The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
3. Results
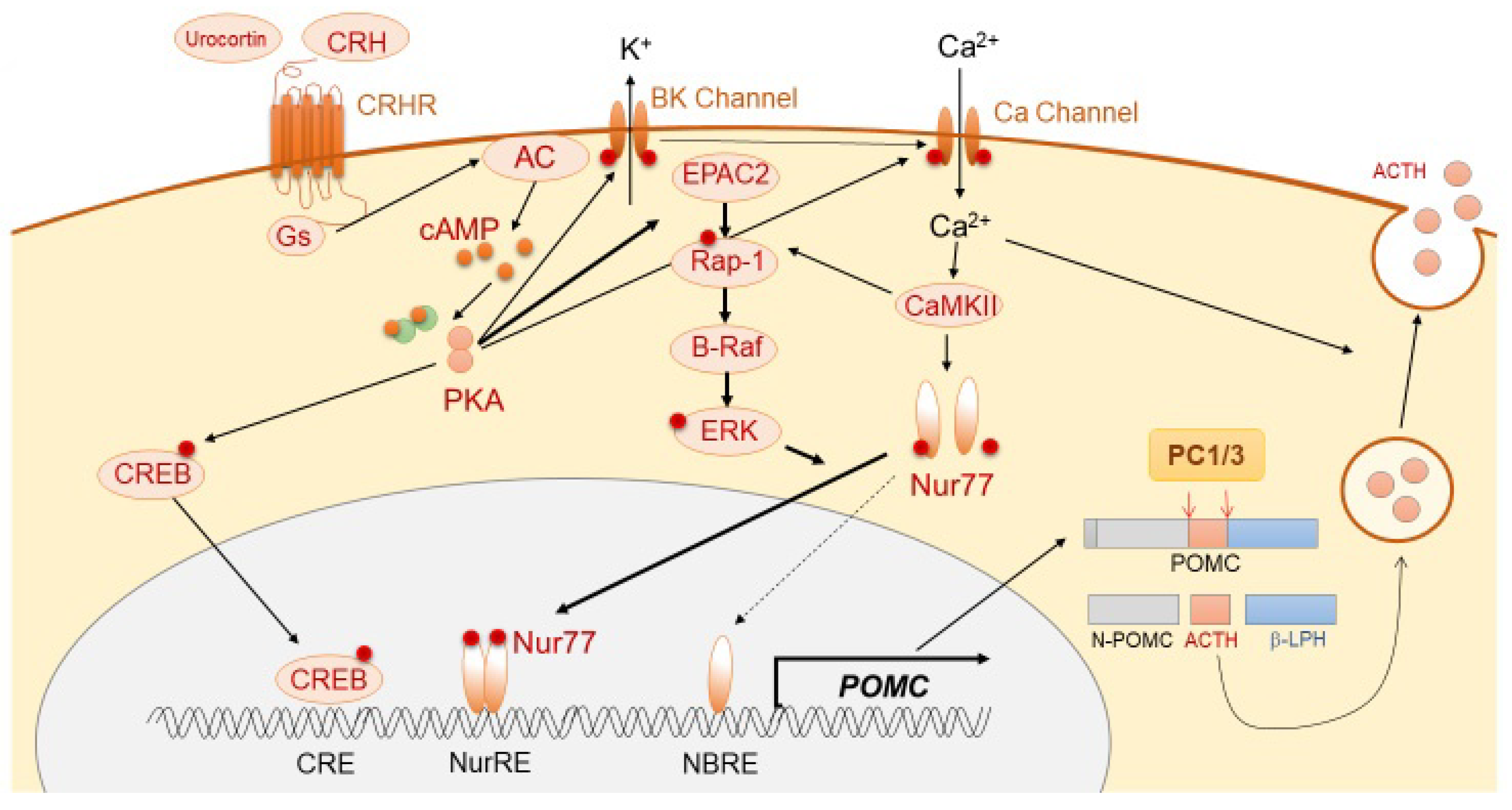
3.1. Physiological Regulation of ACTH
3.1.1. ACTH Synthesis and Secretion
3.1.2. Positive Regulation of ACTH Synthesis and Secretion
3.1.3. Negative Regulation of ACTH Synthesis and Secretion
3.2. Pathological Regulation of ACTH in ACTHomas
3.2.1. ACTHomas Tumorigenesis
3.2.2. ACTH Regulation in ACTHomas
3.3. Pathological Regulation of Acth under Other Conditions
3.3.1. Ectopic ACTH Syndrome
3.3.2. Non-Neoplastic Hypercortisolemia
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AACE | ACTH-converting enzyme |
| ACTH | Adrenocorticotropic hormone |
| ACTHomas | Adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas |
| AgRP | Agouti-related protein |
| AMB | Aminopeptidases B-like |
| ANXA1 | Annexin 1 |
| AVP | Arginine vasopressin |
| β-LPH | β-lipotropin |
| BET | Bromo and extra-terminal domain |
| CaMKII | Calmodulin kinase II |
| CBR | Cannabinoid receptor |
| CPE | Carboxypeptidase E |
| CRH | Corticotrophin-releasing hormone |
| CRHR1 | CRH receptor 1 |
| DDAVP | 1-deamino-8-D-arginine vasopressin |
| Dex | Dexamethasone |
| DST | Dex suppression test |
| EAS | Ectopic ACTH syndrome |
| FIPAs | Familial isolated pituitary adenomas |
| GR | Glucocorticoid receptor |
| GRmb | G protein-coupled glucocorticoid receptor |
| HDAC2 | Histone deacetylase 2 |
| HSP90 | Heat shock protein 90 |
| MEN1 | Multiple endocrine neoplasm type 1 |
| nGRE | Negative glucocorticoid response element |
| NNH | Non-neoplastic hypercortisolemia |
| PAM | Peptidylglycine-amidating monooxygenase |
| PKC | Protein kinase C |
| POMC | Pro-opiomelanocortin |
| PVN | Paraventricular nucleus |
| SgIII | Secretogranin III |
| SON | Supraoptic nuclei |
| SSTR2 | Somatostatin receptor subtype 2 |
| TR4 | Testicular receptor 4 |
References
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, H.; Yamada, S. Cushing’s Disease. J. Clin. Med. 2019, 8, 1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; Montori, V.M.; Edwards, H. The diagnosis of Cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2008, 93, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Drouin, J. 60 Years of POMC: Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef]
- Langlais, D.; Couture, C.; Sylvain-Drolet, G.; Drouin, J. A Pituitary-Specific Enhancer of the POMC Gene with Preferential Activity in Corticotrope Cells. Mol. Endocrinol. 2011, 25, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Vallette-Kasic, S.; Gauthier, Y.; Figarella-Branger, D.; Brue, T.; Berthelet, F.; Lacroix, A.; Batista, D.; Stratakis, C.; Hanson, J.; et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 2006, 20, 2871–2886. [Google Scholar] [CrossRef] [Green Version]
- Cool, D.R.; Normant, E.; Shen, F.; Chen, H.-C.; Pannell, L.; Zhang, Y.; Loh, Y.P. Carboxypeptidase E Is a Regulated Secretory Pathway Sorting Receptor: Genetic Obliteration Leads to Endocrine Disorders in Cpefat Mice. Cell 1997, 88, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Hosaka, M.; Watanabe, T.; Sakai, Y.; Uchiyama, Y.; Takeuchi, T. Identification of a Chromogranin A Domain That Mediates Binding to Secretogranin III and Targeting to Secretory Granules in Pituitary Cells and Pancreatic β-Cells. Mol. Biol. Cell 2002, 13, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Watanabe, T.; Bochimoto, H.; Sakai, Y.; Torii, S.; Takeuchi, T.; Hosaka, M. Multiple Sorting Systems for Secretory Granules Ensure the Regulated Secretion of Peptide Hormones. Traffic 2013, 14, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Cawley, N.X.; Rathod, T.; Young, S.; Lou, H.; Birch, N.; Loh, Y.P. Carboxypeptidase E and Secretogranin III Coordinately Facilitate Efficient Sorting of Proopiomelanocortin to the Regulated Secretory Pathway in AtT20 Cells. Mol. Endocrinol. 2016, 30, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateno, T.; Kato, M.; Tani, Y.; Yoshimoto, T.; Oki, Y.; Hirata, Y. Processing of high-molecular-weight form adrenocorticotropin in human adrenocorticotropin-secreting tumor cell line (DMS-79) after transfection of prohormone convertase 1/3 gene. J. Endocrinol. Investig. 2010, 33, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Marcinkiewicz, M.; Benjannet, S.; Gaspar, L.; Beaubien, G.; Mattei, M.G.; Lazure, C.; Mbikay, M.; Chrétien, M. Cloning and Primary Sequence of a Mouse Candidate Prohormone Convertase PC1 Homologous to PC2, Furin, and Kex2: Distinct Chromosomal Localization and Messenger RNA Distribution in Brain and Pituitary Compared to PC2. Mol. Endocrinol. 1991, 5, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Korner, J.; Chun, J.; Harter, D.; Axel, R. Isolation and functional expression of a mammalian prohormone processing enzyme, murine prohormone convertase 1. Proc. Natl. Acad. Sci. USA 1991, 88, 6834–6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawley, N.X.; Li, Z.; Loh, Y.P. 60 YEARS OF POMC: Biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J. Mol. Endocrinol. 2016, 56, T77–T97. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Mather, J.P.; Morris, P.L.; Bardin, C.W. Expression of pro-opiomelanocortin-like gene in the testis and Leydig cell lines. Ann. N. Y. Acad. Sci. 1984, 438, 659–662. [Google Scholar] [CrossRef]
