How Far Are We from Prescribing Fasting as Anticancer Medicine?


Abstract
:1. Introduction
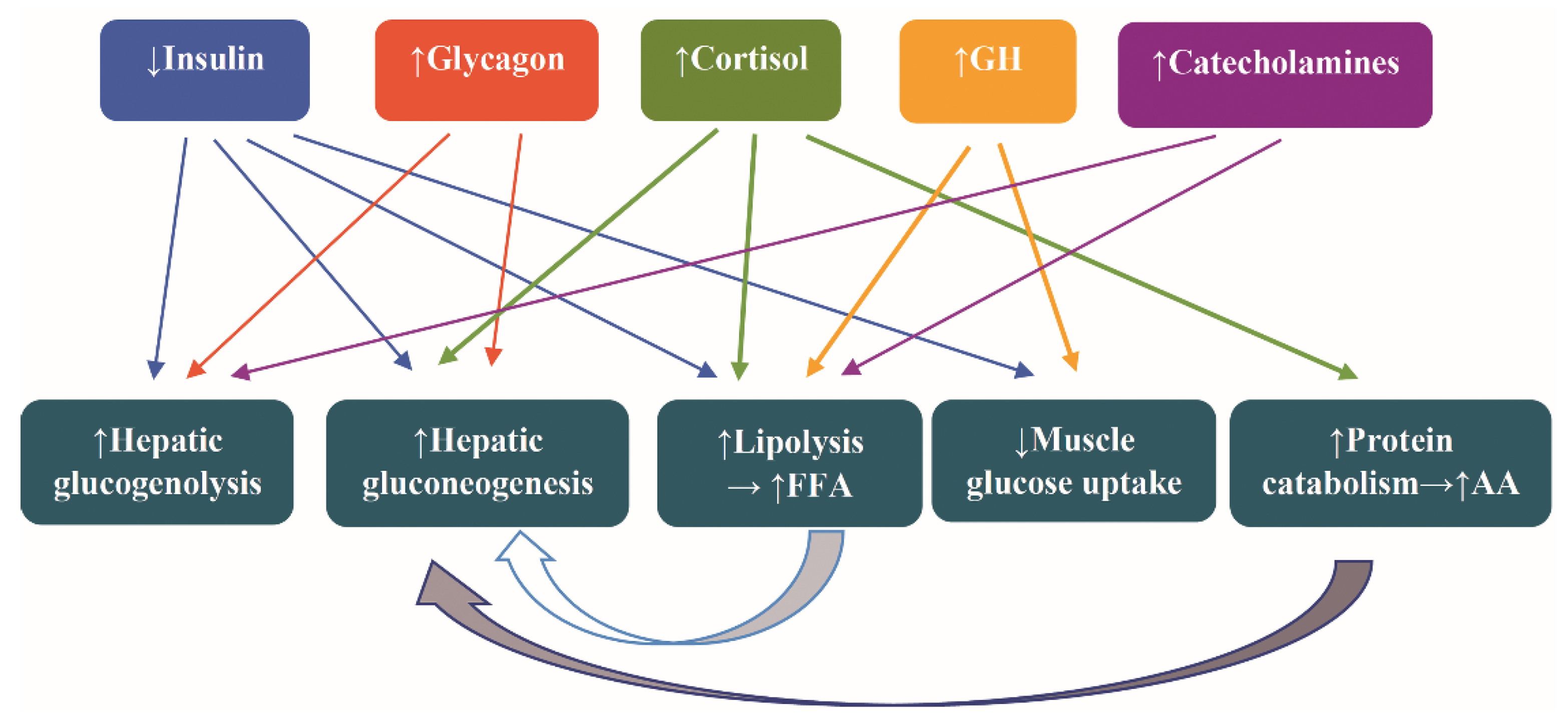
2. The Response to Fasting
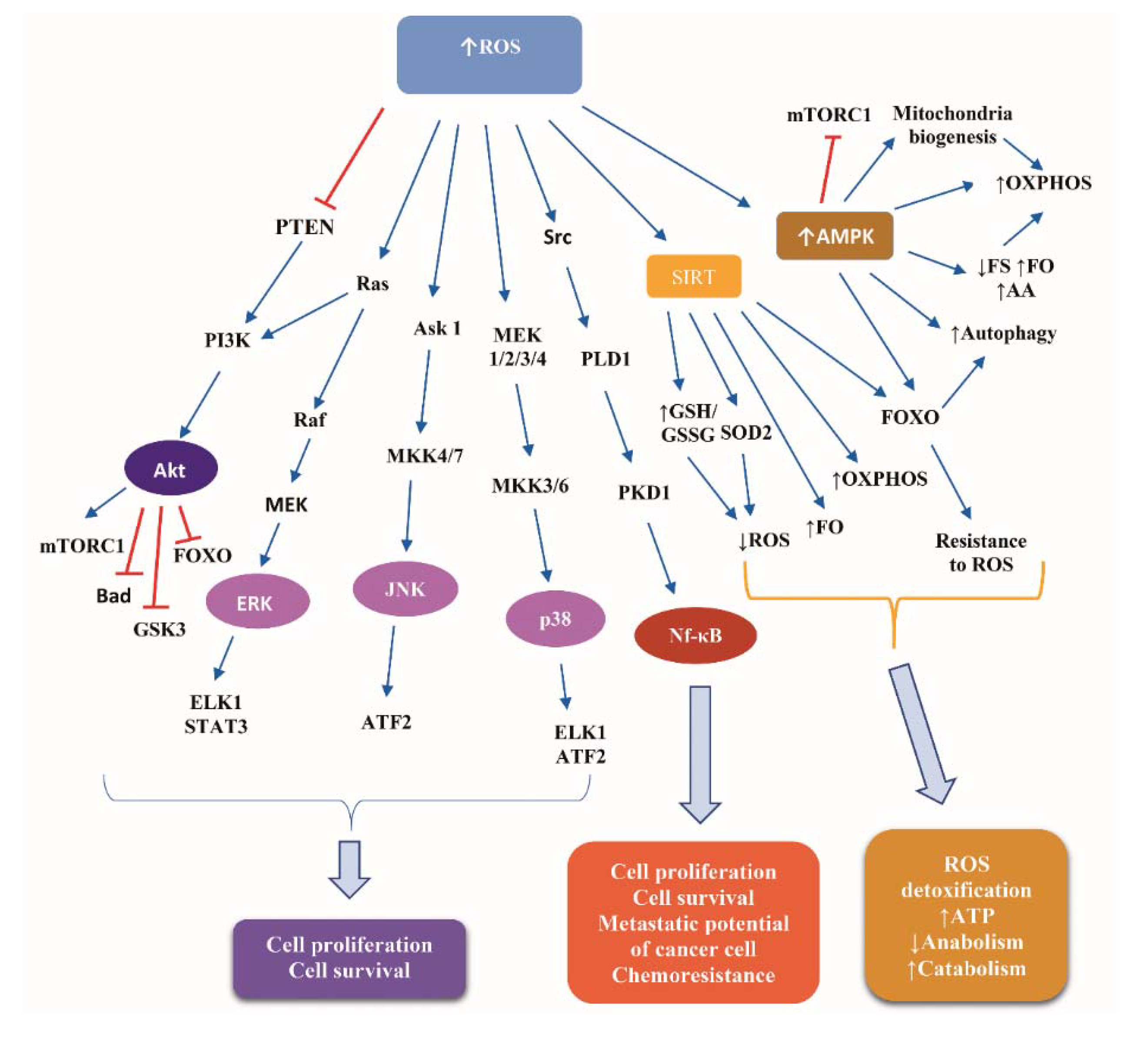
3. The Interrelationship of Fasting with Oxidative Stress: Exemplification of Hormesis
4. Fasting Versus CR
5. Fasting-Induced Increase of the Tolerability of Chemotherapy
5.1. Lessons from Nonclinical Data
5.2. Lessons from Clinical Data
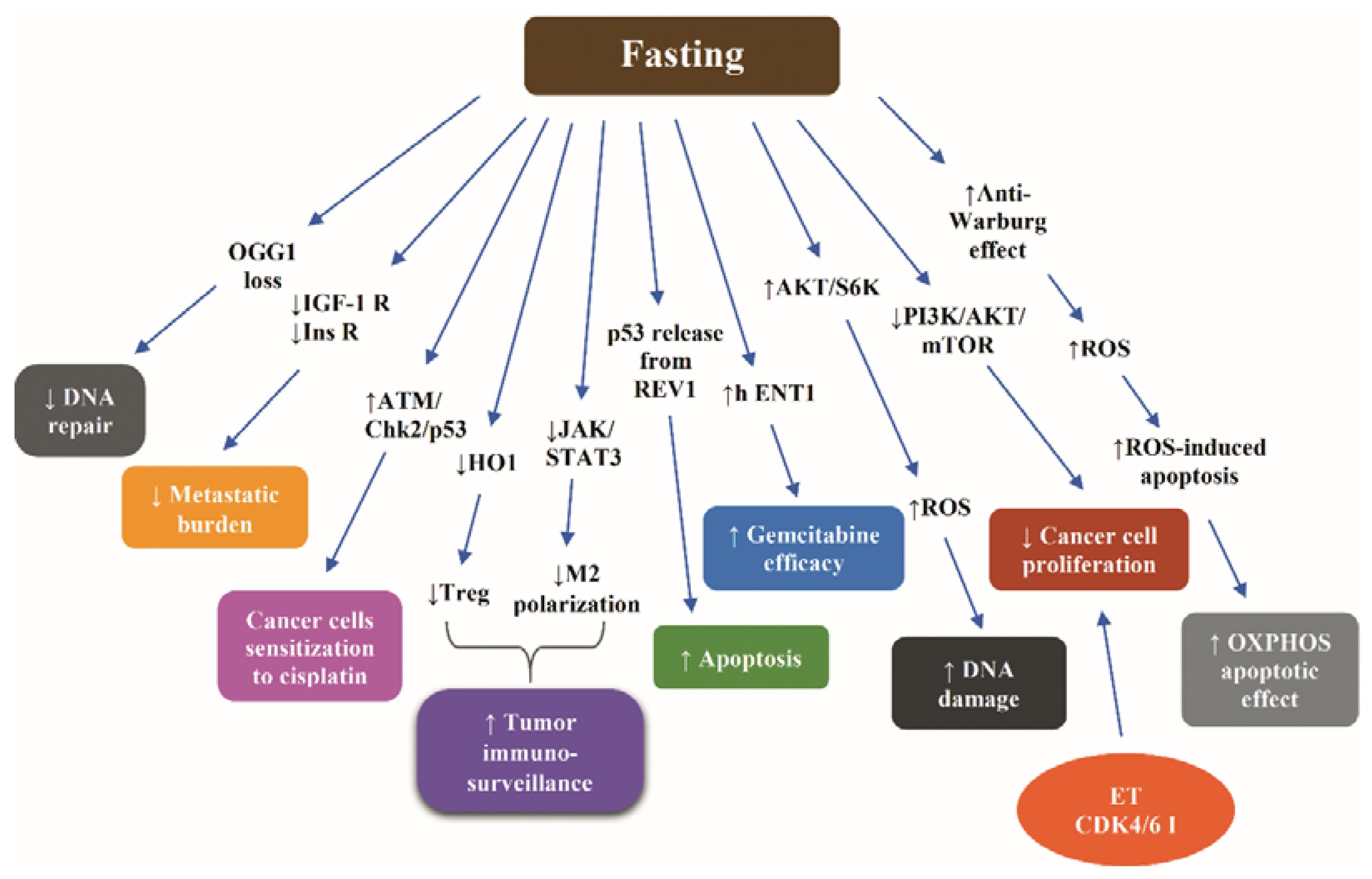
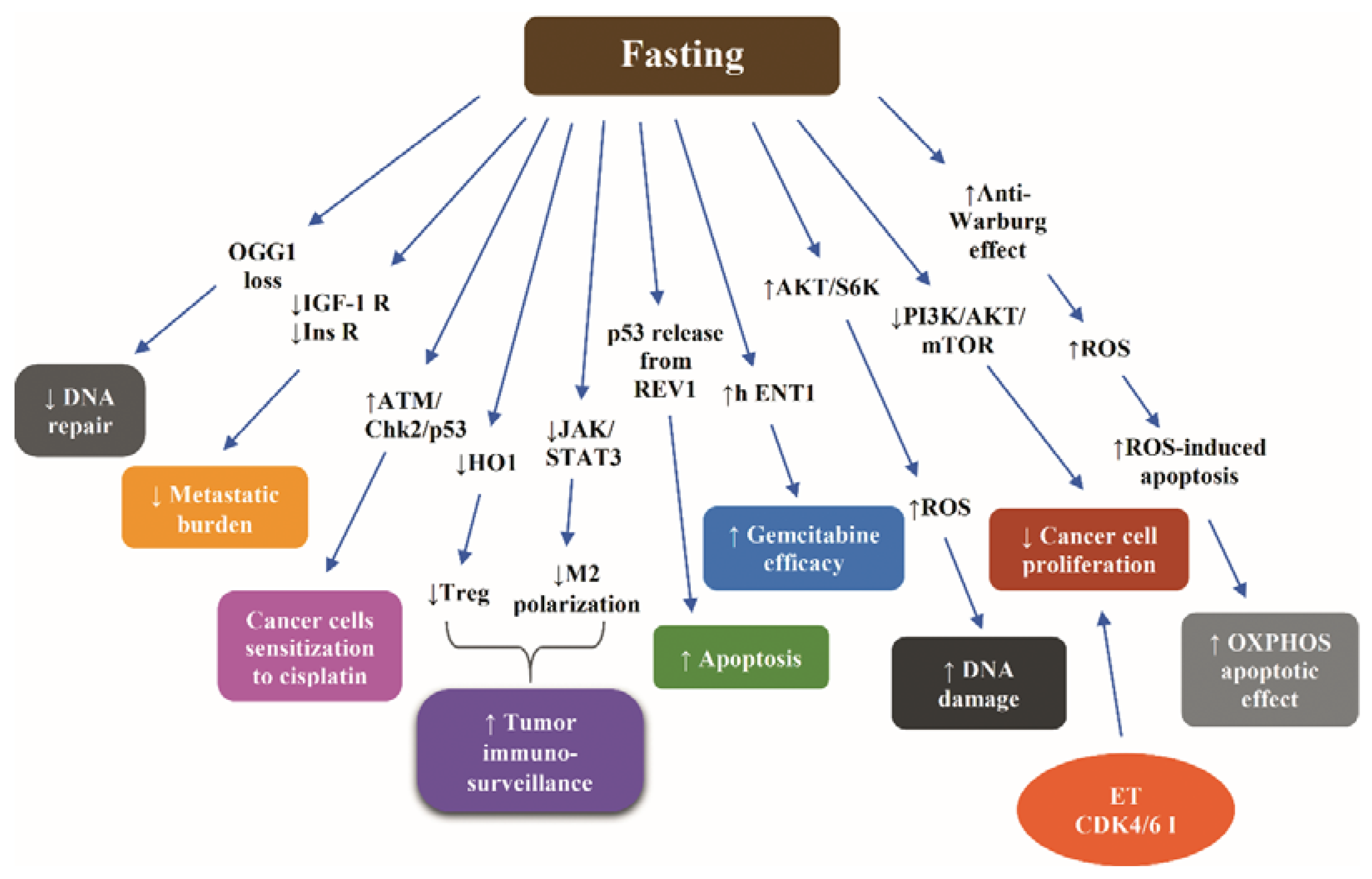
6. Fasting-Induced Increase of the Efficacy of Chemotherapy
6.1. Lessons from Nonclinical Data
6.2. Lessons from Clinical Data
