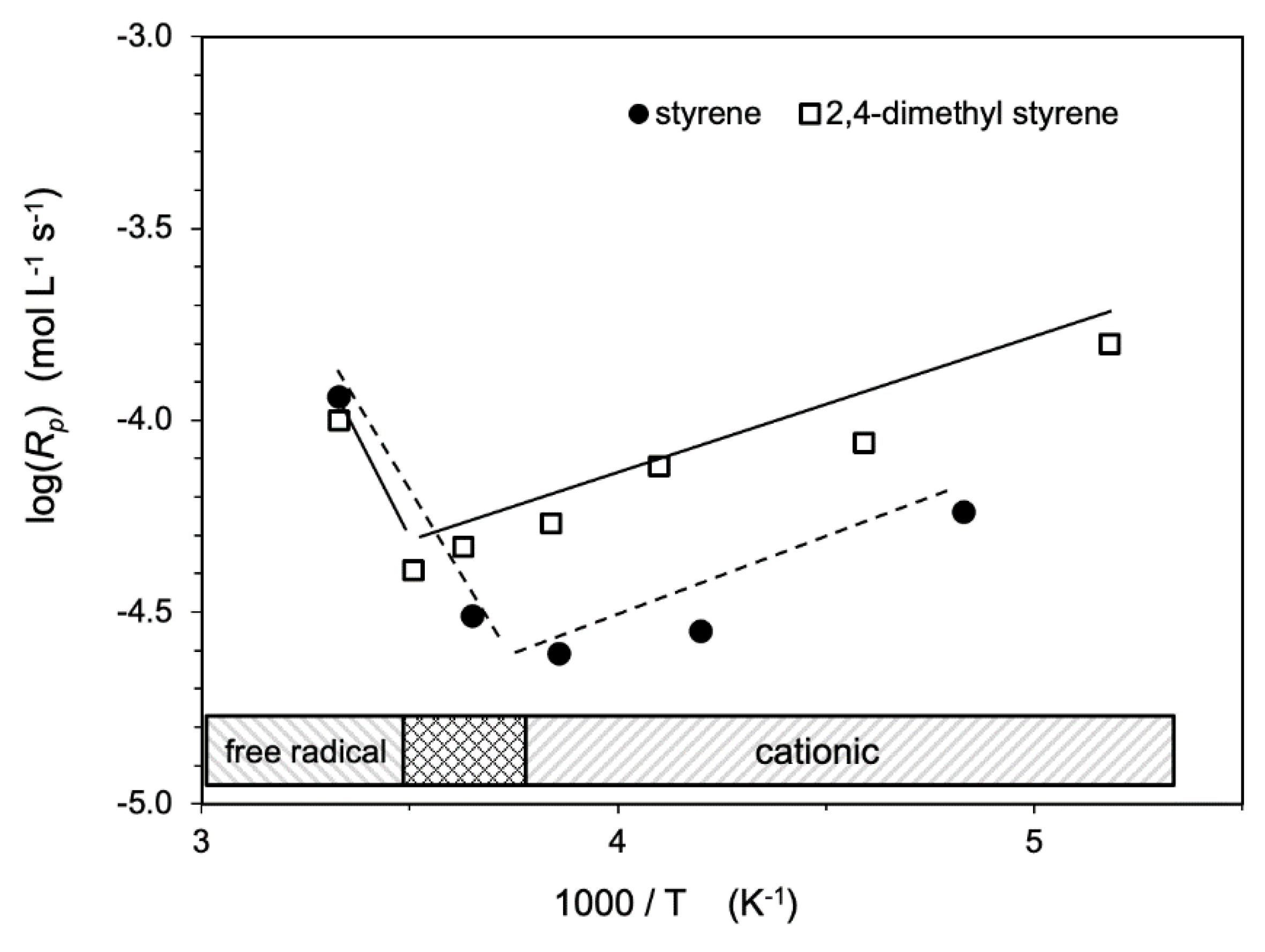{kind=link}
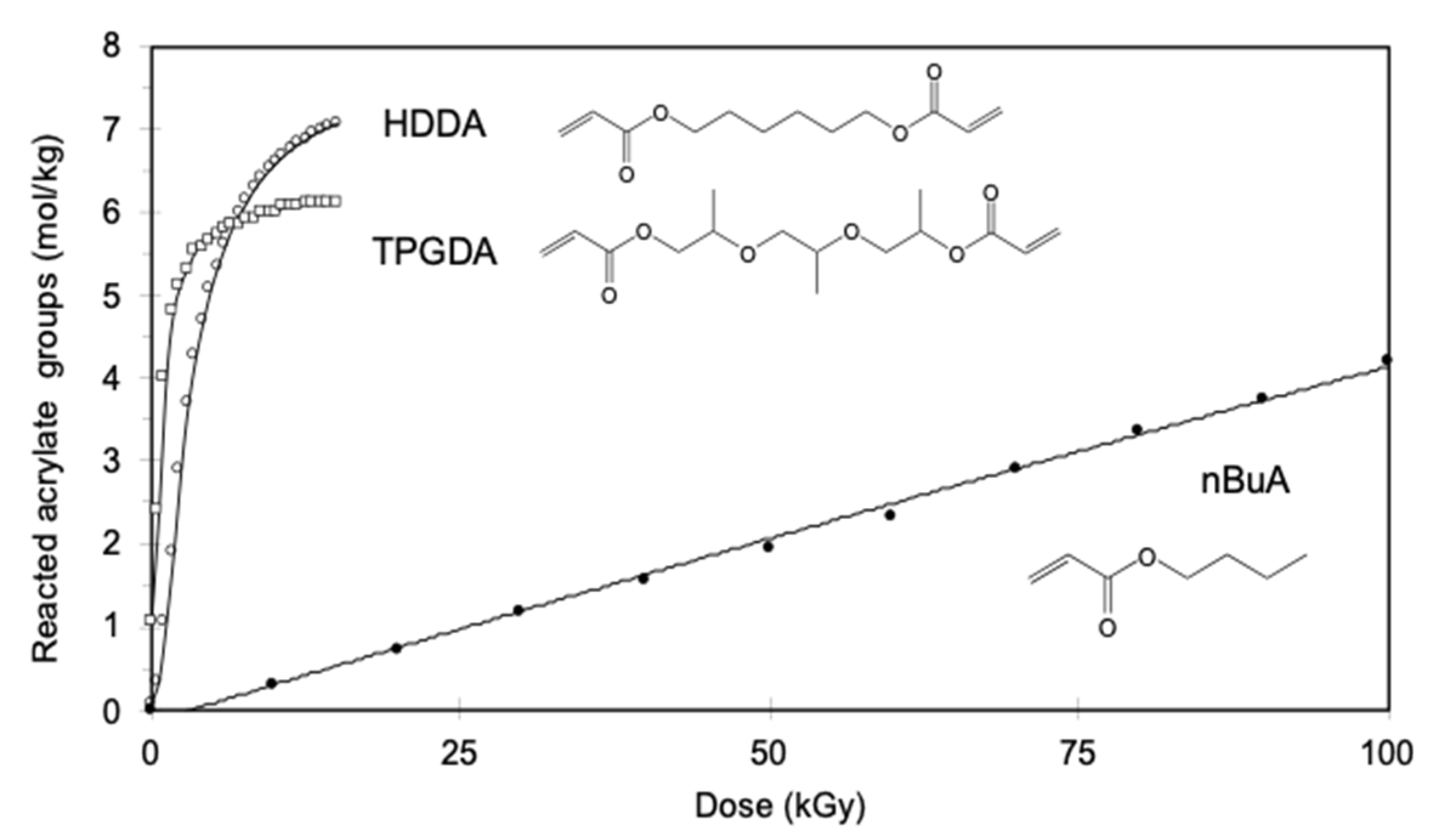{kind=link}
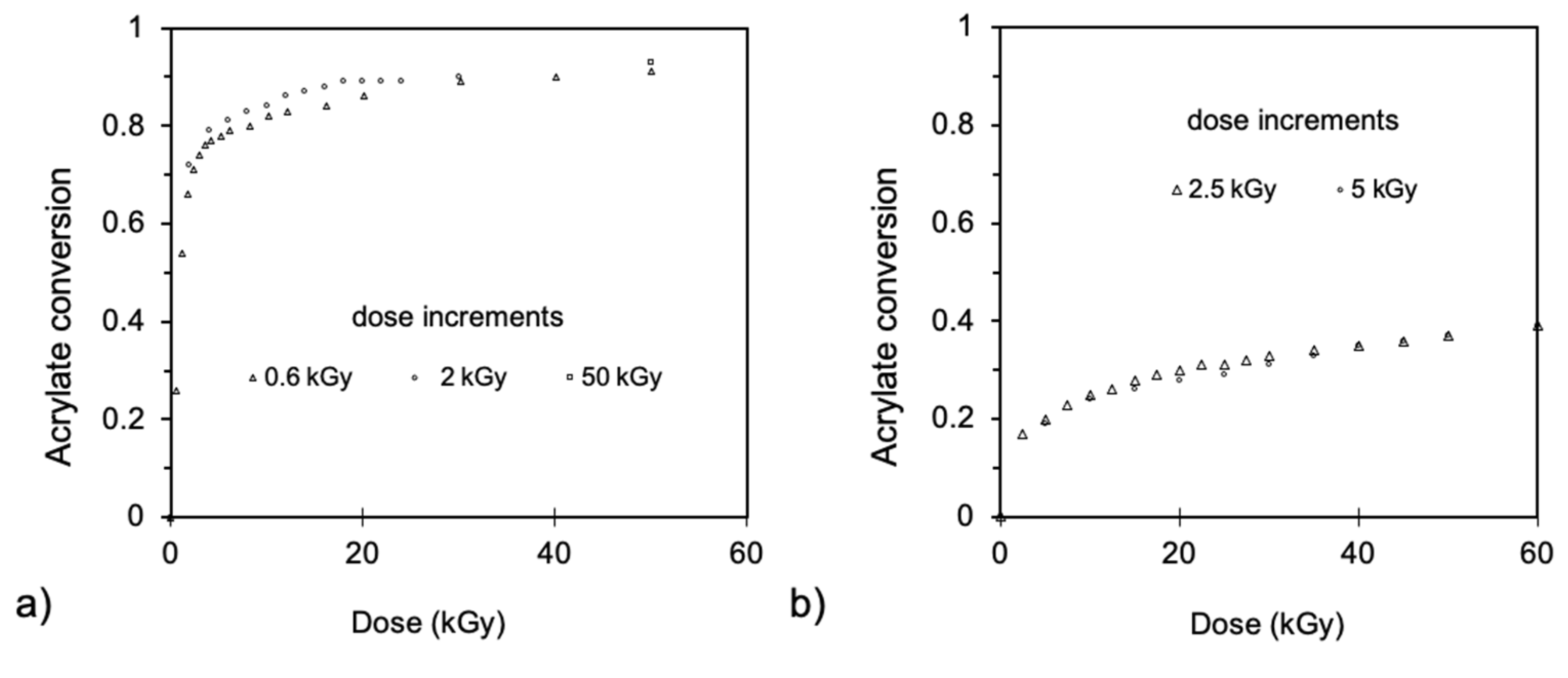{kind=link}
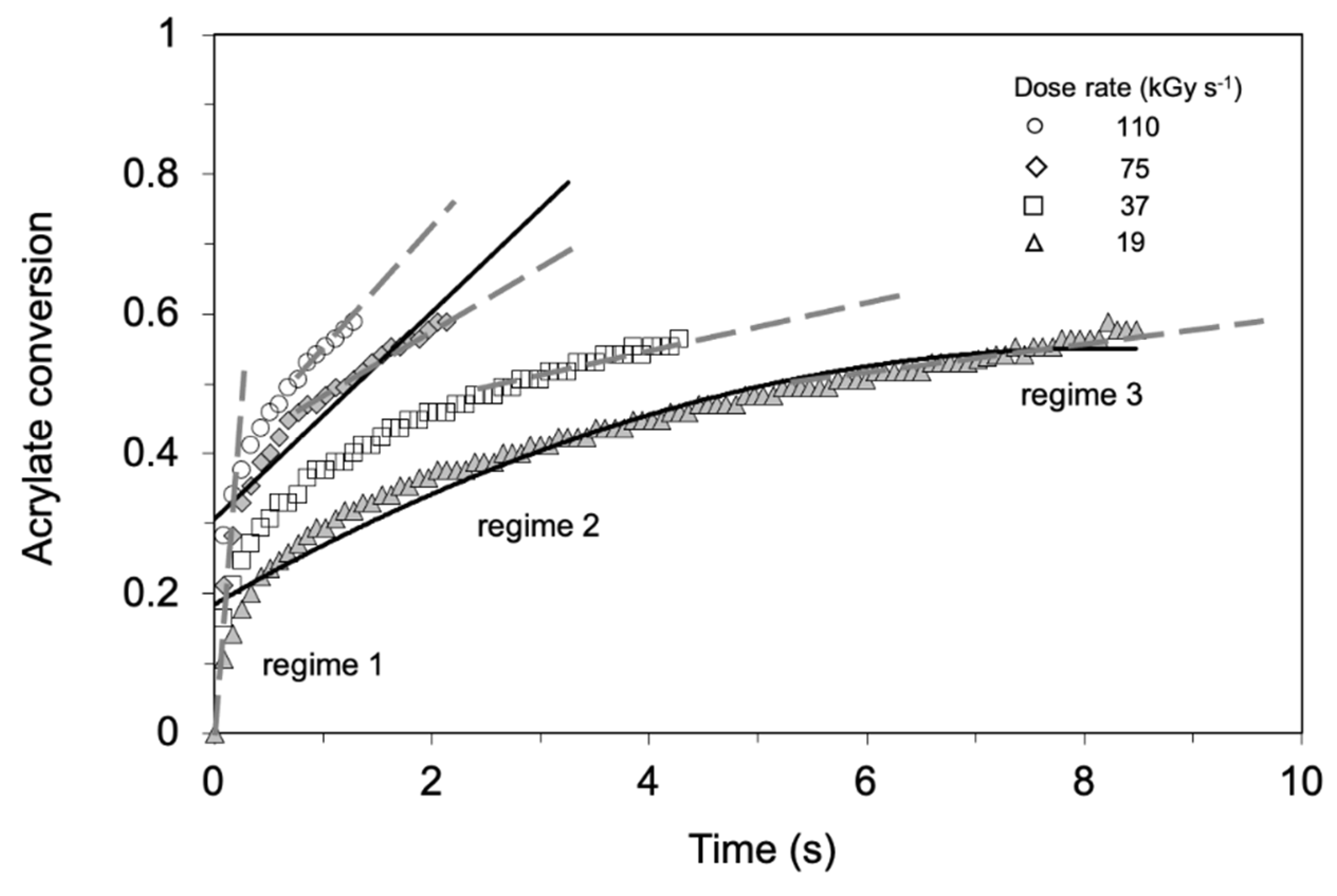{kind=link}
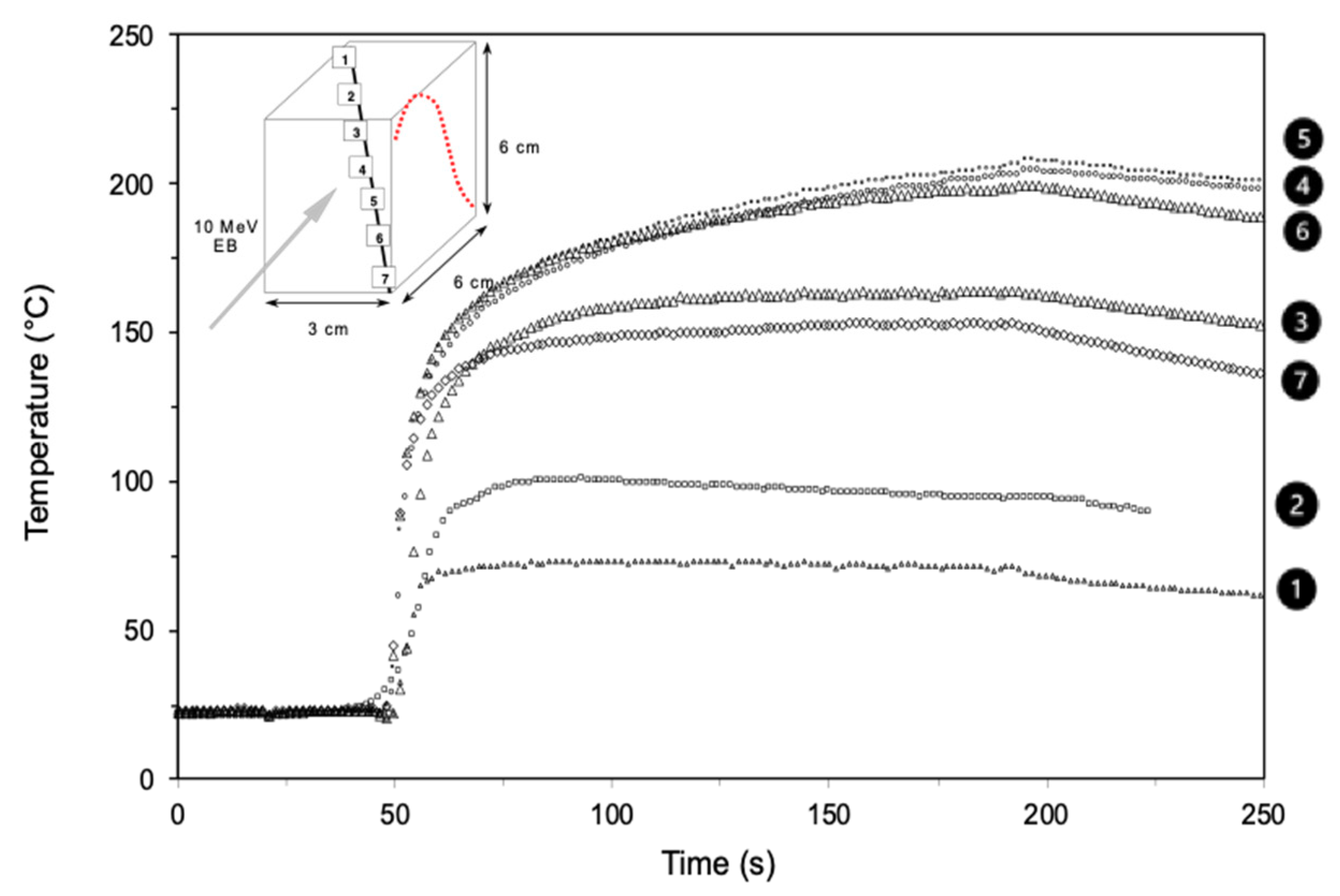{kind=link}
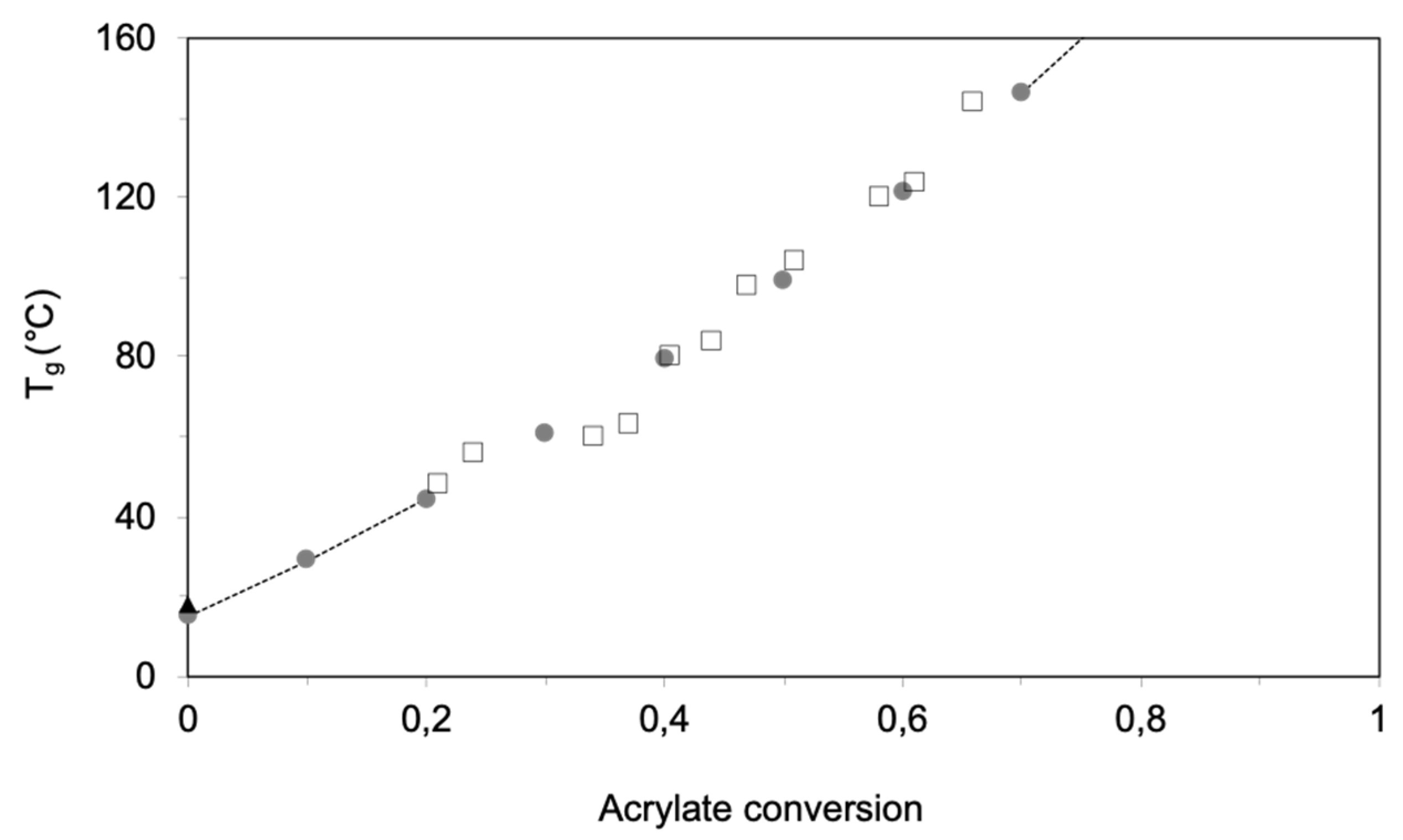{kind=link}
{kind=link}
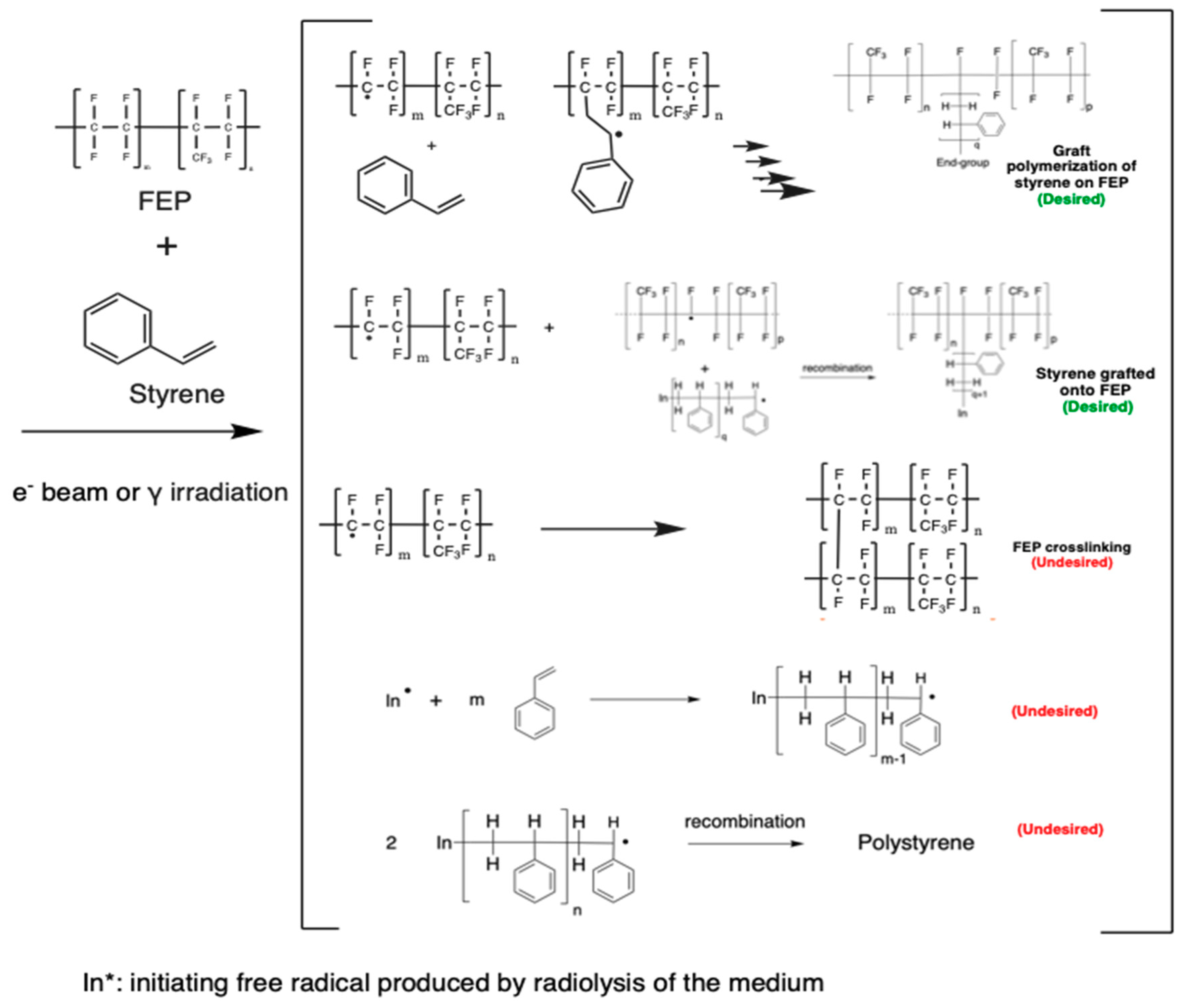{kind=link}