- Chen, C.L.; Chang, C.C.; Krieger, D.T.; Bardin, C.W. Expression and regulation of proopiomelanocortin-like gene in the ovary and placenta: Comparison with the testis. Endocrinology 1986, 118, 2382–2389. [Google Scholar] [CrossRef]
- Pintar, J.; Schachter, B.; Herman, A.; Durgerian, S.; Krieger, D. Characterization and localization of proopiomelanocortin messenger RNA in the adult rat testis. Science 1984, 225, 632–634. [Google Scholar] [CrossRef]
- Lolait, S.J.; Autelitano, D.J.; Markwick, A.J.; Toh, B.H.; Funder, J.W. Co-expression of vasopressin with β-endorphin and dynorphin in individual cells from the ovaries of Brattleboro and Long-Evans rats: Immunocytochemical studies. Peptides 1986, 7, 267–276. [Google Scholar] [CrossRef]
- DeBold, C.R.; Menefee, J.K.; Nicholson, W.E.; Orth, D.N. Proopiomelanocortin Gene is Expressed in Many Normal Human Tissues and in Tumors not Associated with Ectopic Adrenocorticotropin Syndrome. Mol. Endocrinol. 1988, 2, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.L. 60 YEARS OF POMC: The proopiomelanocortin gene: Discovery, deletion and disease. J. Mol. Endocrinol. 2016, 56, T27–T37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, P.J.; Şengül, S.; Tabak, J.; Ruth, P.; Bertram, R.; Shipston, M.J. Large conductance Ca2+ -activated K+ (BK) channels promote secretagogue-induced transition from spiking to bursting in murine anterior pituitary corticotrophs. J. Physiol. 2015, 593, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaneva-Atanasova, K.; Sherman, A.; van Goor, F.; Stojilkovic, S.S. Mechanism of Spontaneous and Receptor-Controlled Electrical Activity in Pituitary Somatotrophs: Experiments and Theory. J. Neurophysiol. 2007, 98, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Veilleux, R.; Lefevre, G.; Coy, D.H.; Sueiras-Diaz, J.; Schally, A.V. Corticotropin-releasing factor stimulates accumulation of adenosine 3′, 5′-monophosphate in rat pituitary corticotrophs. Science 1982, 216, 1007–1008. [Google Scholar] [CrossRef]
- Abou-Samra, A.B.; Harwood, J.P.; Catt, K.J.; Aguilera, G. Mechanisms of action of CRF and other regulators of ACTH release in pituitary corticotrophs. Ann. N. Y. Acad. Sci. 1987, 512, 67–84. [Google Scholar] [CrossRef]
- Boutillier, A.L.; Gaiddon, C.; Lorang, D.; Roberts, J.L.; Loeffler, J.P. Transcriptional activation of the proopiomelanocortin gene by cyclic AMP-responsive element binding protein. Pituitary 1998, 1, 33–43. [Google Scholar] [CrossRef]
- Maira, M.; Martens, C.; Batsché, E.; Gauthier, Y.; Drouin, J. Dimer-Specific Potentiation of NGFI-B (Nur77) Transcriptional Activity by the Protein Kinase A Pathway and AF-1-Dependent Coactivator Recruitment. Mol. Cell. Biol. 2003, 23, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Jenks, B.G. Regulation of Proopiomelanocortin Gene Expression. Ann. N. Y. Acad. Sci. 2009, 1163, 17–30. [Google Scholar] [CrossRef]
- Lee, A.K.; Tse, A. Mechanism underlying corticotropin-releasing hormone (CRH) triggered cytosolic Ca2+ rise in identified rat corticotrophs. J. Physiol. 1997, 504, 367–378. [Google Scholar] [CrossRef]
- Philips, A.; Lesage, S.; Gingras, R.; Maira, M.H.; Gauthier, Y.; Hugo, P.; Drouin, J. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol. Cell. Biol. 1997, 17, 5946–5951. [Google Scholar] [CrossRef] [Green Version]
- Kovalovsky, D.; Refojo, D.; Liberman, A.C.; Hochbaum, D.; Pereda, M.P.; Coso, O.A.; Stalla, G.K.; Holsboer, F.; Arzt, E. Activation and Induction of NUR77/NURR1 in Corticotrophs by CRH/cAMP: Involvement of Calcium, Protein Kinase A, and MAPK Pathways. Mol. Endocrinol. 2002, 16, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Tabak, J.; Tomaiuolo, M.; Gonzalez-Iglesias, A.E.; Milescu, L.S.; Bertram, R. Fast-Activating Voltage- and Calcium-Dependent Potassium (BK) Conductance Promotes Bursting in Pituitary Cells: A Dynamic Clamp Study. J. Neurosci. 2011, 31, 16855–16863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knepper, M.A.; Kwon, T.-H.; Nielsen, S. Molecular Physiology of Water Balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Wotjak, C.T.; Kubota, M.; Kohl, G.; Landgraf, R. Release of vasopressin from supraoptic neurons within the median eminence in vivo. A combined microdialysis and push-pull perfusion study in the rat. Brain Res. 1996, 726, 237–241. [Google Scholar] [CrossRef]