7. Challenges and Future Perspectives in the Clinical Translation of Fasting in Oncology
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moreschi, C. Beziehung zwischen ernahrung and tumorwachstum. ZImmunitatsforsch 1909, 2, 651–675. [Google Scholar]
- McCay, C.M.; Crowel, M.F.; Maynard, L.A. The effect of retarded growth upon the length of the life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Kritchevsky, D. Caloric restriction and cancer. J. Nutr. Sci. Vitaminol. 2001, 47, 13–19. [Google Scholar] [CrossRef] [Green Version]
- McDonald, R.B.; Ramsey, J.J. Honoring Clive McCay and 75 years of calorie restriction research. J. Nutr. 2010, 140, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmi de Toledo, F.; Buchinger, A.; Burggrabe, H.; Hölz, G.; Kuhn, C.; Lischka, E.; Lischka, N.; Lützner, H.; May, W.; Ritzmann-Widderich, M.; et al. Fasting Therapy—An Expert Panel Update of the 2002 Consensus Guidelines. Forsch. Komplementmed. 2013, 20, 434–443. [Google Scholar] [CrossRef]
- Buono, R.; Longo, V.D. Starvation, Stress Resistance, and Cancer. Trends Endocrinol. Metab. 2018, 9, 271–280. [Google Scholar] [CrossRef]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Cancer statistics. Available online: https://seer.cancer.gov/statfacts/html/all.html (accessed on 30 July 2020).