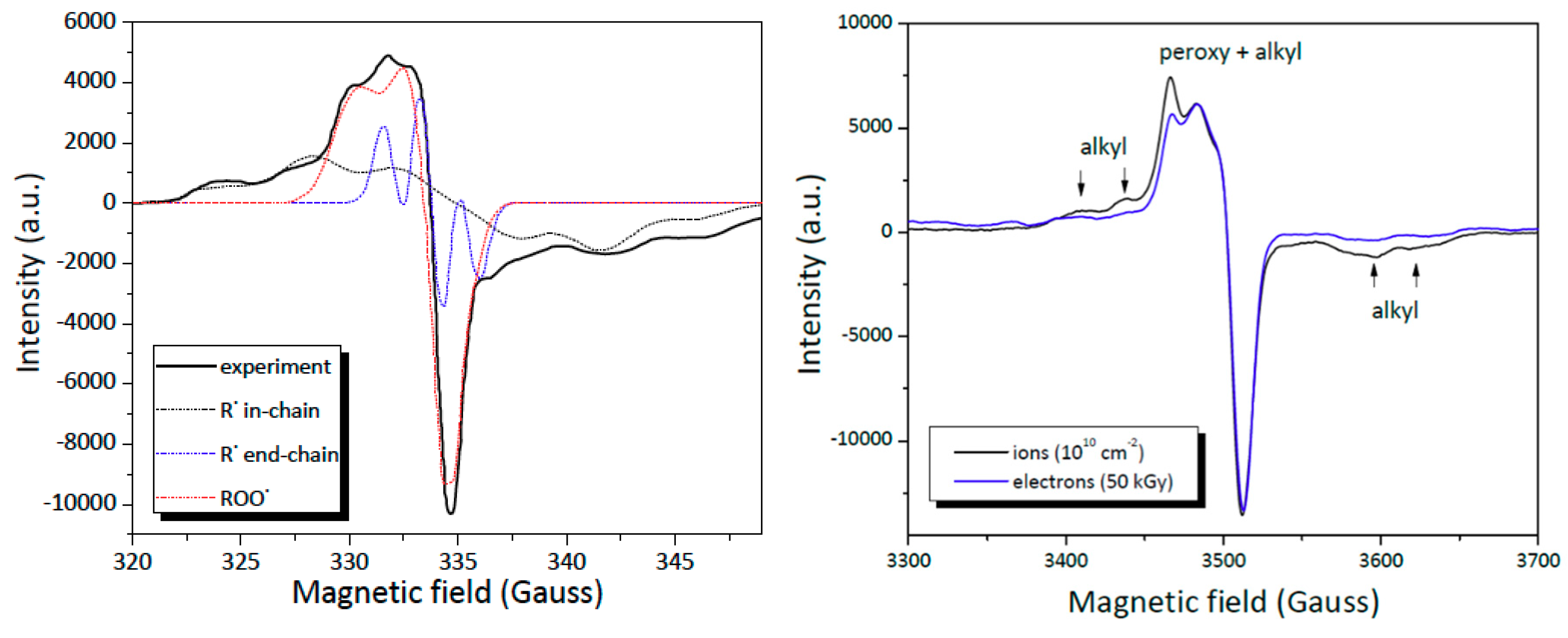{kind=link}
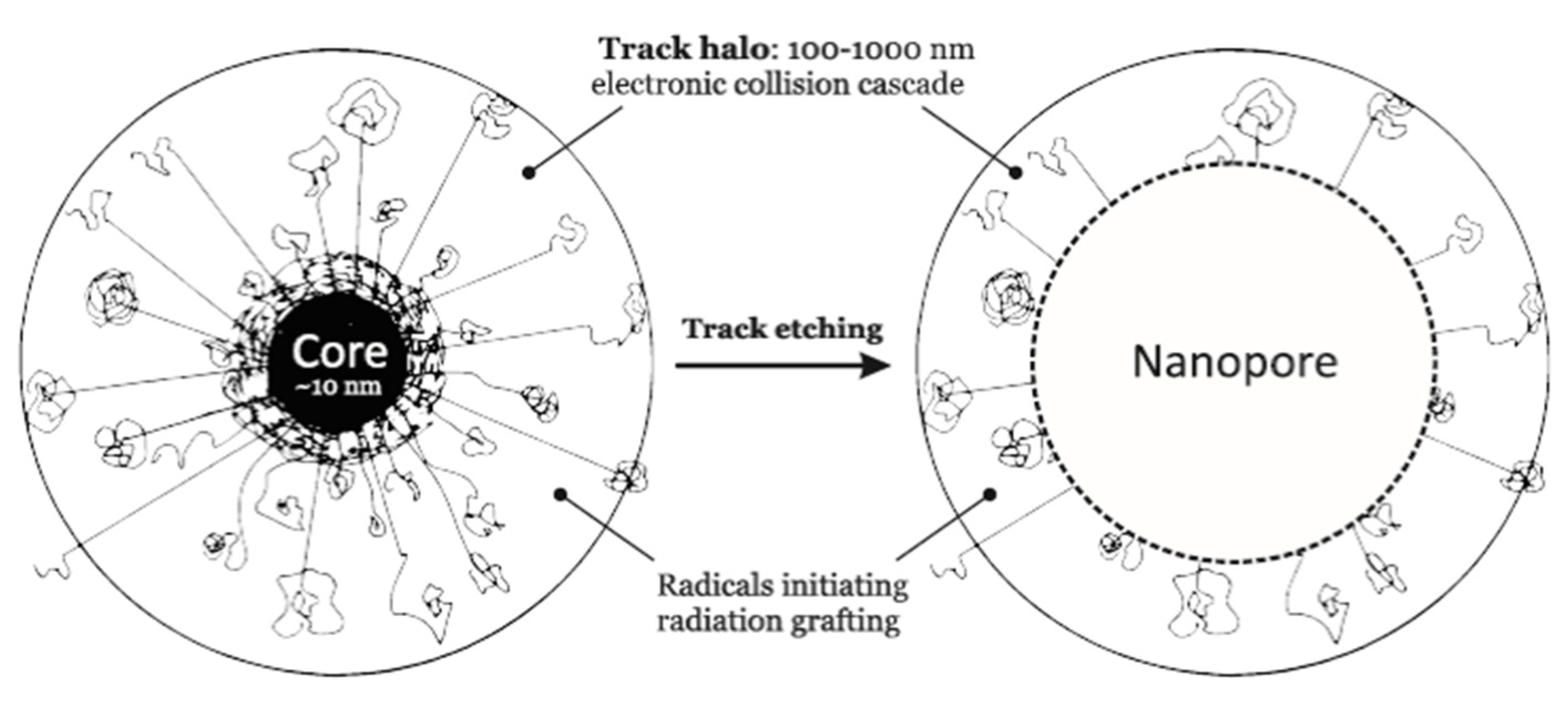{kind=link}
{kind=link}
{kind=link}
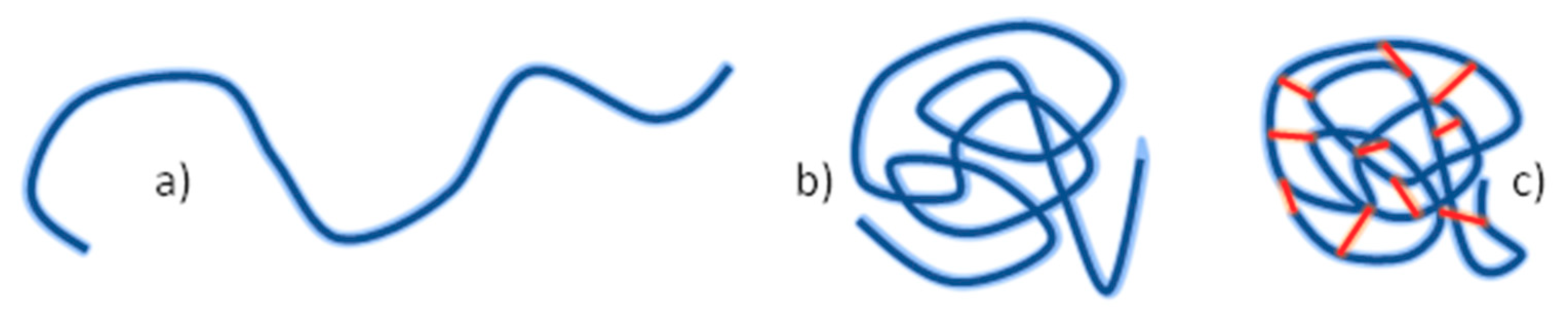{kind=link}
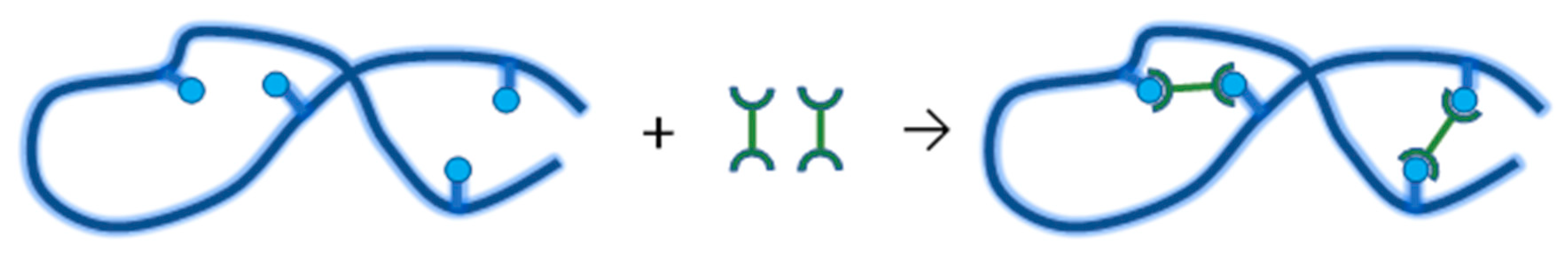{kind=link}
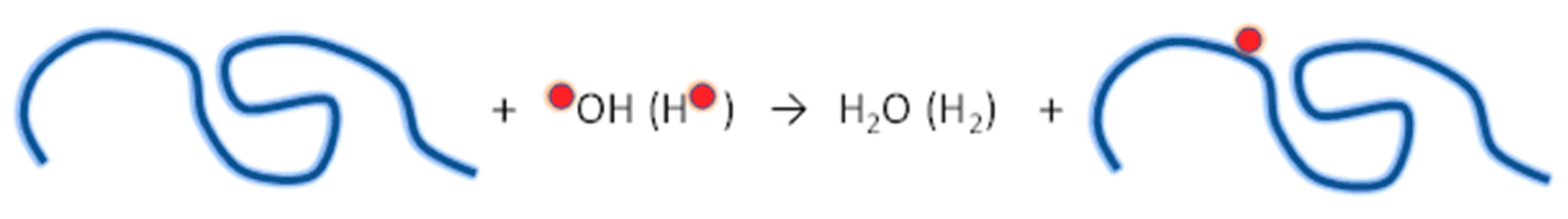{kind=link}
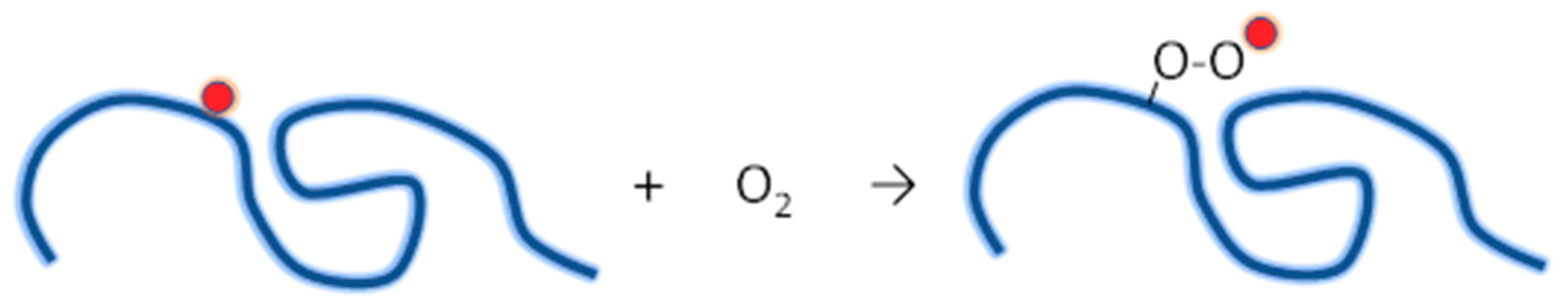{kind=link}