- Tse, A.; Lee, A.K. Arginine Vasopressin Triggers Intracellular Calcium Release, a Calcium-Activated Potassium Current and Exocytosis in Identified Rat Corticotropes 1. Endocrinology 1998, 139, 2246–2252. [Google Scholar] [CrossRef]
- Raymond, V.; Leung, P.C.K.; Veilleux, R.; Labrie, F. Vasopressin rapidly stimulates phosphatidic acid-phosphatidylinositol turnover in rat anterior pituitary cells. FEBS Lett. 1985, 182, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Makara, G.B.; Mergl, Z.; Zelena, D. The role of vasopressin in hypothalamo-pituitary-adrenal axis activation during stress: An assessment of the evidence. Ann. N. Y. Acad. Sci. 2004, 1018, 151–161. [Google Scholar] [CrossRef]
- Roper, J.; O’Carroll, A.-M.; Young, W.; Lolait, S. The vasopressin Avpr1b receptor: Molecular and pharmacological studies. Stress 2011, 14, 98–115. [Google Scholar] [CrossRef]
- DeBold, C.R.; Sheldon, W.R.; DeCherney, G.S.; Jackson, R.V.; Alexander, A.N.; Vale, W.; Rivier, J.; Orth, D.N. Arginine vasopressin potentiates adrenocorticotropin release induced by ovine corticotropin-releasing factor. J. Clin. Investig. 1984, 73, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Lamberts, S.W.; Verleun, T.; Oosterom, R.; de Jong, F.; Hackeng, W.H. Corticotropin-releasing factor (ovine) and vasopressin exert a synergistic effect on adrenocorticotropin release in man. J. Clin. Endocrinol. Metab. 1984, 58, 298–303. [Google Scholar] [CrossRef]
- Turnbull, A.V.; Rivier, C.L. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: Actions and mechanisms of action. Physiol. Rev. 1999, 79, 1–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ren, S.G.; Melmed, S. Hypothalamic and pituitary leukemia inhibitory factor gene expression in vivo: A novel endotoxin-inducible neuro-endocrine interface. Endocrinology 1996, 137, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, C.; Melmed, S. Critical Role for STAT3 in Murine Pituitary Adrenocorticotropin Hormone Leukemia Inhibitory Factor Signaling. J. Biol. Chem. 1999, 274, 10723–10730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousquet, C.; Zatelli, M.C.; Melmed, S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 2000, 106, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Kameda, H.; Yamamoto, M.; Tone, Y.; Tone, M.; Melmed, S. Proton Sensitivity of Corticotropin-Releasing Hormone Receptor 1 Signaling to Proopiomelanocortin in Male Mice. Endocrinology 2019, 160, 276–291. [Google Scholar] [CrossRef] [Green Version]
- Asaba, K.; Makino, S.; Hashimoto, K. Effect of urocortin on ACTH secretion from rat anterior pituitary in vitro and in vivo: Comparison with corticotropin-releasing hormone. Brain Res. 1998, 806, 95–103. [Google Scholar] [CrossRef]
- Vaughan, J.; Donaldson, C.; Bittencourt, J.; Perrin, M.H.; Lewis, K.; Sutton, S.; Chan, R.; Turnbull, A.V.; Lovejoy, D.; Rivier, C.; et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature 1995, 378, 287–292. [Google Scholar] [CrossRef]
- Spiga, F.; Walker, J.J.; Gupta, R.; Terry, J.R.; Lightman, S.L. Glucocorticoid dynamics: Insights from mathematical, experimental and clinical studies. J. Endocrinol. 2015, 226, T55–T66. [Google Scholar] [CrossRef] [Green Version]
- Di, S.; Malcher-Lopes, R.; Halmos, K.C.; Tasker, J.G. Nongenomic Glucocorticoid Inhibition via Endocannabinoid Release in the Hypothalamus: A Fast Feedback Mechanism. J. Neurosci. 2003, 23, 4850–4857. [Google Scholar] [CrossRef] [Green Version]
- Malkoski, S.P.; Dorin, R.I. Composite Glucocorticoid Regulation at a Functionally Defined Negative Glucocorticoid Response Element of the Human Corticotropin-Releasing Hormone Gene. Mol. Endocrinol. 1999, 13, 1629–1644. [Google Scholar] [CrossRef]
- Yamamori, E.; Iwasaki, Y.; Taguchi, T.; Nishiyama, M.; Yoshida, M.; Asai, M.; Oiso, Y.; Itoi, K.; Kambayashi, M.; Hashimoto, K. Molecular mechanisms for corticotropin-releasing hormone gene repression by glucocorticoid in BE(2)C neuronal cell line. Mol. Cell. Endocrinol. 2007, 264, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Duncan, P.J.; Tabak, J.; Ruth, P.; Bertram, R.; Shipston, M.J. Glucocorticoids Inhibit CRH/AVP-Evoked Bursting Activity of Male Murine Anterior Pituitary Corticotrophs. Endocrinology 2016, 157, 3108–3121. [Google Scholar] [CrossRef] [Green Version]
- Chapman, L.P.; Epton, M.J.; Buckingham, J.C.; Morris, J.F.; Christian, H.C. Evidence for a Role of the Adenosine 5′-Triphosphate-Binding Cassette Transporter A1 in the Externalization of Annexin I from Pituitary Folliculo-Stellate Cells. Endocrinology 2003, 144, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Drouin, J.; Trifiro, M.A.; Plante, R.K.; Nemer, M.; Eriksson, P.; Wrange, O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol. Cell. Biol. 1989, 9, 5305–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, R.; Saito-Hakoda, A.; Shimada, H.; Shimizu, K.; Noro, E.; Iwasaki, Y.; Fujiwara, K.; Yokoyama, A.; Sugawara, A. Role of NeuroD1 on the negative regulation of Pomc expression by glucocorticoid. PLoS ONE 2017, 12, e0175435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.J.M.; Cidlowski, J.A. Molecular mechanisms of glucocorticoid action and resistance. J. Steroid Biochem. Mol. Biol. 2002, 83, 37–48. [Google Scholar] [CrossRef]