- Xu, M.; Pu, Y.; Weichselbaum, R.R.; Fu, Y.X. Integrating conventional and antibody-based targeted anticancer treatment into immunotherapy. Oncogene 2016, 6, 585–592. [Google Scholar]
- Laviano, A.; Molfino, A.; Fanelli, F.R. Cancer-treatment toxicity: Can nutrition help? Nat. Rev. Clin. Oncol. 2012, 9, 605. [Google Scholar] [CrossRef]
- Costa, A.S.H.; Frezza, C. Metabolic Reprogramming and Oncogenesis: One Hallmark, Many Organelles. Int. Rev. Cell Mol. Biol. 2017, 332, 213–231. [Google Scholar]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017, 858, 686–699. [Google Scholar] [CrossRef]
- Caro, M.M.; Laviano, A.; Pichard, C. Nutritional intervention and quality of life in adult oncology patients. Clin. Nutr. 2007, 6, 289–301. [Google Scholar] [CrossRef]
- Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Fearon, K.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN guidelines on nutrition in cancer patients. Clin. Nutr. 2017, 36, 11–48. [Google Scholar] [CrossRef] [Green Version]
- Emmons, K.M.; Colditz, G.A. Realizing the Potential of Cancer Prevention—The Role of Implementation Science. N. Engl. J. Med. 2017, 376, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 106. [Google Scholar] [CrossRef]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Simone, B.A.; Champ, C.E.; Rosenberg, A.L.; Berger, A.C.; Monti, D.A.; Dicker, A.P.; Simone, N.L. Selectively starving cancer cells through dietary manipulation: Methods and clinical implications. Future Oncol. 2013, 9, 959–976. [Google Scholar] [CrossRef]
- Lee, C.; Longo, V. Fasting vs. dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra27. [Google Scholar] [CrossRef] [Green Version]
- Varady, K.A.; Hellerstein, M.K. Alternate-day fasting and chronic disease prevention: A review of human and animal trials. Am. J. Clin. Nutr. 2007, 86, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Goodrick, C.L.; Ingram, D.K.; Reynolds, M.A.; Freeman, J.R.; Cider, N.L. Differential effects of intermittent feeding and voluntary exercise on body weight and lifespan in adult rats. J. Gerontol. 1983, 38, 36–45. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting- mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9, eaai8700. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, C.; Lu, M.; Dong, Q.; Wang, Z.; Wang, Z.; Xiong, W.; Zhang, N.; Zhou, J.; Liu, Q.; et al. Calorie restriction is the most reasonable anti-ageing intervention: A meta-analysis of survival curves. Sci. Rep. 2018, 8, 5779. [Google Scholar] [CrossRef]
- Swindell, W.R. Dietary restriction in rats and mice: A meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res. Rev. 2012, 11, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Lagisz, M.; Hector, K.L.; Spencer, H.G. Comparative and meta-analytic insights into life extension via dietary restriction. Aging Cell 2012, 11, 401–409. [Google Scholar] [CrossRef]
- Jensen, K.; McClure, C.; Priest, N.K.; Hunt, J. Sex-specific effects of protein and carbohydrate intake on reproduction but not lifespan in Drosophila melanogaster. Aging Cell 2015, 14, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group (2016). Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Aspects Med. 2011, 32, 159–221. [Google Scholar] [CrossRef]
- Kopeina, G.S.; Senichkin, V.V.; Zhivotovsky, B. Caloric restriction—A promising anti-cancer approach: From molecular mechanisms to clinical trials. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 29–41. [Google Scholar] [CrossRef]
- Redman, L.M.; Heilbronn, L.K.; Martin, C.K.; Alfonso, A.; Smith, S.R.; Ravussin, E.; Pennington CALERIE Team. Effect of calorie restriction with or without exercise on body composition and fat distribution. J. Clin. Endocrinol. Metab. 2007, 92, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Rochon, J.; Bales, C.W.; Ravussin, E.; Redman, L.M.; Holloszy, J.O.; Racette, S.B.; Roberts, S.B.; Das, S.K.; Romashkan, S.; Galan, K.M.; et al. Design and conduct of the CALERIE study: Comprehensive assessment of the long-term effects of reducing intake of energy. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 97–108. [Google Scholar] [CrossRef]
- Fontana, L.; Klein, S. Aging, adiposity, and calorie restriction. JAMA 2007, 297, 986–994. [Google Scholar] [CrossRef]
- Lope, V.; Martín, M.; Castelló, A.; Ruiz, A.; Casas, A.M.; Baena-Cañada, J.M.; Antolín, S.; Ramos-Vázquez, M.; García-Sáenz, J.Á.; Muñoz, M.; et al. Overeating, caloric restriction and breast cancer risk by pathologic subtype: The EPIGEICAM study. Sci. Rep. 2019, 9, 3904. [Google Scholar] [CrossRef] [Green Version]
- Dirx, M.J.; Zeegers, M.P.; Dagnelie, P.C.; van den Bogaard, T.; van den Brandt, P.A. Energy restriction and the risk of spontaneous mammary tumors in mice: A meta-analysis. Int. J. Cancer 2003, 106, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhu, X.; Wang, H.; Wang, F.; Guan, W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: A systematic review and meta-analysis. PLoS ONE 2014, 9, e115147. [Google Scholar] [CrossRef]
- Masharani, U.; Gitelman, S.E. Hypoglemic Disorders. In Greenspan’s Basic & Clinical Endocrinology, 9th ed.; Mc GrawHill Lange: New York, NY, USA, 2011; pp. 657–773. ISBN 978-0-07-176743-9. [Google Scholar]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Naveed, S.; Aslam, M.; Ahmad, A. Starvation based differential chemotherapy: A novel approach for cancer treatment. Oman Med. J. 2014, 29, 391–398. [Google Scholar] [CrossRef]
- Gupta, R.; Ma, Y.; Wang, M.; Whim, M.D. AgRP-Expressing Adrenal Chromaffin Cells Are Involved in the Sympathetic Response to Fasting. Endocrinology 2017, 158, 2572–2584. [Google Scholar] [CrossRef]