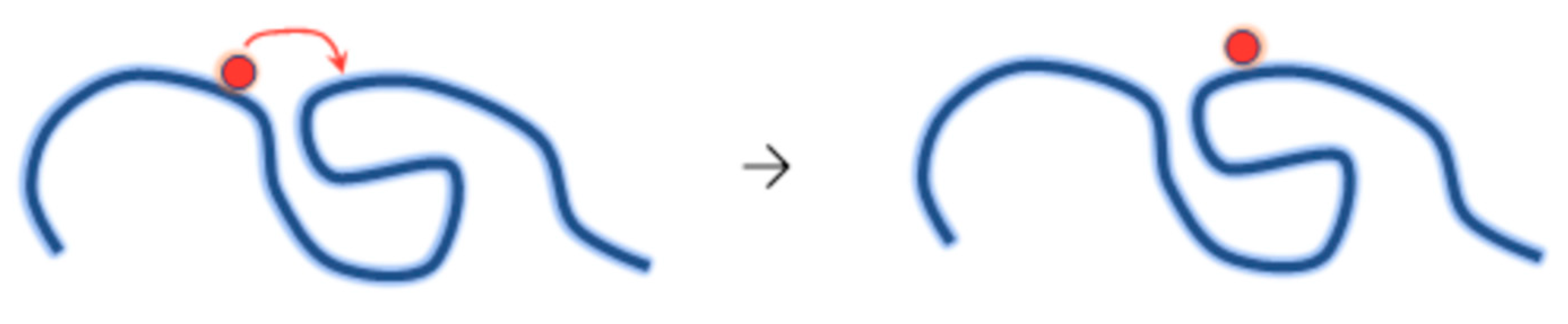{kind=link}
{kind=link}
{kind=link}
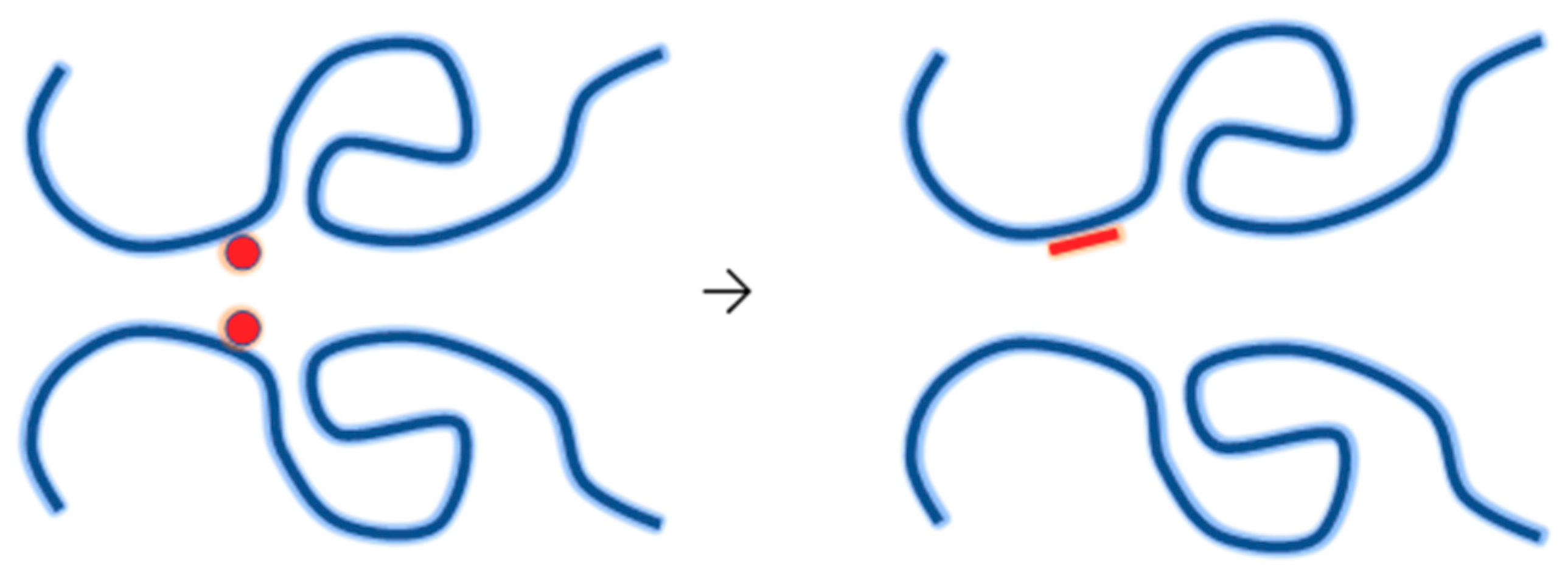{kind=link}
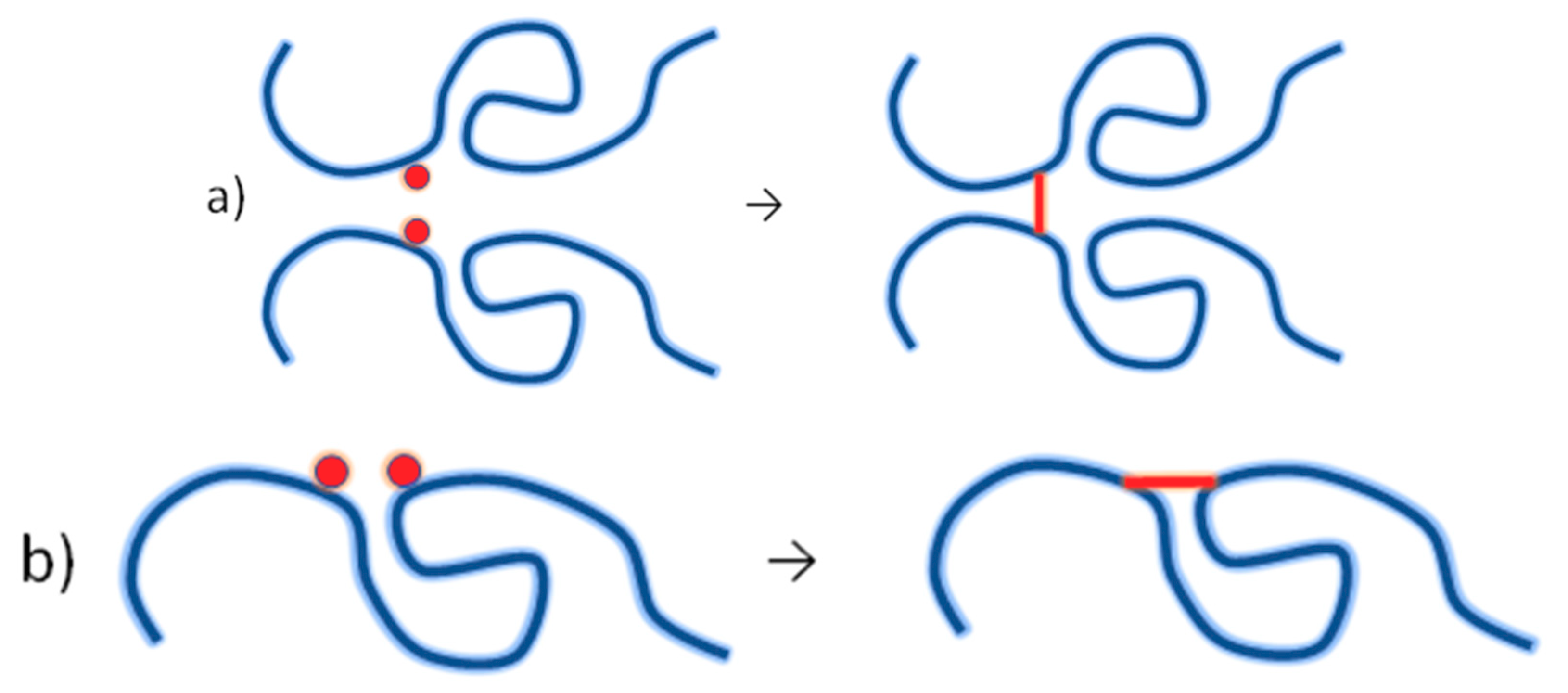{kind=link}
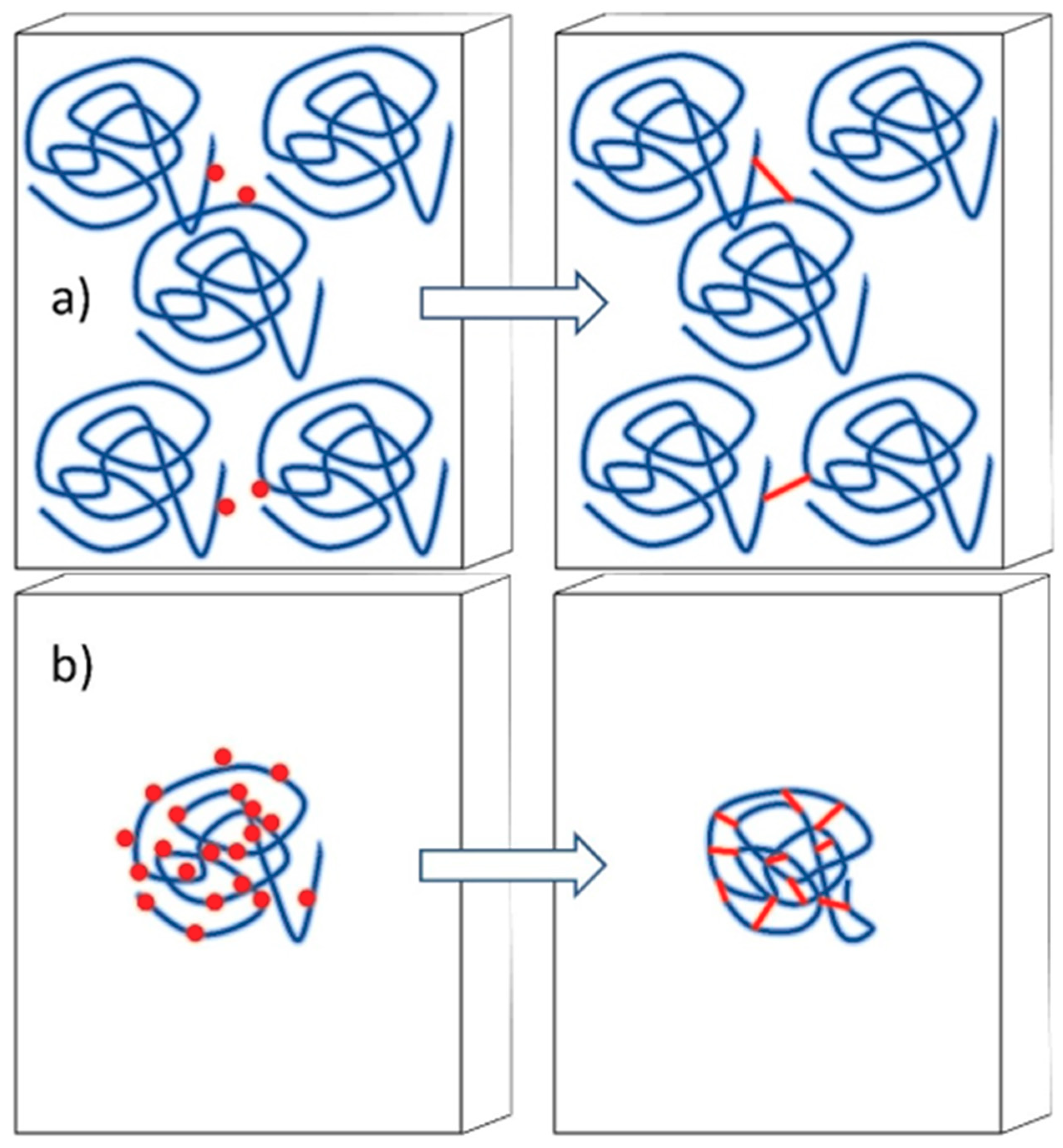{kind=link}
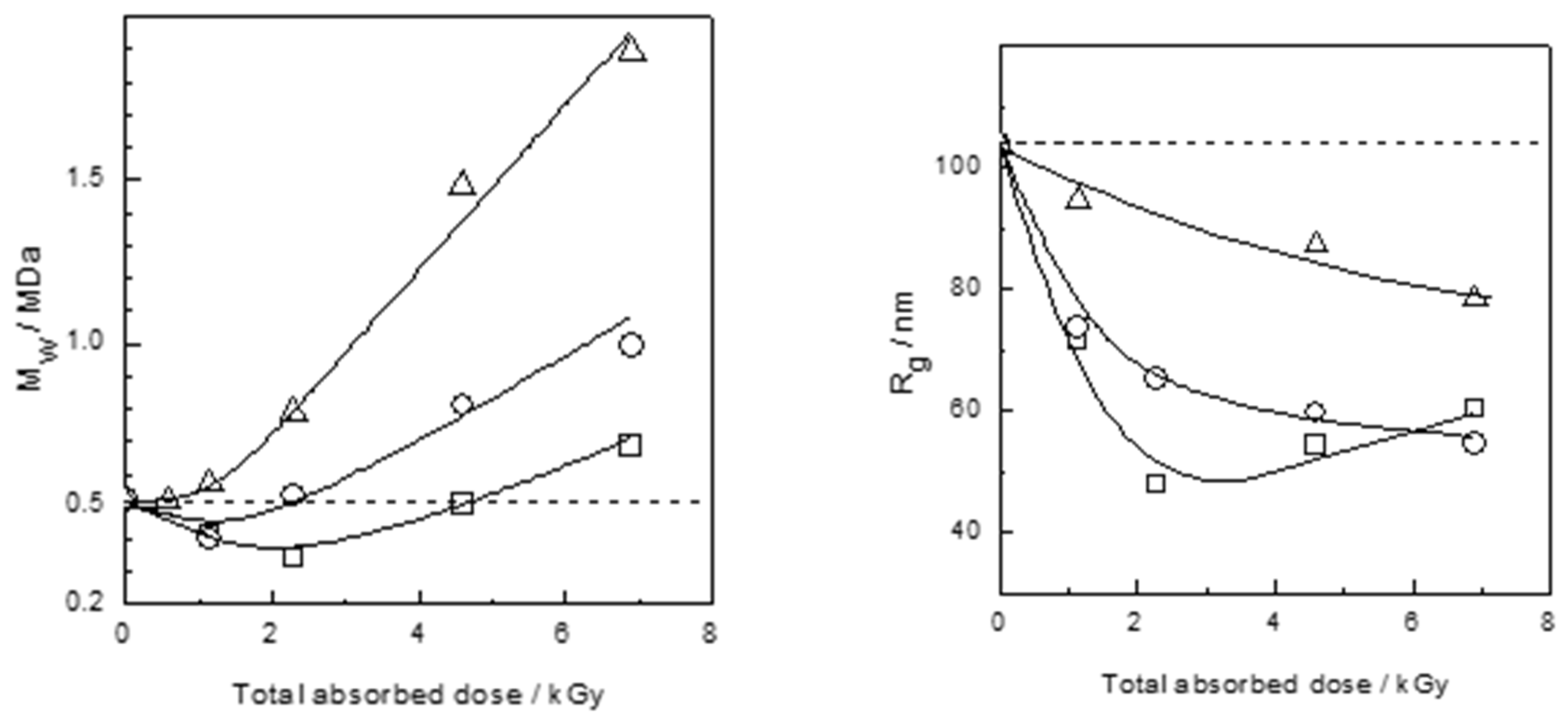{kind=link}
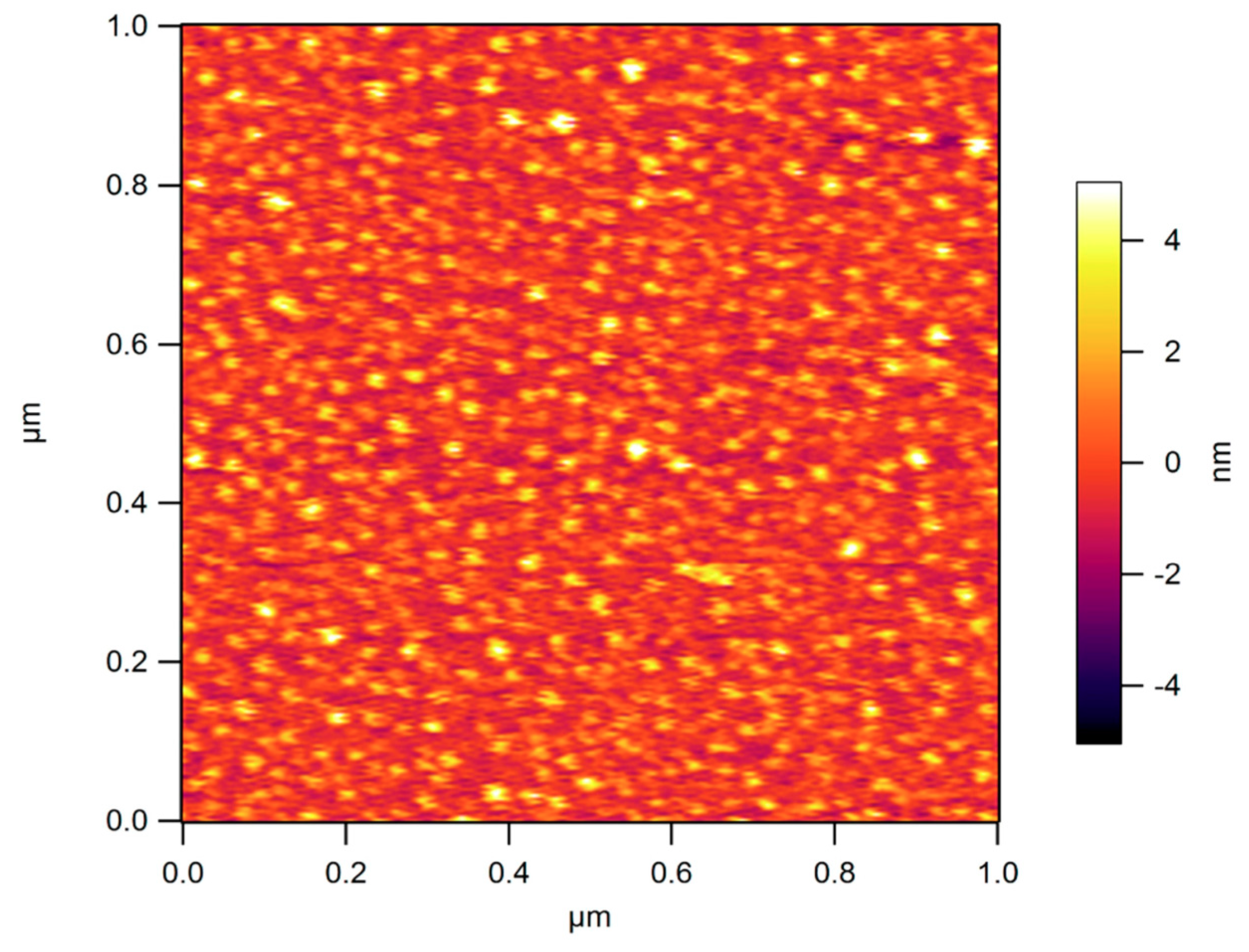{kind=link}
{kind=link}
{kind=link}