- Martens, C.; Bilodeau, S.; Maira, M.; Gauthier, Y.; Drouin, J. Protein-Protein Interactions and Transcriptional Antagonism between the Subfamily of NGFI-B/Nur77 Orphan Nuclear Receptors and Glucocorticoid Receptor. Mol. Endocrinol. 2005, 19, 885–897. [Google Scholar] [CrossRef]
- Giacomini, D.; Páez-Pereda, M.; Theodoropoulou, M.; Labeur, M.; Refojo, D.; Gerez, J.; Chervin, A.; Berner, S.; Losa, M.; Buchfelder, M.; et al. Bone Morphogenetic Protein-4 Inhibits Corticotroph Tumor Cells: Involvement in the Retinoic Acid Inhibitory Action. Endocrinology 2006, 147, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Nudi, M.; Ouimette, J.-F.; Drouin, J. Bone Morphogenic Protein (Smad)-Mediated Repression of Proopiomelanocortin Transcription by Interference with Pitx/Tpit Activity. Mol. Endocrinol. 2005, 19, 1329–1342. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, N.; Otsuka, F.; Ogura-Ochi, K.; Inagaki, K.; Nakamura, E.; Toma, K.; Terasaka, T.; Iwasaki, Y.; Makino, H. Melatonin receptor activation suppresses adrenocorticotropin production via BMP-4 action by pituitary AtT20 cells. Mol. Cell. Endocrinol. 2013, 375, 1–9. [Google Scholar] [CrossRef]
- Brown, M.R.; Rivier, C.; Vale, W. Central Nervous System Regulation of Adrenocorticotropin Secretion: Role of Somatostatins *. Endocrinology 1984, 114, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Kraicer, J.; Gajewski, T.C.; Moor, B.C. Release of Pro-Opiomelanocortin-Derived Peptides from the Pars intermedia and Pars distalis of the Rat Pituitary: Effect of Corticotrophin-Releasing Factor and Somatostatin. Neuroendocrinology 1985, 41, 363–373. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, C.; Feelders, R.A.; Lamberts, S.W.J.; Hofland, L.J. Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s Syndrome. Rev. Endocr. Metab. Disord. 2009, 10, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocrinol. 1999, 20, 157–198. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Schonbrunn, A.; Armstrong, D.L. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature 1991, 351, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B.; Dahl, R.; Sherman, N.; Sussman, K.E.; Staehelin, L.A. Exocytosis in normal anterior pituitary cells. Quantitative correlation between growth hormone release and the morphological features of exocytosis. J. Clin. Investig. 1988, 81, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Ben-Shlomo, A.; Kameda, H.; Fukuoka, H.; Deng, N.; Ding, Y.; Melmed, S. Somatostatin receptor subtype 5 modifies hypothalamic-pituitary-adrenal axis stress function. JCI Insight 2018, 3, e122932. [Google Scholar] [CrossRef]
- Schulte, H.M.; Oldfield, E.H.; Allolio, B.; Katz, D.A.; Berkman, R.A.; Ali, I.U. Clonal composition of pituitary adenomas in patients with Cushing’s disease: Determination by X-chromosome inactivation analysis. J. Clin. Endocrinol. Metab. 1991, 73, 1302–1308. [Google Scholar] [CrossRef]
- Gicquel, C.; Le Bouc, Y.; Luton, J.P.; Girard, F.; Bertagna, X. Monoclonality of corticotroph macroadenomas in Cushing’s disease. J. Clin. Endocrinol. Metab. 1992, 75, 472–475. [Google Scholar]
- Biller, B.M.; Alexander, J.M.; Zervas, N.T.; Hedley-Whyte, E.T.; Arnold, A.; Klibanski, A. Clonal origins of adrenocorticotropin-secreting pituitary tissue in Cushing’s disease. J. Clin. Endocrinol. Metab. 1992, 75, 1303–1309. [Google Scholar]
- Guru, S.C.; Manickam, P.; Crabtree, J.S.; Olufemi, S.E.; Agarwal, S.K.; Debelenko, L.V. Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. J. Intern. Med. 1998, 243, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Vanbellinghen, J.-F.; Khoo, S.K.; Jaffrain-Rea, M.-L.; Naves, L.A.; Guitelman, M.A.; Murat, A.; Emy, P.; Gimenez-Roqueplo, A.-P.; Tamburrano, G.; et al. Aryl Hydrocarbon Receptor-Interacting Protein Gene Mutations in Familial Isolated Pituitary Adenomas: Analysis in 73 Families. J. Clin. Endocrinol. Metab. 2007, 92, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Chahal, H.S.; King, P.; Bolger, G.B.; Srirangalingam, U.; Guasti, L.; Chapple, J.P.; Trivellin, G.; Gueorguiev, M.; Guegan, K.; et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum. Mutat. 2010, 31, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-Y.; Song, Z.-J.; Chen, J.-H.; Wang, Y.-F.; Li, S.-Q.; Zhou, L.-F.; Mao, Y.; Li, Y.-M.; Hu, R.-G.; Zhang, Z.-Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: Evidence for a role in corticotropinoma cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, H.; Cooper, O.; Ben-Shlomo, A.; Mamelak, A.; Ren, S.