- Wang, Q.; Whim, M.D. Stress-induced changes in adrenal neuropeptide Y expression are regulated by a negative feedback loop. J. Neurochem. 2013, 125, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Ray, A.; Cleary, M.P. The influence of different calorie restriction protocols on serum pro-inflammatory cytokines, adipokines and IGF-I levels in female C57BL6 mice: Short term and long term diet effects. Meta Gene 2017, 12, 22–32. [Google Scholar] [CrossRef]
- Messaoudi, I.; Warner, J.; Fischer, M.; Park, B.; Hill, B.; Mattison, J.; Lane, M.A.; Roth, G.S.; Ingram, D.K.; Picker, L.J.; et al. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc. Natl. Acad. Sci. USA 2006, 103, 19448–19453. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Khoo, A.; Wei, J.; Bowser, R.K.; Weathington, N.M.; Xiao, S.; Zhang, L.; Ma, H.; Zhao, Y.; Zhao, J. Serum starvation regulates E-cadherin upregulation via activation of c-Src in non-small-cell lung cancer A549 cells. Am. J. Physiol. Cell Physiol. 2014, 307, C893–C899. [Google Scholar] [CrossRef] [Green Version]
- Orgel, E.; Mittelman, S.D. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Sokolovska, N.; Brazeilles, R.; Affeldt, S.; Pelloux, V.; Prifti, E.; Chilloux, J.; Verger, E.O.; Kayser, B.D.; Aron-Wisnewsky, J.; et al. A Data Integration Multi-Omics Approach to Study Calorie Restriction-Induced Changes in Insulin Sensitivity. Front. Physiol. 2019, 9, 1958. [Google Scholar] [CrossRef]
- Lu, C.; Shi, Y.; Wang, Z.; Song, Z.; Zhu, M.; Cai, Q.; Chen, T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 2008, 582, 2703–2708. [Google Scholar] [CrossRef] [Green Version]
- Braun, F.; Bertin-Ciftci, J.; Gallouet, A.S.; Millour, J.; Juin, P. Serum-nutrient starvation induces cell death mediated by Bax and Puma that is counteracted by p21 and unmasked by Bcl-x(L) inhibition. PLoS ONE 2011, 6, e23577. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, B.; Xie, X.; Xiao, Y.F.; Yang, S.M.; Zhang, J.W. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol. Ther. 2015, 16, 1005–1013. [Google Scholar] [CrossRef]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; de Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar]
- Huang, Q.; Shen, H.M. To die or to live: The dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 2009, 5, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef]
- Liao, Z.; Damien, C.; Tan, N. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Gredilla, R.; Barja, G. Minireview: The role of oxidative stress in relation to caloric restriction and longevity. Endocrinology 2005, 146, 3713–3717. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E.J.; McCarthy, M.E.; Kenyon, E. The occurrence of chemically induced hormesis. Health Phys. 1987, 52, 531–541. [Google Scholar] [CrossRef]
- Zimmermann, A.; Bauer, M.A.; Kroemer, G.; Madeo, F.; Carmona-Gutierrez, D. When less is more: Hormesis against stress and disease. Microb. Cell. 2014, 1, 150–153. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B. Strategies for reducing or preventing the generation of oxidative stress. Oxid. Med. Cell Longev. 2011, 2011, 194586. [Google Scholar] [CrossRef] [Green Version]
- Martucci, M.; Ostan, R.; Biondi, F.; Bellavista, E.; Fabbri, C.; Bertarelli, C.; Salvioli, S.; Capri, M.; Franceschi, C.; Santoro, A. Mediterranean diet and inflammaging within the hormesis paradigm. Nutr. Rev. 2017, 5, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Ferre, P.; Azzout-Marniche, D.; Foufelle, F. AMP-activated protein kinase and hepatic genes involved in glucose metabolism. Biochem. Soc. Trans. 2003, 31, 220–223. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Proud, C.G. Role of mTOR signalling in the control of translation initiation and elongation by nutrients. Curr. Top Microbiol. Immunol. 2004, 279, 215–244. [Google Scholar]
- Anthony, T.G.; McDaniel, B.J.; Byerley, R.L.; McGrath, B.C.; Cavener, D.R.; McNurlan, M.A.; Wek, R.C. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J. Biol. Chem. 2004, 279, 36553–36561. [Google Scholar] [CrossRef] [Green Version]
- Dever, T.E.; Hinnebusch, A.G. GCN2 whets the appetite for amino acids. Mol. Cell 2005, 18, 141–142. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Towle, H.C. The metabolic sensor GCN2 branches out. Cell Metab. 2007, 5, 85–87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–946. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Masharani, U.; German, M.S. Pancreatic Hormones and Diabetes Mellitus. In Greenspan’s Basic & Clinical Endocrinology, 9th ed.; Mc GrawHill Lange: New York, NY, USA, 2011; pp. 573–655. ISBN 978-0-07-176743-9. [Google Scholar]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Kim, H.; Oh, S.; Lee, J.G.; Kee, M.; Ko, H.J.; Kweon, M.N.; Won, K.J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Dali-Youcef, N.; Lagouge, M.; Froelich, S.; Koehl, C.; Schoonjans, K.; Auwerx, J. Sirtuins: The ‘magnificent seven’, function, metabolism and longevity. The sirtuin family of histone deacetylases (HDACs). Ann. Med. 2007, 39, 335–345. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, A287, 4078–4086. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.C.; et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–534. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Moley, K.H.; Mueckler, M.M. Glucose transport and apoptosis. Apoptosis 2000, 5, 99–105. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Chaube, B.; MalvI, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [Green Version]