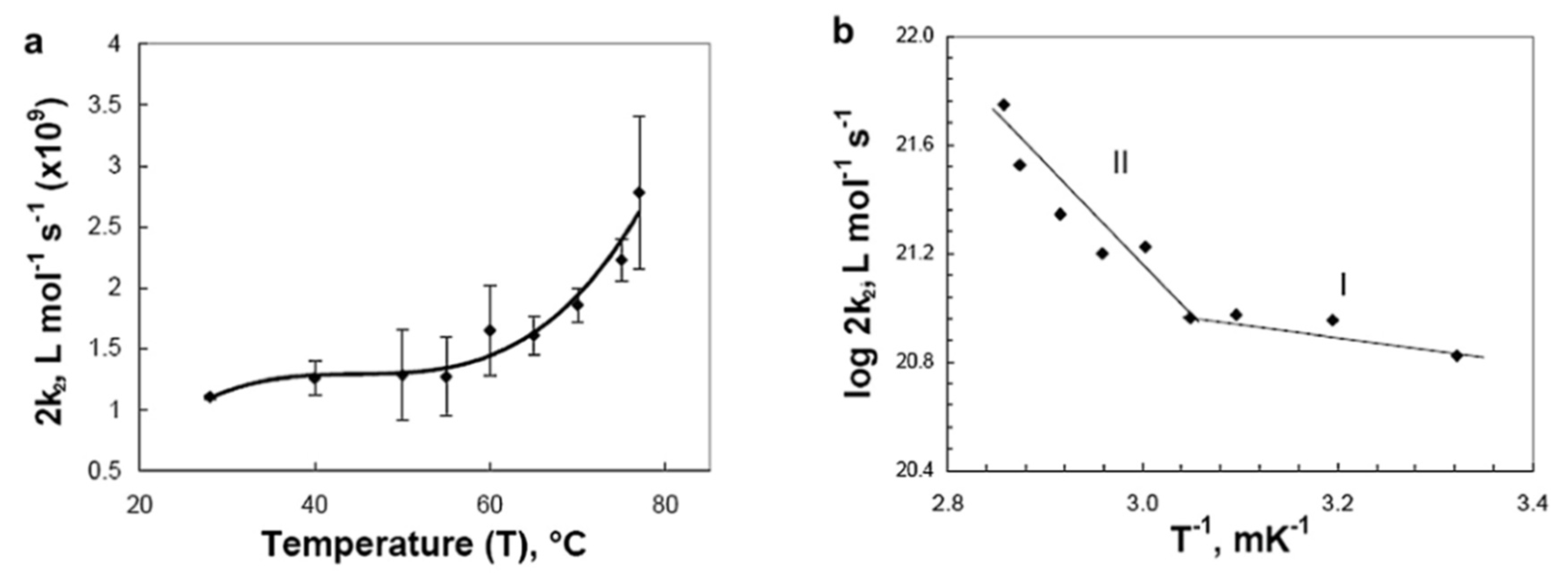{kind=link}
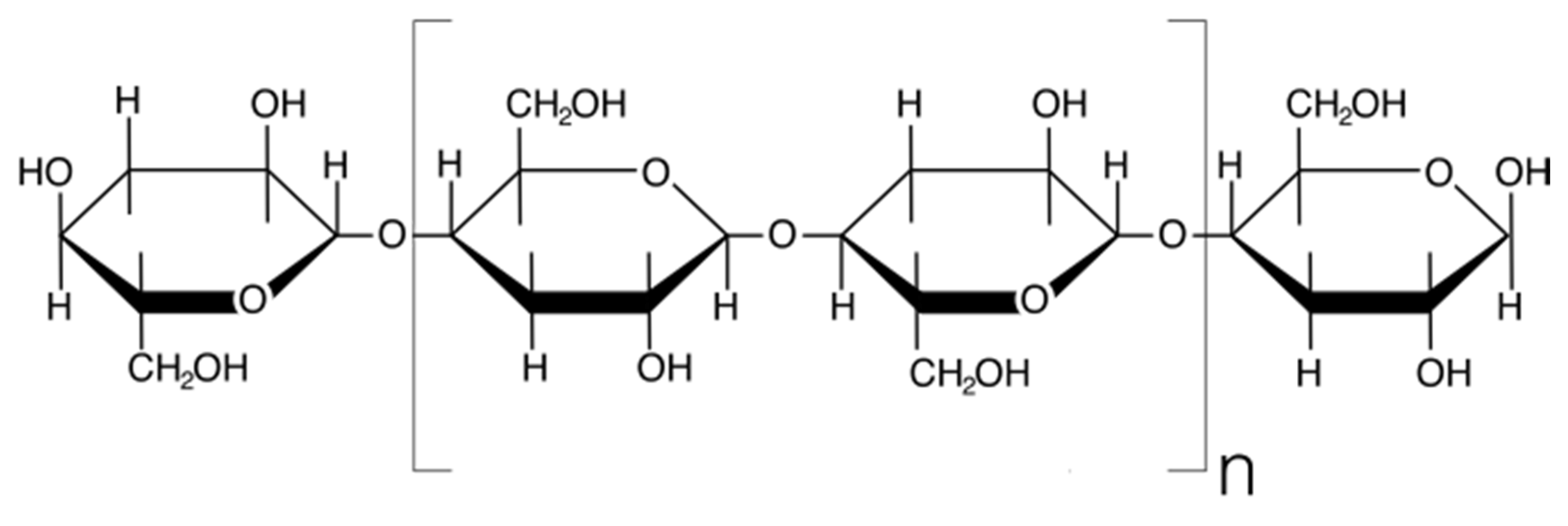{kind=link}
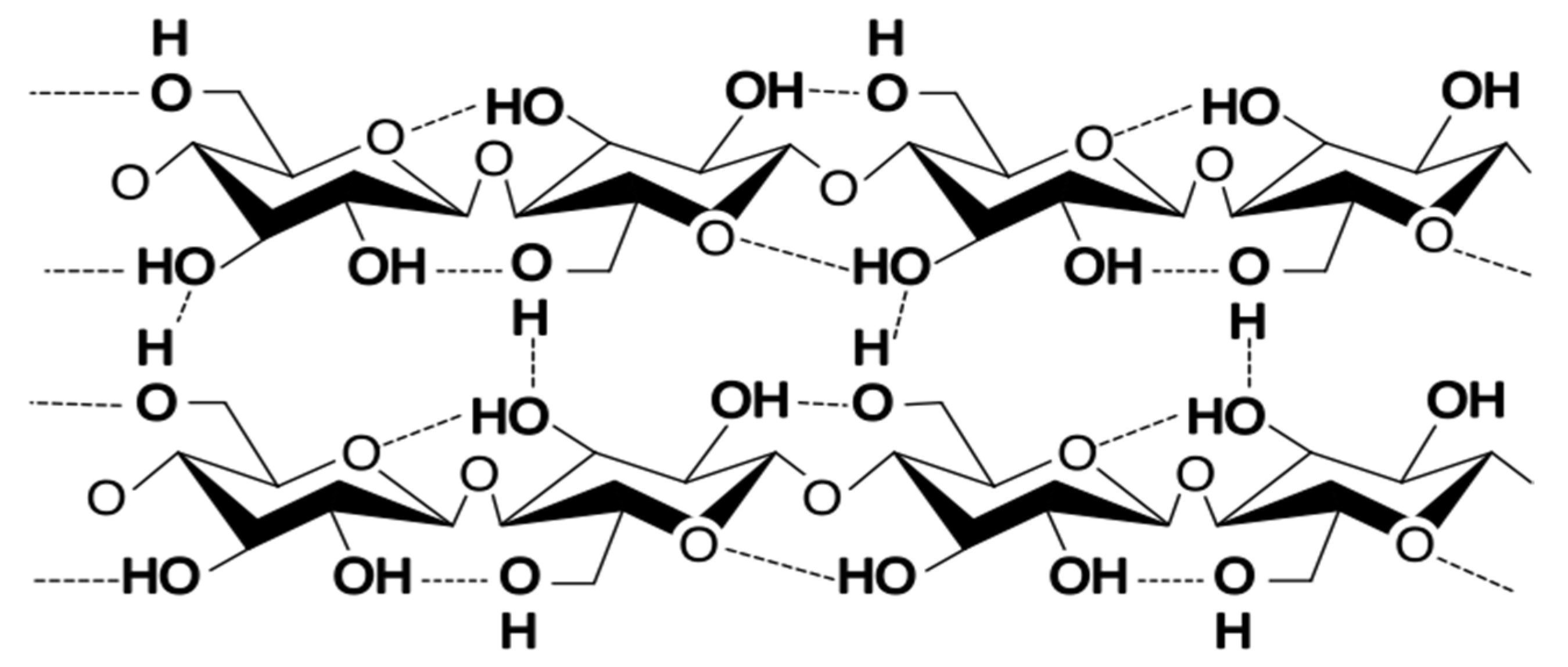{kind=link}
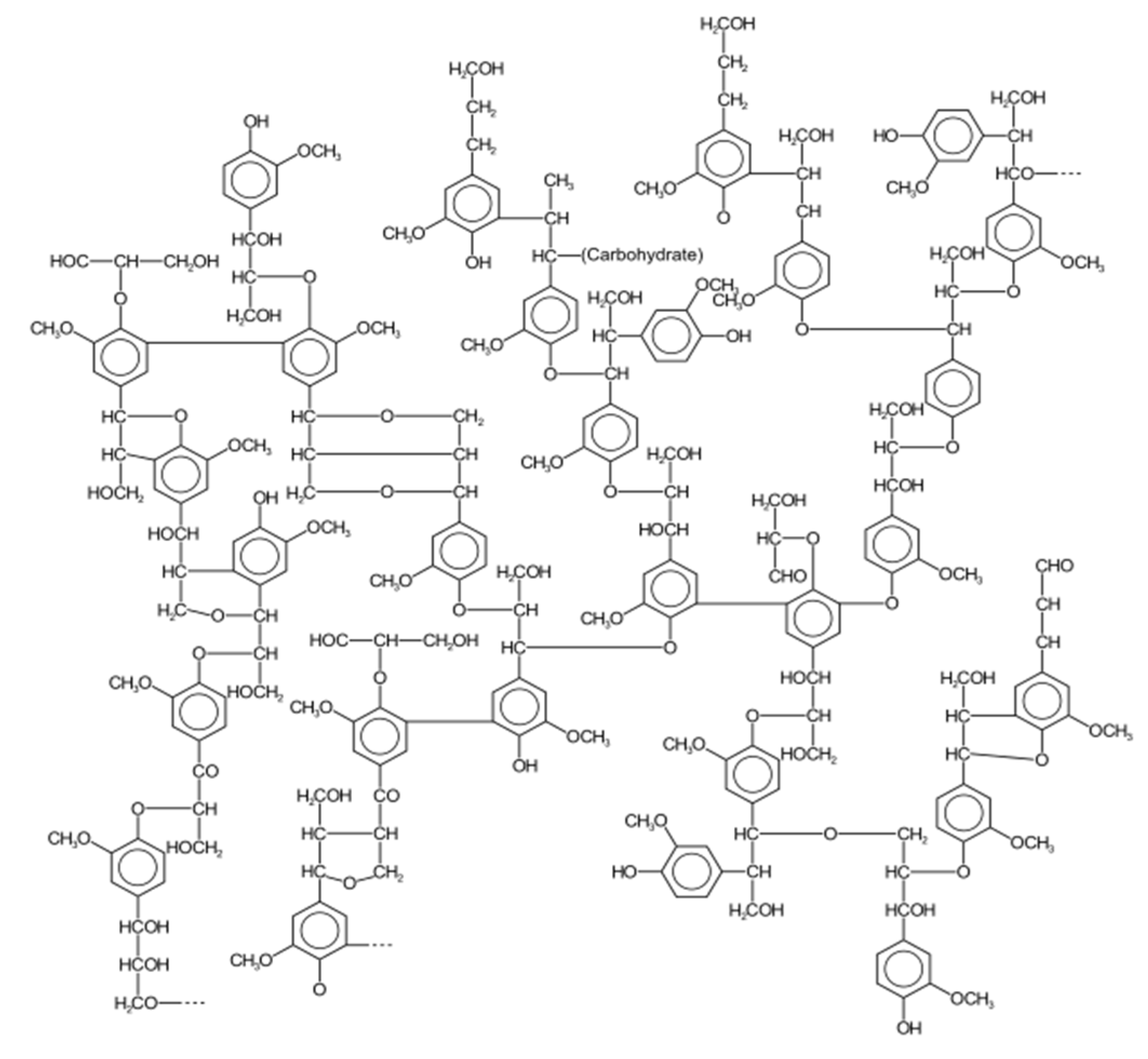{kind=link}
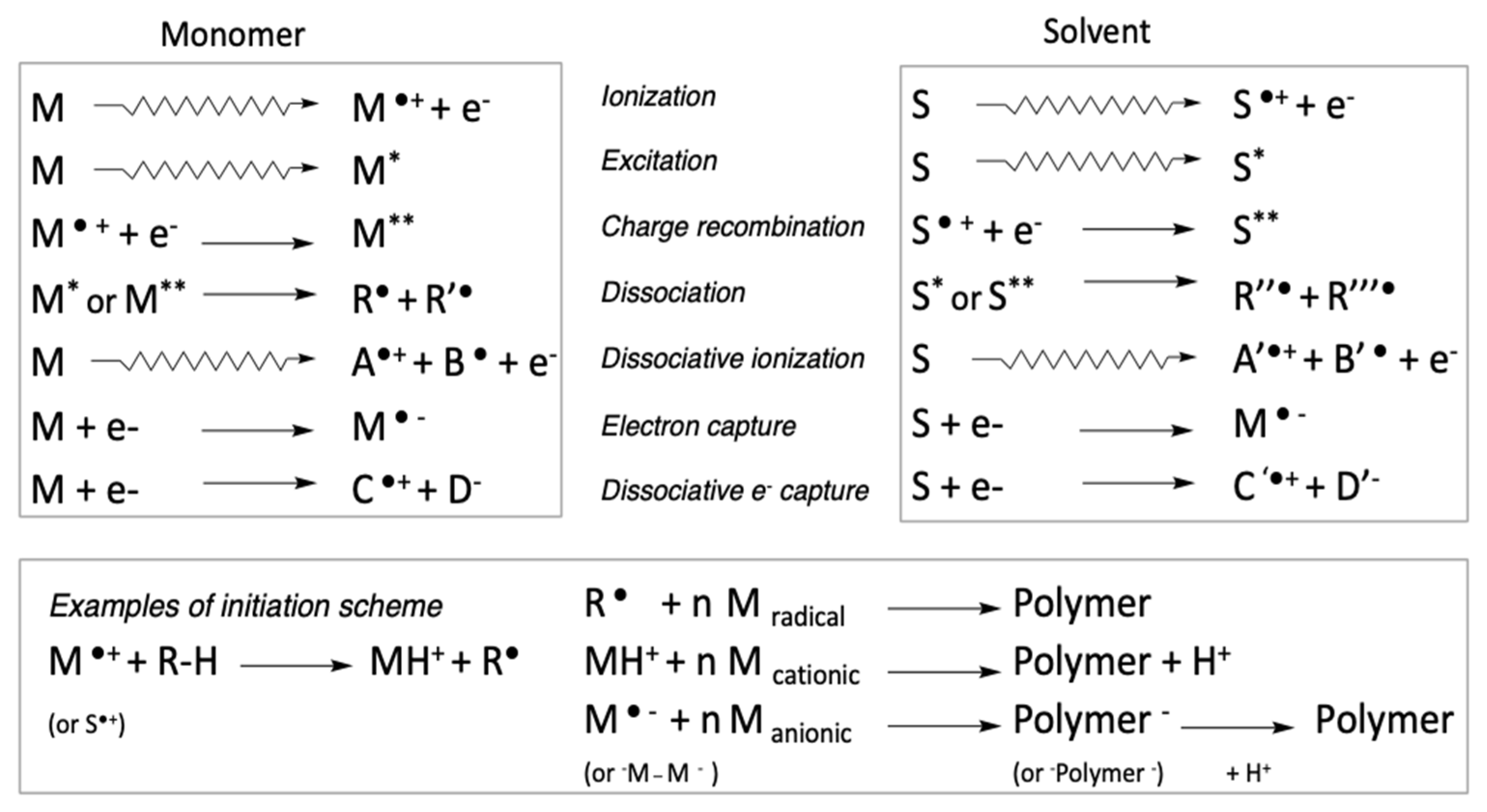{kind=link}
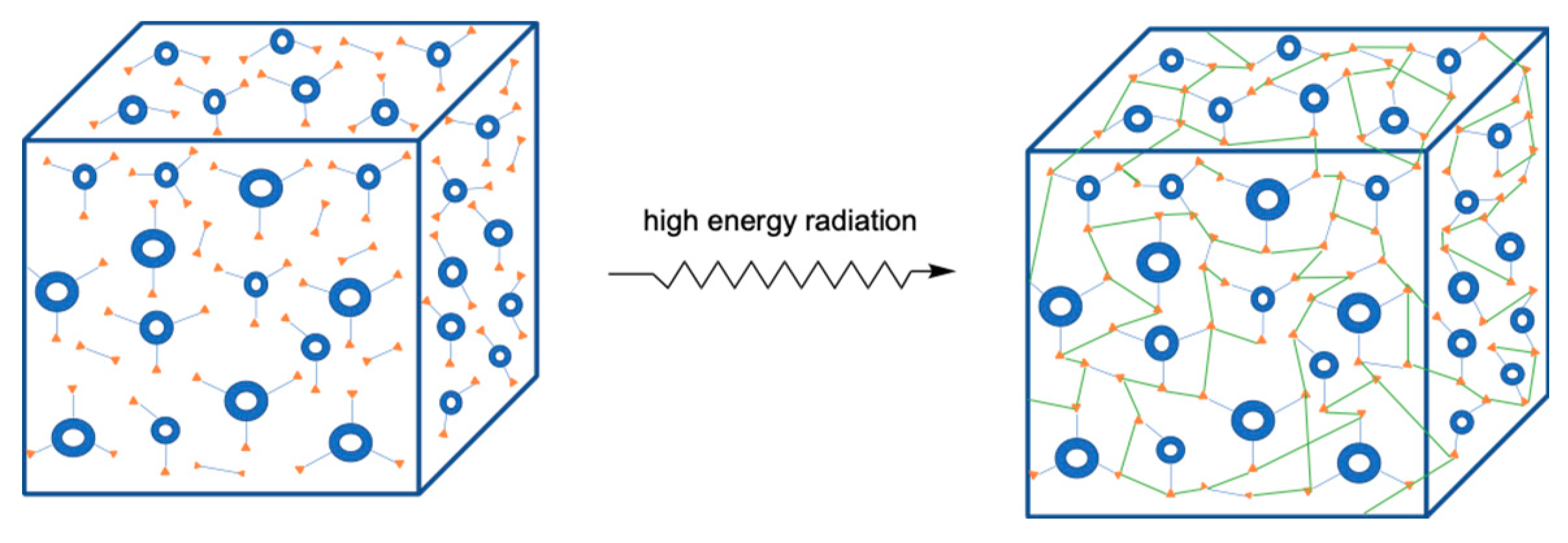{kind=link}
{kind=link}
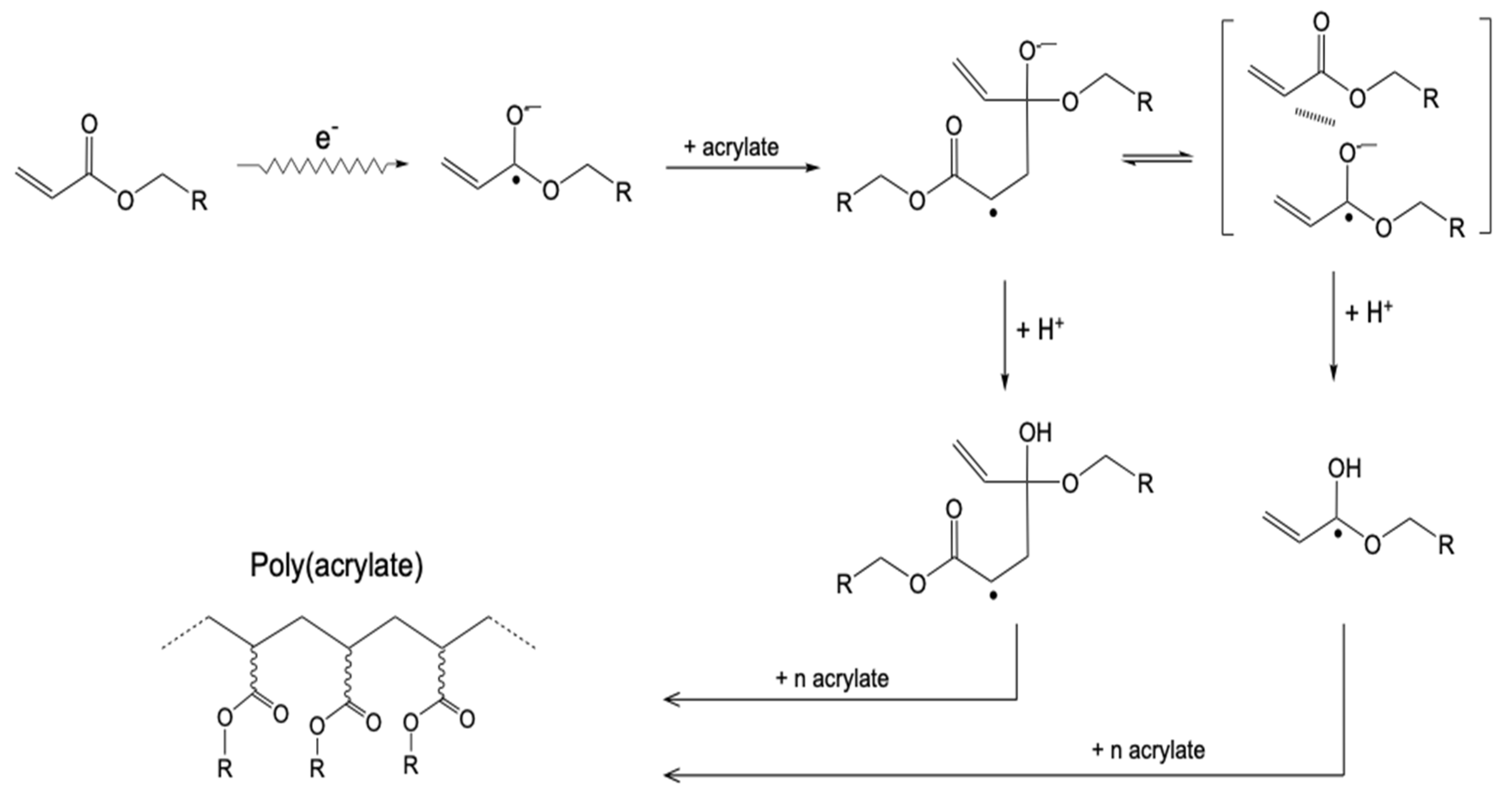{kind=link}