-G.; Bruyette, D.; Melmed, S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Investig. 2011, 121, 4712–4721. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Liu, X.; Kameda, H.; Tone, Y.; Fukuoka, H.; Tone, M.; Melmed, S. EGFR Induces E2F1-Mediated Corticotroph Tumorigenesis. J. Endocr. Soc. 2017, 1, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Persky, R.; Stegemann, R.; Hernández-Ramírez, L.C.; Zeltser, D.; Lodish, M.B.; Chen, A.; Keil, M.F.; Tatsi, C.; Faucz, F.R.; et al. Germline USP8 Mutation Associated With Pediatric Cushing Disease and Other Clinical Features: A New Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4676–4682. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [Green Version]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.-M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro. Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Kunz, M.; Weigand, I.; Deutschbein, T.; Dandekar, T.; Fassnacht, M. The New Genetic Landscape of Cushing’s Disease: Deubiquitinases in the Spotlight. Cancers 2019, 11, 1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, H.L.; Merchant, T.E.; Warmuth-Metz, M.; Martinez-Barbera, J.-P.; Puget, S. Craniopharyngioma. Nat. Rev. Dis. Prim. 2019, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Haugh, A.M.; Johnson, D.B. Management of V600E and V600K BRAF-Mutant Melanoma. Curr. Treat. Options Oncol. 2019, 20, 81. [Google Scholar] [CrossRef]
- Fugazzola, L.; Muzza, M.; Pogliaghi, G.; Vitale, M. Intratumoral Genetic Heterogeneity in Papillary Thyroid Cancer: Occurrence and Clinical Significance. Cancers 2020, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Maraka, S.; Janku, F. BRAF alterations in primary brain tumors. Discov. Med. 2018, 26, 51–60. [Google Scholar]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Ramírez, L.C.; Gam, R.; Valdés, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R.; Trivellin, G.; Chittiboina, P.; et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing’s disease. Endocr. Relat. Cancer 2017, 24, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Solarski, M.; Rotondo, F.; Foulkes, W.D.; Priest, J.R.; Syro, L.V.; Butz, H.; Cusimano, M.D.; Kovacs, K. DICER1 gene mutations in endocrine tumors. Endocr. Relat. Cancer 2018, 25, R197–R208. [Google Scholar] [CrossRef]
- De Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.-H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014, 128, 111–122. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.; Tichomirowa, M.; Boikos, S.; Azevedo, M.; Lodish, M.; Martari, M.; Verma, S.; Daly, A.; Raygada, M.; Keil, M.; et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin. Genet. 2010, 78, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin, J.; Bilodeau, S.; Vallette, S. Of old and new diseases: Genetics of pituitary ACTH excess (Cushing) and deficiency. Clin. Genet. 2007, 72, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S.W. Glucocorticoid receptors and Cushing’s disease. Mol. Cell. Endocrinol. 2002, 197, 69–72. [Google Scholar] [CrossRef]
- Korbonits, M.; Bujalska, I.; Shimojo, M.; Nobes, J.; Jordan, S.; Grossman, A.B.; Stewart, P.M. Expression of 11β-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: Induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. J. Clin. Endocrinol. Metab. 2001, 86, 2728–2733. [Google Scholar] [CrossRef] [Green Version]
- Tateno, T.; Izumiyama, H.; Doi, M.; Yoshimoto, T.; Shichiri, M.; Inoshita, N.; Oyama, K.; Yamada, S.; Hirata, Y. Differential gene expression in ACTH -secreting and non-functioning pituitary tumors. Eur. J. Endocrinol. 2007, 157, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Riebold, M.; Kozany, C.; Freiburger, L.; Sattler, M.; Buchfelder, M.; Hausch, F.; Stalla, G.K.; Paez-Pereda, M. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat. Med. 2015, 21, 276–280. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 641–649. [Google Scholar] [CrossRef]
- Pratt, W.B.; Galigniana, M.D.; Harrell, J.M.; DeFranco, D.B. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell. Signal. 2004, 16, 857–872. [Google Scholar] [CrossRef]
- Lu, J.; Chatain, G.P.; Bugarini, A.; Wang, X.; Maric, D.; Walbridge, S.; Zhuang, Z.; Chittiboina, P. Histone deacetylase inhibitor SAHA is a promising treatment of Cushing disease. J. Clin. Endocrinol. Metab. 2017, 102, 2825–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Du, L.; Heaney, A.P. Testicular Receptor-4: Novel Regulator of Glucocorticoid Resistance. J. Clin. Endocrinol. Metab. 2016, 101, 3123–3133. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Bergsneider, M.; Mirsadraei, L.; Young, S.H.; Jonker, J.W.; Downes, M.; Yong, W.H.; Evans, R.M.; Heaney, A.P. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8555–8560. [Google Scholar] [CrossRef] [Green Version]