- Suwa, M.; Nakano, H.; Kumagai, S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J. Appl. Physiol. 2003, 95, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Terada, S.; Goto, M.; Kato, M.; Kawanaka, K.; Shimokawa, T.; Tabata, I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem. Biophys. Res. Commun. 2002, 96, 350–354. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009, 5, 1030–1033. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 92–203. [Google Scholar] [CrossRef]
- Moatt, J.P.; Nakagawa, S.; Lagisz, M.; Walling, C.A. The effect of dietary restriction on reproduction: A meta-analytic perspective. BMC Evol. Biol. 2016, 16, 199. [Google Scholar] [CrossRef] [Green Version]
- Villareal, D.T.; Fontana, L.; Weiss, E.P.; Racette, S.B.; Steger-May, K.; Schechtman, K.B.; Klein, S.; Holloszy, J.O. Bone mineral density response to caloric restriction-induced weight loss or exercise-induced weight loss: A randomized controlled trial. Arch. Intern. Med. 2006, 166, 502–510. [Google Scholar] [CrossRef]
- Pifferi, F.; Terrien, J.; Marchal, J.; Dal-Pan, A.; Djelti, F.; Hardy, I.; Chahory, S.; Cordonnier, N.; Desquilbet, L.; Hurion, M.; et al. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun. Biol. 2018, 1, 30. [Google Scholar] [CrossRef] [Green Version]
- De Groot, S.; Lugtenberg, R.T.; Cohen, D.; Welters, M.J.P.; Ehsan, I.; Vreeswijk, M.; Smit, V.; de Graaf, H.; Heijns, J.B.; Portielje, J.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 3083. [Google Scholar] [CrossRef]
- De Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W.; et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef] [Green Version]
- Dorff, T.B.; Groshen, S.; Garcia, A.; Shah, M.; Tsao-Wei, D.; Pham, H.; Cheng, C.W.; Brandhorst, S.; Cohen, P.; Wei, M.; et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 2016, 16, 360. [Google Scholar] [CrossRef] [Green Version]
- Safdie, F.M.; Dorff, T.; Quinn, D.; Fontana, L.; Wei, M.; Lee, C.; Cohen, P.; Longo, V.D. Fasting and cancer treatment in humans: A case series report. Aging 2009, 1, 988–1007. [Google Scholar] [CrossRef] [Green Version]
- Bauersfeld, S.P.; Kessler, C.S.; Wischnewsky, M.; Jaensch, A.; Steckhan, N.; Stange, R.; Kunz, B.; Brückner, B.; Sehouli, J.; Michalsen, A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: A randomized cross-over pilot study. BMC Cancer 2018, 18, 476. [Google Scholar] [CrossRef]
- Wilhelmi de Toledo, F.; Grundler, F.; Bergouignan, A.; Drinda, S.; Michalsen, A. Safety, health improvement and well-being during a 4 to 21-day fasting period in an observational study including 1422 subjects. PLoS ONE 2019, 14, e0209353. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.; Mitrofanis, J.; Johnstone, D.M.; Falsini, B.; Bisti, S.; Adam, P.; Nuevo, A.B.; George-Weinstein, M.; Mason, R.; Eells, J. Acquired Resilience: An Evolved System of Tissue Protection in Mammals. Dose Response 2018, 16, 1559325818803428. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Verweij, M.; Brand, K.; van de Ven, M.; Goemaere, N.; van den Engel, S.; Chu, T.; Forrer, F.; Müller, C.; de Jong, M.; et al. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell 2009, 9, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Van Ginhoven, T.M.; Mitchell, J.R.; Verweij, M.; Hoeijmakers, J.H.; Ijzermans, J.N.; de Bruin, R.W. The use of preoperative nutritional interventions to protect against hepatic ischemia-reperfusion injury. Liver Transp. 2009, 15, 1183–1191. [Google Scholar] [CrossRef]
- Varendi, K.; Airavaara, M.; Anttila, J.; Vose, S.; Planken, A.; Saarma, M.; Mitchell, J.R.; Andressoo, J.O. Short-term preoperative dietary restriction is neuroprotective in a rat focal stroke model. PLoS ONE 2014, 9, e93911. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef]
- Zhong, F.; Jiang, Y. Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front. Endocrinol. (Lausanne) 2019, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.F.; Mattson, M.P. Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: Evidence for a preconditioning mechanism. J. Neurosci. Res. 1999, 57, 830–839. [Google Scholar] [CrossRef]
- Lee, C.; Safdie, F.M.; Raffaghello, L.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010, 70, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Berryman, D.E.; Christiansen, J.S.; Johannsson, G.; Thorner, M.O.; Kopchick, J.J. Role of the GH/IGF-1 axis in lifespan and healthspan: Lessons from animal models. Growth Horm. IGF Res. 2008, 18, 455–471. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D. Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol. Aging 1999, 20, 479–486. [Google Scholar] [CrossRef]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [Green Version]
- Swindell, W.R. Gene expression profiling of long-lived dwarf mice: Longevity-associated genes and relationships with diet, gender and aging. BMC Genomics 2007, 8, 353. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Mitra, M.S.; Donthamsetty, S.; White, B.; Latendresse, J.R.; Mehendale, H.M. Mechanism of protection of moderately diet restricted rats against doxorubicin-induced acute cardiotoxicity. Toxicol. Appl. Pharmacolol. 2007, 225, 90–101. [Google Scholar] [CrossRef]
- Di Biase, S.; Shim, H.S.; Kim, K.H.; Vinciguerra, M.; Rappa, F.; Rappa, F.; Wei, M.; Brandhorst, S.; Cappello, F.; Mirzaei, H.; et al. Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 2017, 15, e2001951. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic stem cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 4, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Orlowski, K.; Pruschy, M.; Stahel, R.A. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer 2012, 12, 571. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Pardee, A.B. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res. 2001, 61, 4301–4305. [Google Scholar]