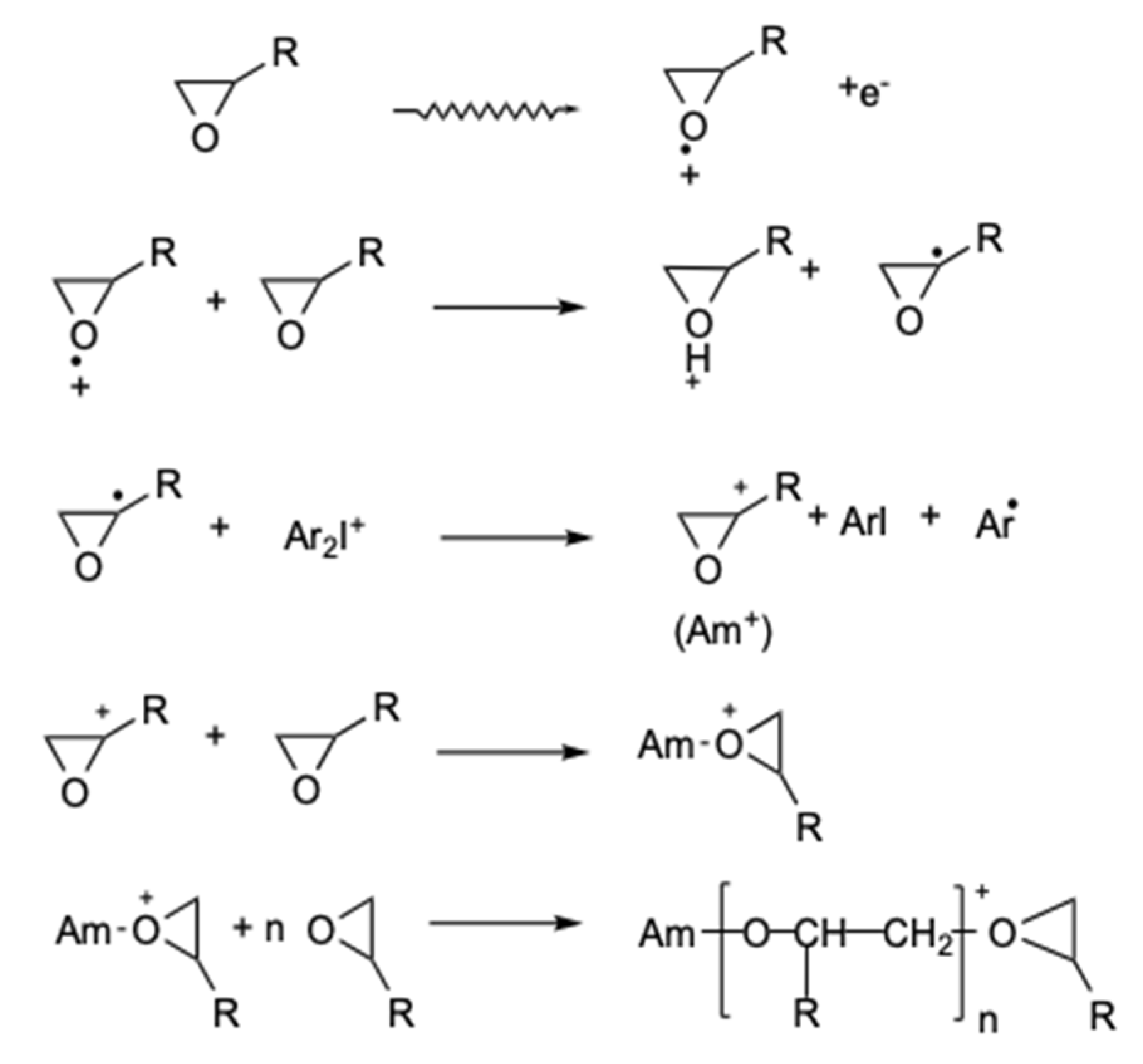{kind=link}
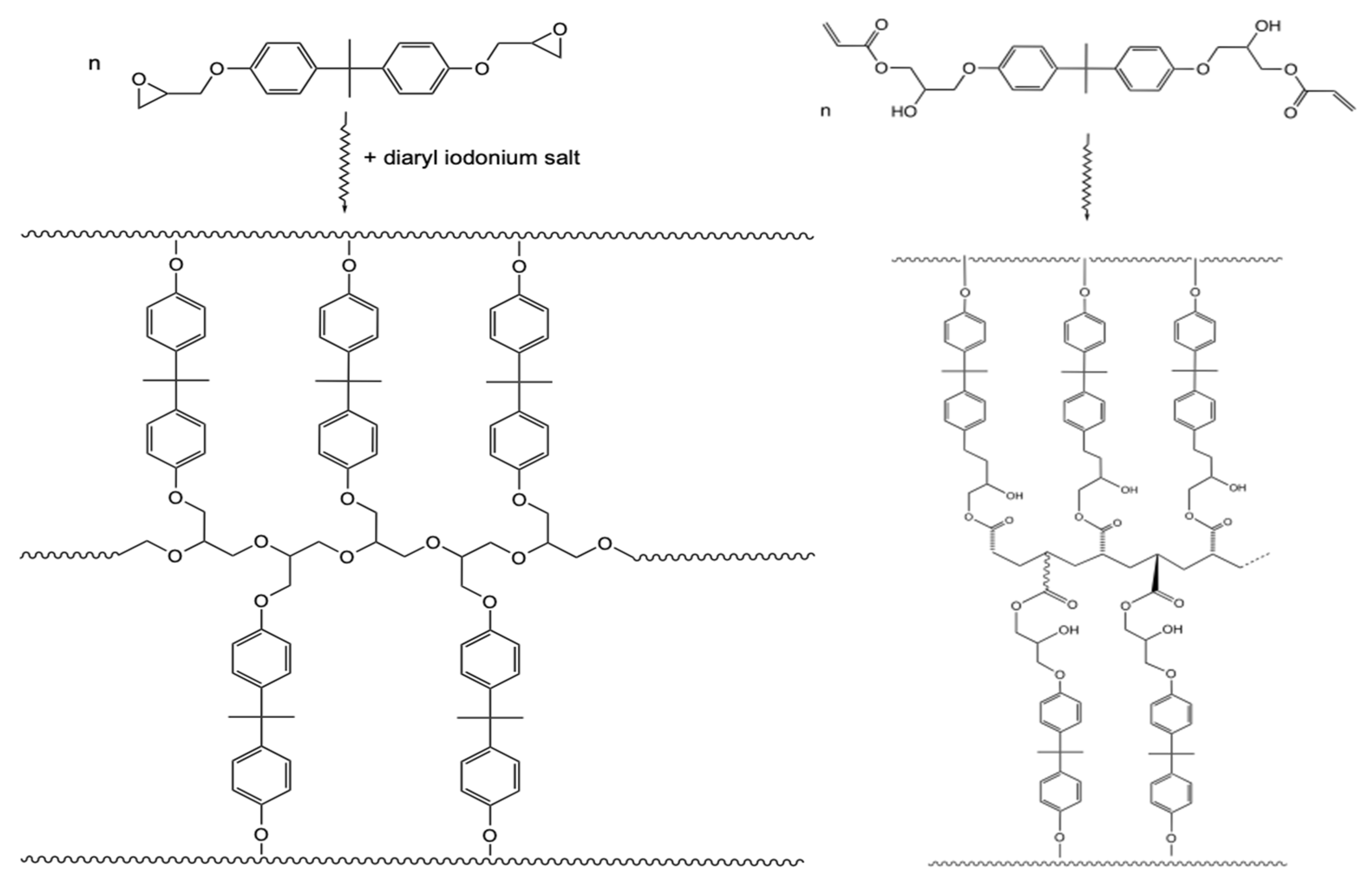{kind=link}
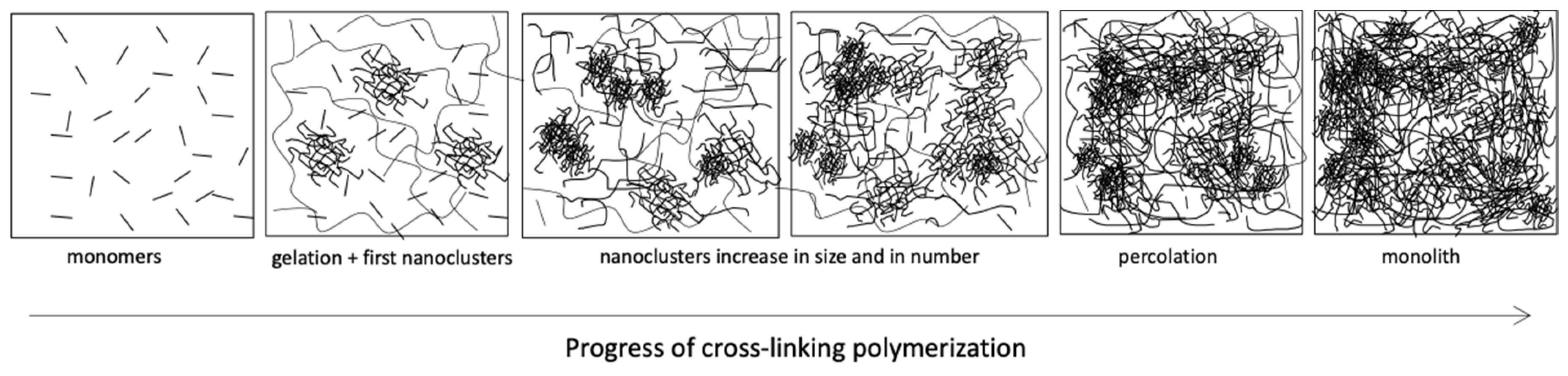{kind=link}
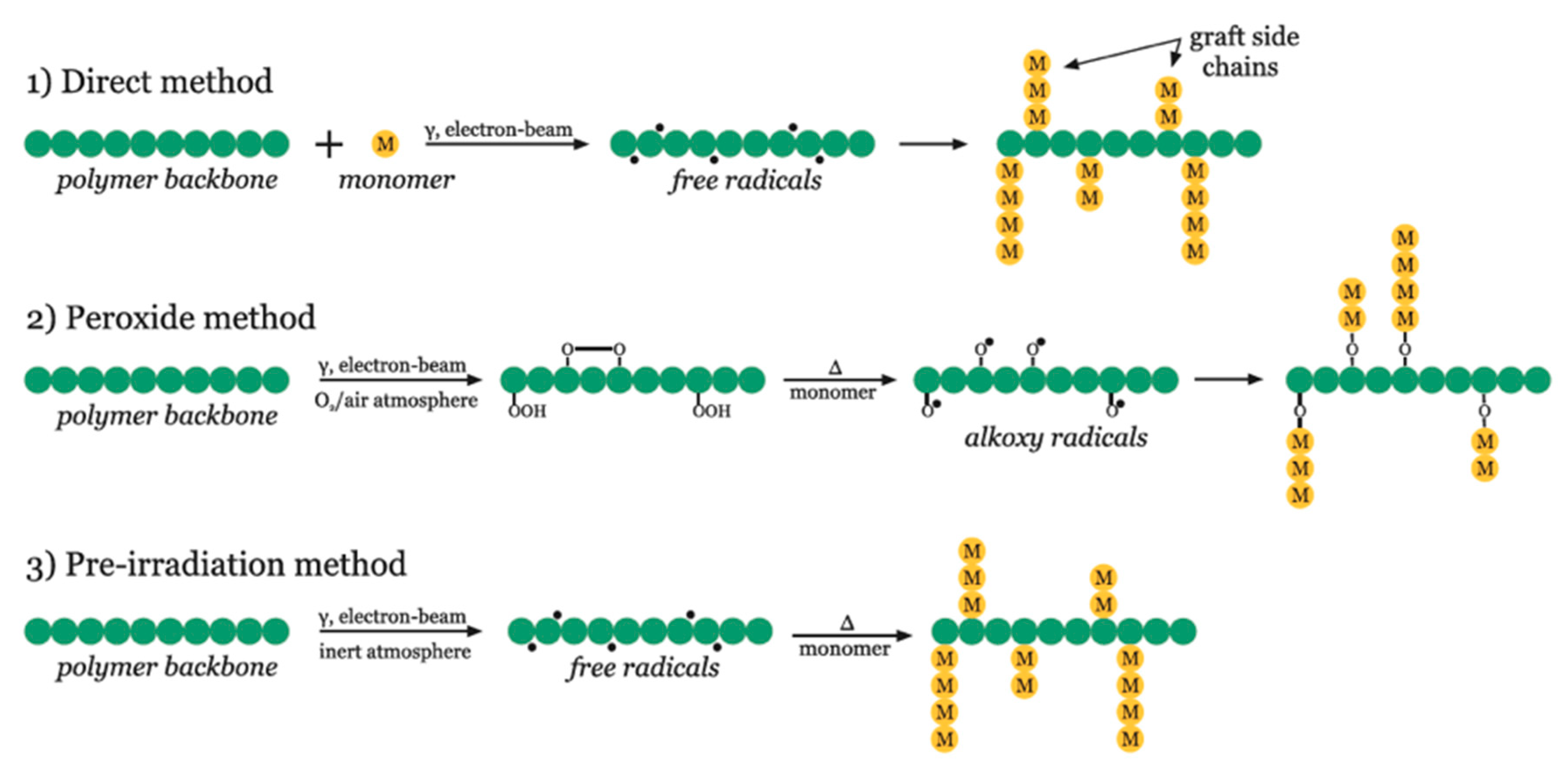{kind=link}
| Radical | –CH2–CFOO.–CH2– | –CF2–C·H–CF2 | –CF2–C·H2 |
|---|---|---|---|
| , | |||
| , | |||