- Roussel-Gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-Branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque, R.M.; Ibáñez-Costa, A.; López-Sánchez, L.M.; Jiménez-Reina, L.; Venegas-Moreno, E.; Gálvez, M.A.; Villa-Osaba, A.; Madrazo-Atutxa, A.M.; Japón, M.A.; de la Riva, A.; et al. A Cellular and Molecular Basis for the Selective Desmopressin-Induced ACTH Release in Cushing Disease Patients: Key Role of AVPR1b Receptor and Potential Therapeutic Implications. J. Clin. Endocrinol. Metab. 2013, 98, 4160–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.-F.; Tang, K.-T.; Yen, Y.-S.; Ho, D.M.-T.; Yang, A.-H.; Hwang, C.-I.; Lin, H.-D.; Won, J.G.S. Plasma corticotrophin response to desmopressin in patients with Cushing’s disease correlates with the expression of vasopressin receptor 2, but not with that of vasopressin receptor 1 or 3, in their pituitary tumours. Clin. Endocrinol. 2012, 76, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Damoiseaux, R.; Babayan, L.; Rivera-Meza, E.K.; Yang, Y.; Bergsneider, M.; Wang, M.B.; Yong, W.H.; Kelly, K.; Heaney, A.P. Targeting Corticotroph HDAC and PI3-Kinase in Cushing Disease. J. Clin. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Lines, K.E.; Filippakopoulos, P.; Stevenson, M.; Müller, S.; Lockstone, H.E.; Wright, B.; Knapp, S.; Buck, D.; Bountra, C.; Thakker, R. V Effects of epigenetic pathway inhibitors on corticotroph tumour AtT20 cells. Endocr. Relat. Cancer 2020, 27, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Sharma, S.T.; Nieman, L.K.; Feelders, R.A. Cushing’s syndrome: Epidemiology and developments in disease management. Clin. Epidemiol. 2015, 7, 281–293. [Google Scholar]
- Isidori, A.M.; Kaltsas, G.A.; Pozza, C.; Frajese, V.; Newell-Price, J.; Reznek, R.H.; Jenkins, P.J.; Monson, J.P.; Grossman, A.B.; Besser, G.M. Extensive clinical experience—The ectopic adrenocorticotropin syndrome: Clinical features, diagnosis, management, and long-term follow-up. J. Clin. Endocrinol. Metab. 2006, 91, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Aniszewski, J.P.; Young, W.F.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A. Gushing syndrome doe to ectopic adrenocorticotropic hormone secretion. World J. Surg. 2001, 25, 934–940. [Google Scholar] [CrossRef]
- Kent Jex, R.; van Heerden, J.A.; Carpenter, P.C.; Grant, C.S. Ectopic ACTH syndrome. Am. J. Surg. 1985, 149, 276–282. [Google Scholar] [CrossRef]
- Howlett, T.A.; Drury, P.L.; Perry, L.; Doniach, I.; Rees, L.H.; Besser, G.M. Diagnosis and management of ACTH-dependent Cushing’s syndrome: Comparison of the features in ectopic and pituitary ACTH production. Clin. Endocrinol. 1986, 24, 699–713. [Google Scholar] [CrossRef]
- Doppman, J.L.; Nieman, L.; Miller, D.L.; Pass, H.I.; Chang, R.; Cutler, G.B.; Schaaf, M.; Chrousos, G.P.; Norton, J.A.; Ziessman, H.A. Ectopic adrenocorticotropic hormone syndrome: Localization studies in 28 patients. Radiology 1989, 172, 115–124. [Google Scholar] [CrossRef]
- Tabarin, A.; Valli, N.; Chanson, P.; Bachelot, Y.; Rohmer, V.; Bex-Bachellerie, V.; Catargi, B.; Roger, P.; Laurent, F. Usefulness of Somatostatin Receptor Scintigraphy in Patients with Occult Ectopic Adrenocorticotropin Syndrome. J. Clin. Endocrinol. Metab. 1999, 84, 1193–1202. [Google Scholar] [CrossRef]
- Torpy, D.J.; Mullen, N.; Ilias, I.; Nieman, L.K. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: A series of 58 cases. Ann. N. Y. Acad. Sci. 2002, 970, 134–144. [Google Scholar] [CrossRef]
- Ilias, I.; Torpy, D.J.; Pacak, K.; Mullen, N.; Wesley, R.A.; Nieman, L.K. Cushing’s Syndrome Due to Ectopic Corticotropin Secretion: Twenty Years’ Experience at the National Institutes of Health. J. Clin. Endocrinol. Metab. 2005, 90, 4955–4962. [Google Scholar] [CrossRef] [Green Version]
- Hernández, I.; Espinosa-de-los-Monteros, A.L.; Mendoza, V.; Cheng, S.; Molina, M.; Sosa, E.; Mercado, M. Ectopic ACTH-Secreting Syndrome: A Single Center Experience Report with a High Prevalence of Occult Tumor. Arch. Med. Res. 2006, 37, 976–980. [Google Scholar] [CrossRef]
- Salgado, L.R.; Fragoso, M.C.B.V.; Knoepfelmacher, M.; Machado, M.C.; Domenice, S.; Pereira, M.A.A.; de Mendonça, B.B. Ectopic ACTH syndrome: Our experience with 25 cases. Eur. J. Endocrinol. 2006, 155, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Wajchenberg, B.L.; Mendonca, B.B.; Liberman, B.; Pereira, M.A.; Carneiro, P.C.; Wakamatsu, A.; Kirschner, M.A. Ectopic adrenocorticotropic hormone syndrome. Endocr. Rev. 1994, 15, 752–787. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Jiang, X.; Cheng, Y.; Zhou, W.; Su, T.; Xie, J.; Zhong, X.; Song, D.; Wu, L.; et al. Whole exome sequencing of thymic neuroendocrine tumor with ectopic ACTH syndrome. Eur. J. Endocrinol. 2017, 176, 187–194. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Liu, N.-A.; Tone, Y.; Cuevas-Ramos, D.; Heltsley, R.; Tone, M.; Melmed, S. E2F1-mediated human POMC expression in ectopic Cushing’s syndrome. Endocr. Relat. Cancer 2016, 23, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, I.; Higuchi, S.; Fujimoto, M.; Takiguchi, T.; Nakayama, A.; Tamura, A.; Kohno, T.; Komai, E.; Shiga, A.; Nagano, H.; et al. Cushing Syndrome Due to ACTH-Secreting Pheochromocytoma, Aggravated by Glucocorticoid-Driven Positive-Feedback Loop. J. Clin. Endocrinol. Metab. 2016, 101, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Oliver, R.L.; Davis, J.R.E.; White, A. Characterisation of ACTH Related Peptides in Ectopic Cushing’s Syndrome. Pituitary 2003, 6, 119–126. [Google Scholar] [CrossRef]