- Jongbloed, F.; Huisman, S.A.; van Steeg, H.; Pennings, J.L.A.; IJzermans, J.N.M.; Dollé, M.; de Bruin, R. The transcriptomic response to irinotecan in colon carcinoma bearing mice preconditioned by fasting. Oncotarget 2019, 10, 2224–2234. [Google Scholar] [CrossRef] [Green Version]
- Huisman, S.A.; Bijman-Lagcher, W.; IJzermans, J.N.; Smits, R.; de Bruin, R.W. Fasting protects against the side effects of irinotecan but preserves its anti-tumor effect in Apc15lox mutant mice. Cell Cycle 2015, 14, 2333–2339. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Takemura, G.; Kanamori, H.; Takeyama, T.; Watanabe, T.; Morishita, K.; Ogino, A.; Tsujimoto, A.; Goto, K.; Maruyama, R.; et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 2012, 96, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, S.; Wei, M.; Hwang, S.; Morgan, T.E.; Longo, V.D. Short-term calorie and protein restriction provide partial protection from chemotoxicity but do not delay glioma progression. Exp. Gerontol. 2013, 48, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Tinkum, K.L.; Stemler, K.M.; White, L.S.; Loza, A.J.; Jeter-Jones, S.; Michalski, B.M.; Kuzmicki, C.; Pless, R.; Stappenbeck, T.S.; Piwnica-Worms, D.; et al. Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proc. Natl. Acad. Sci. USA 2015, 112, E7148–E7154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. S1), 35–48. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Qasim, M.; Kim, J.H. Nanoparticle-Mediated Combination Therapy: Two-in-One Approach for Cancer. Int. J. Mol. Sci. 2018, 9, 3264. [Google Scholar] [CrossRef] [Green Version]
- Simone, B.A.; Palagani, A.; Strickland, K.; Ko, K.; Jin, L.; Lim, M.K.; Dan, T.D.; Sarich, M.; Monti, D.A.; Cristofanilli, M.; et al. Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle 2018, 17, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Safdie, F.; Brandhorst, S.; Wei, M.; Safdie, F.; Brandhorst, S.; Hwang, S.; Conti, P.S.; Chen, T.C.; Longo, V.D. Fasting enhances the response of glioma to chemo- and radiotherapy. PLoS ONE 2012, 7, e44603. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A.; et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 2015, 6, 11806–11819. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 47–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Maiuri, M.C.; Madeo, F.; Kroemer, G. Autophagy induction for the treatment of cancer. Autophagy 2016, 12, 1962–1964. [Google Scholar] [CrossRef] [Green Version]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Wang, H.; He, Z.; Chen, X.; Wu, Q.; Chen, W.; Sun, Z.; Weng, M.; Zhu, M.; Ma, D.; et al. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 2017, 8, 74649–74660. [Google Scholar] [CrossRef] [Green Version]
- D’Aronzo, M.; Vinciguerra, M.; Mazza, T.; Panebianco, C.; Saracino, C.; Pereira, S.P.; Graziano, P.; Pazienza, V. Fasting cycles potentiate the efficacy of gemcitabine treatment in in vitro and in vivo pancreatic cancer models. Oncotarget 2015, 6, 18545–18557. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.S.; Wei, M.; Brandhorst, S.; Longo, V.D. Starvation promotes REV1 SUMOylation and p53-dependent sensitization of melanoma and breast cancer cells. Cancer Res. 2015, 75, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xie, J.; Wu, G.; Shen, J.; Collins, R.; Chen, W.; Kang, X.; Luo, M.; Zou, Y.; Huang, L.J.; et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat. Med. 2017, 23, 79–90. [Google Scholar] [CrossRef]
- Siggens, L.; Figg, N.; Bennett, M.; Foo, R. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. FASEB J. 2012, 26, 2117–2124. [Google Scholar] [CrossRef] [Green Version]
- Witkamp, R.F.; van Norren, K. Let thy food be thy medicin when possible. Eur. J. Pharmacol. 2018, 836, 102–114. [Google Scholar] [CrossRef]
- Vernieri, C.; Casola, S.; Foiani, M.; Pietrantonio, F.; de Braud, F.; Longo, V. Targeting Cancer Metabolism: Dietary and Pharmacologic Interventions. Cancer Discov. 2016, 6, 1315–1333. [Google Scholar] [CrossRef] [Green Version]
- Tannenbaum, A. Effects of varying caloric intake upon tumor incidence and tumor growth. Ann. N. Y. Acad. Sci. 1947, 49, 5–18. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L. Dietary restriction in mice beginning at one year of age. Effects on life span and spontaneous cancer incidence. Science 1982, 215, 1415–1418. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Erickson, N.; Boscheri, A.; Linke, B.; Huebner, J. Systematic review: Isocaloric ketogenic dietary regimes for cancer patients. Med. Oncol. 2017, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Demark-Wahnefried, W.; Peterson, C.M.; Norian, L.A. Targeting Glucose Metabolism to Enhance Immunotherapy: Emerging Evidence on Intermittent Fasting and Calorie Restriction Mimetics. Front. Immunol. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell. 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Adelaiye, R.M.; Rastelli, A.L.; Miles, K.M.; Ciamporcero, E.; Longo, V.D.; Nguyen, H.; Vessella, R.; Pilli, R. Dietary protein restriction inhibits tumor growth in human xenograft models. Oncotarget 2013, 4, 2451–2461. [Google Scholar] [CrossRef]