| - | , |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashfaq, A.; Clochard, M.-C.; Coqueret, X.; Dispenza, C.; Driscoll, M.S.; Ulański, P.; Al-Sheikhly, M. Polymerization Reactions and Modifications of Polymers by Ionizing Radiation. Polymers 2020, 12, 2877. https://doi.org/10.3390/polym12122877
Ashfaq A, Clochard M-C, Coqueret X, Dispenza C, Driscoll MS, Ulański P, Al-Sheikhly M. Polymerization Reactions and Modifications of Polymers by Ionizing Radiation. Polymers. 2020; 12(12):2877. https://doi.org/10.3390/polym12122877
Chicago/Turabian StyleAshfaq, Aiysha, Marie-Claude Clochard, Xavier Coqueret, Clelia Dispenza, Mark S. Driscoll, Piotr Ulański, and Mohamad Al-Sheikhly. 2020. "Polymerization Reactions and Modifications of Polymers by Ionizing Radiation" Polymers 12, no. 12: 2877. https://doi.org/10.3390/polym12122877
APA StyleAshfaq, A., Clochard, M.-C., Coqueret, X., Dispenza, C., Driscoll, M. S., Ulański, P., & Al-Sheikhly, M. (2020). Polymerization Reactions and Modifications of Polymers by Ionizing Radiation. Polymers, 12(12), 2877. https://doi.org/10.3390/polym12122877