- Raffin-Sanson, M.L.; Massias, J.F.; Dumont, C.; Raux-Demay, M.C.; Proeschel, M.F.; Luton, J.P.; Bertagna, X. High plasma proopiomelanocortin in aggressive adrenocorticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1996, 81, 4272–4277. [Google Scholar]
- Scopsi, L.; Gullo, M.; Rilke, F.; Martin, S.; Steiner, D.F. Proprotein convertases (PC1/PC3 and PC2) in normal and neoplastic human tissues: Their use as markers of neuroendocrine differentiation. J. Clin. Endocrinol. Metab. 1995, 80, 294–301. [Google Scholar]
- Vieau, D.; Seidah, N.G.; Mbikay, M.; Chrétien, M.; Bertagna, X. Expression of the prohormone convertase PC2 correlates with the presence of corticotropin-like intermediate lobe peptide in human adrenocorticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1994, 79, 1503–1506. [Google Scholar]
- Kimura, N.; Ishikawa, T.; Sasaki, Y.; Sasano, N.; Onodera, K.; Shimizu, Y.; Kimura, I.; Steiner, D.F.; Nagura, H. Expression of prohormone convertase, PC2, in adrenocorticotropin-producing thymic carcinoid with elevated plasma corticotropin-releasing hormone. J. Clin. Endocrinol. Metab. 1996, 81, 390–395. [Google Scholar] [PubMed] [Green Version]
- Page-Wilson, G.; Freda, P.U.; Jacobs, T.P.; Khandji, A.G.; Bruce, J.N.; Foo, S.T.; Meece, K.; White, A.; Wardlaw, S.L. Clinical Utility of Plasma POMC and AgRP Measurements in the Differential Diagnosis of ACTH-Dependent Cushing’s Syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E1838–E1845. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.W.; Littlewood, A.C.; Clark, A.J.; Davis, J.R.; White, A. Human small cell lung cancer cell lines expressing the proopiomelanocortin gene have aberrant glucocorticoid receptor function. J. Clin. Investig. 1994, 93, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Tozawa, F.; Dobashi, I.; Horiba, N.; Ohmori, N.; Yamakado, M.; Yamada, M.; Demura, H. Corticotropin-releasing hormone, proopiomelanocortin, and glucocorticoid receptor gene expression in adrenocorticotropin-producing tumors in vitro. J. Clin. Investig. 1993, 92, 2790–2795. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.C.; Valeria de Sa, S.; Correa-Giannella, M.L.; Giorgi, R.R.; Pereira, M.A.A.; Cescato, V.A.S.; Giannella-Neto, D.; Salgado, L.R. Association between tumoral GH-releasing peptide receptor type 1a mRNA expression and in vivo response to GH-releasing peptide-6 in ACTH-dependent Cushing’s syndrome patients. Eur. J. Endocrinol. 2008, 158, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Tsagarakis, S.; Tsigos, C.; Vasiliou, V.; Tsiotra, P.; Kaskarelis, J.; Sotiropoulou, C.; Raptis, S.A.; Thalassinos, N. The Desmopressin and Combined CRH-Desmopressin Tests in the Differential Diagnosis of ACTH-Dependent Cushing’s Syndrome: Constraints Imposed by the Expression of V2 Vasopressin Receptors in Tumors with Ectopic ACTH Secretion. J. Clin. Endocrinol. Metab. 2002, 87, 1646–1653. [Google Scholar]
- De Keyzer, Y.; Lenne, F.; Auzan, C.; Jégou, S.; René, P.; Vaudry, H.; Kuhn, J.M.; Luton, J.P.; Clauser, E.; Bertagna, X. The pituitary V3 vasopressin receptor and the corticotroph phenotype in ectopic ACTH syndrome. J. Clin. Investig. 1996, 97, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L.; Ahmed-Shuaib, A.; Jacobs, R.A.; Chew, S.L.; Honegger, J.; Fahlbusch, R.; Besser, G.M.; Grossman, A.B. Vasopressin receptor expression and mutation analysis in corticotropin-secreting tumors. J. Clin. Endocrinol. Metab. 1996, 81, 1768–1771. [Google Scholar] [PubMed] [Green Version]
- Arlt, W.; Dahia, P.L.M.; Callies, F.; Nordmeyer, J.P.; Allolio, B.; Grossman, A.B.; Reincke, M. Ectopic ACTH production by a bronchial carcinoid tumour responsive to desmopressin in vivo and in vitro. Clin. Endocrinol. 1997, 47, 623–627. [Google Scholar] [CrossRef]
- Tani, Y.; Sugiyama, T.; Hirooka, S.; Izumiyama, H.; Hirata, Y. Ectopic ACTH syndrome caused by bronchial carcinoid tumor indistinguishable from Cushing’s disease. Endocr. J. 2010, 57, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Findling, J.W.; Raff, H. Diagnosis of Endocrine Disease: Differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur. J. Endocrinol. 2017, 176, R205–R216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wand, G.S.; Dobs, A.S. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J. Clin. Endocrinol. Metab. 1991, 72, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Imaki, T.; Vale, W. Prolonged exposure to alcohol: Effect on CRF mRNA levels, and CRF- and stress-induced ACTH secretion in the rat. Brain Res. 1990, 520, 1–5. [Google Scholar] [CrossRef]
- Ogilvie, K.M.; Lee, S.; Rivier, C. Role of arginine vasopressin and corticotropin-releasing factor in mediating alcohol-induced adrenocorticotropin and vasopressin secretion in male rats bearing lesions of the paraventricular nuclei. Brain Res. 1997, 744, 83–95. [Google Scholar] [CrossRef]
- Rees, L.; Besser, G.; Jeffcoate, W.; Goldie, D.; Marks, V. Alcohol-Induced Pseudo-Cushing’s Syndrome. Lancet 1977, 309, 726–728. [Google Scholar] [CrossRef]
- Jacobson, L. Hypothalamic-pituitary-adrenocortical axis: Neuropsychiatric aspects. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 4, pp. 715–738. [Google Scholar]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef]
- Nelson, J.C.; Davis, J.M. DST Studies in Psychotic Depression: A Meta-Analysis. Am. J. Psychiatry 1997, 154, 1497–1503. [Google Scholar] [CrossRef]
- Holsboer, F. How can we realize the promise of personalized antidepressant medicines? Nat. Rev. Neurosci. 2008, 9, 638–646. [Google Scholar] [CrossRef]
- Reynolds, P.D.; Ruan, Y.; Smith, D.F.; Scammell, J.G. Glucocorticoid Resistance in the Squirrel Monkey Is Associated with Overexpression of the Immunophilin FKBP51 1. J. Clin. Endocrinol. Metab. 1999, 84, 663–669. [Google Scholar] [CrossRef]
- Scammell, J.G.; Denny, W.B.; Valentine, D.L.; Smith, D.F. Overexpression of the FK506-Binding Immunophilin FKBP51 Is the Common Cause of Glucocorticoid Resistance in Three New World Primates. Gen. Comp. Endocrinol. 2001, 124, 152–165. [Google Scholar] [CrossRef]
- Binder, E.B.; Salyakina, D.; Lichtner, P.; Wochnik, G.M.; Ising, M.; Pütz, B.; Papiol, S.; Seaman, S.; Lucae, S.; Kohli, M.A.; et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 2004, 36, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Hackett, R.A.; Steptoe, A.; Kumari, M. Association of Diurnal Patterns in Salivary Cortisol with Type 2 Diabetes in the Whitehall II Study. J. Clin. Endocrinol. Metab. 2014, 99, 4625–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Bravata, D.M.; Cabaccan, J.; Raff, H.; Ryzen, E. Elevated late-night salivary cortisol levels in elderly male type 2 diabetic veterans. Clin. Endocrinol. 2005, 63, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Constantinopoulos, P.; Michalaki, M.; Kottorou, A.; Habeos, I.; Psyrogiannis, A.; Kalfarentzos, F.; Kyriazopoulou, V. Cortisol in tissue and systemic level as a contributing factor to the development of metabolic syndrome in severely obese patients. Eur. J. Endocrinol. 2015, 172, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Rabadan-Diehl, C.; Makara, G.; Kiss, A.; Lolait, S.; Zelena, D.; Ochedalski, T.; Aguilera, G. Regulation of Pituitary V1b Vasopressin Receptor Messenger Ribonucleic Acid by Adrenalectomy and Glucocorticoid Administration. Endocrinology 1997, 138, 5189–5194. [Google Scholar] [CrossRef]
- Dahia, P.L.M.; Grossman, A.B. The Molecular Pathogenesis of Corticotroph Tumors. Endocr. Rev. 1999, 20, 136–155. [Google Scholar] [CrossRef]
- Pecori Giraldi, F.; Pivonello, R.; Ambrogio, A.G.; De Martino, M.C.; De Martin, M.; Scacchi, M.; Colao, A.; Toja, P.M.; Lombardi, G.; Cavagnini, F. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test and the desmopressin test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. Clin. Endocrinol. 2007, 66, 251–257. [Google Scholar] [CrossRef]
- Hundt, W.; Zimmermann, U.; Pöttig, M.; Spring, K.; Holsboer, F. The combined dexamethasone-suppression/CRH-stimulation test in alcoholics during and after acute withdrawal. Alcohol. Clin. Exp. Res. 2001, 25, 687–691. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuoka, H.; Shichi, H.; Yamamoto, M.; Takahashi, Y. The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. Int. J. Mol. Sci. 2020, 21, 9132. https://doi.org/10.3390/ijms21239132
Fukuoka H, Shichi H, Yamamoto M, Takahashi Y. The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. International Journal of Molecular Sciences. 2020; 21(23):9132. https://doi.org/10.3390/ijms21239132
Chicago/Turabian StyleFukuoka, Hidenori, Hiroki Shichi, Masaaki Yamamoto, and Yutaka Takahashi. 2020. "The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease" International Journal of Molecular Sciences 21, no. 23: 9132. https://doi.org/10.3390/ijms21239132
APA StyleFukuoka, H., Shichi, H., Yamamoto, M., & Takahashi, Y. (2020). The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. International Journal of Molecular Sciences, 21(23), 9132. https://doi.org/10.3390/ijms21239132