- Yu, D.; Yang, S.E.; Miller, B.R.; Wisinski, J.A.; Sherman, D.S.; Brinkman, J.A.; Tomasiewicz, J.L.; Cummings, N.E.; Kimple, M.E.; Cryns, V.L.; et al. Lamming Short-term methionine deprivation improves metabolic health via sexually dimorphic, mTORC1-independent mechanisms. FASEB J. 2018, 32, 3471–3482. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Champ, C.E.; Otto, C.; Kämmerer, U. Anti-Tumor Effects of Ketogenic Diets in Mice: A Meta-Analysis. PLoS ONE 2016, 11, e0155050. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.T.; Staels, B. Fasting the Microbiota to Improve Metabolism? Cell Metab. 2017, 26, 584–585. [Google Scholar] [CrossRef] [Green Version]
- Caffa, I.; D’Agostino, V.; Damonte, P.; Soncini, D.; Cea, M.; Monacelli, F.; Odetti, P.; Ballestrero, A.; Provenzani, A.; Longo, V.D.; et al. Fasting potentiates the anticancer activity of tyrosine kinase inhibitors by strengthening MAPK signaling inhibition. Oncotarget 2015, 6, 11820–11832. [Google Scholar] [CrossRef]
- Lo Re, O.; Panebianco, C.; Porto, S.; Cervi, C.; Rappa, F.; Di Biase, S.; Michele Caraglia, M.; Pazienza, V.; Manlio Vinciguerra, M. Fasting inhibits hepatic stellate cells activation and potentiates anti-cancer activity of Sorafenib in hepatocellular cancer cells. Cell Physiol. 2018, 233, 1202–1212. [Google Scholar] [CrossRef]
- Bragazzi, N.L.; Briki, W.; Khabbache, H.; Rammouz, I.; Chamari, K.; Demaj, T.; Re, T.S.; Zouhir, M. Ramadan Fasting and Patients with Cancer: State-of-the-Art and Future Prospects. Front. Oncol. 2016, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Adawi, M.; Watad, A.; Brown, S.; Aazza, K.; Aazza, H.; Zouhir, M.; Sharif, K.; Ghanayem, K.; Farah, R.; Mahagna, H.; et al. Ramadan Fasting Exerts Immunomodulatory Effects: Insights from a Systematic Review. Front. Immunol. 2017, 8, 1144. [Google Scholar] [CrossRef] [Green Version]
- Bragazzi, N.L.; Sellami, M.; Salem, I.; Conic, R.; Kimak, M.; Pigatto, P.D.M.; Damiani, G. Fasting and Its Impact on Skin Anatomy, Physiology, and Physiopathology: A Comprehensive Review of the Literature. Nutrients 2019, 11, 249. [Google Scholar] [CrossRef] [Green Version]
- Damiani, G.; Mahroum, N.; Pigatto, P.D.M.; Pacifico, A.; Malagoli, P.; Tiodorovic, D.; Conic, R.R.; Amital, H.; Bragazzi, N.L.; Watad, A.; et al. The Safety and Impact of a Model of Intermittent, Time-Restricted Circadian Fasting (“Ramadan Fasting”) on Hidradenitis Suppurativa: Insights from a Multicenter, Observational, Cross-Over, Pilot, Exploratory Study. Nutrients 2019, 11, 1781. [Google Scholar] [CrossRef] [Green Version]
- Active Clinical Trials Addressing Fasting in Oncology. Available online: https://clinicaltrials.gov (accessed on 2 August 2020).
- Lévesque, S.; Pol, J.G.; Ferrere, G.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Trial watch: Dietary interventions for cancer therapy. Oncoimmunology 2019, 8, 1591878. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terminology | Dietary Intervention |
|---|---|
| Fasting | Consumption of only water, for a period varying from 12 h to 3 weeks |
| Short term fasting (STF) | Fasting for an average of 3–5 consecutive days |
| Periodic fasting | Fasting repeated every 2 or more weeks |
| Intermittent fasting | Alternate day fasting (≥16 h) or 48 h of fasting/week |
| Fasting mimicking diet (FMD) | A regimen providing low calories, low amounts of proteins, and high amounts of fats. FMD provides 4600 KJ (11% protein, 46% fat, and 43% carbohydrates) for day 1 and 300 KJ (9% protein, 44% fat, and 47% carbohydrates) for days 2–5 |
| Calorie restriction (CR) | 20–40% reduction in calorie intake with reduction of all ingredients without intercepting the intake of vitamins and minerals, usually used by experts as synonym to dietary restriction (DR) |
| Ref. | Materials | Mode of Fasting Plus CT | Outcome of Fasting Plus CT |
|---|---|---|---|
| [126] | A/J, CD-1, athymic (Nude-nu) mice | 48h Starvation prior to etoposide |
|
| Neuroblastoma (NXS2)-bearing mice | |||
| [127] | Male Sprague-Dawley rats | 35% CR prior to DXR | Protection against DXR cardiotoxicity and death |
| [128] | C57BL/6 mice | DXR ± STS (48 h) | Protection of cardiomyocytes against DXR toxicity by conserved PKA/AMPK/transcription factors Msn2/4 (yeast) and Egr1 (mice) pathway |
| [129] | C57BL/6J mice | Fasting prior to CP | Decreased DNA damage in leukocytes and bone marrow cells. Increased self-renewal and regeneration of hematopoietic cells |
| [130] | Primary human mesothelial SDM104 cells | Serum starvation 24 h prior to CDDP | Protection of normal cells against CDDP toxicity via complete arrest of cellular proliferation mediated by AMPK-dependent and ATM-independent p53/p21 activation |
| [132] | Male BALB/c mice were subcutaneously injected with C26 colon carcinoma cells | 3 d Fasting prior to irinotecan | Protection of host, but not of tumor, against irinotecan toxicity by altering the transcriptional response in liver and the hepatic metabolism of irinotecan |
| [133] | FabplCre; Apc(15lox/+) mice spontaneously developing intestinal tumors | 3 d Fasting pior to irinotecan | Protection against irinotecan toxicity |
| [134] | GFP-LC3 transgenic mice | 48 h Fasting prior to DXR | Protection against DXR cardiotoxicity by inducing autophagy via restoring AMPK and ULK1 |
| [135] | CD-1, BalB/C or | CR (60%, 50%, 40%, 20%, and 10% of calorie density of AIN93G) or STS (no food for up to 60 h) | Protection against DXR toxicity |
| C57BL/6N mice | Protection against DXR toxicity by combination of short-term 50% CR with either severe protein-deficiency or KD similarly to 50% CR, but less than STS | ||
| [136] | Bmi1CreER/+;R26R mice | 24 h Fasting prior to etoposide | Protection against etoposide toxicity in SI via preservation of SI stem cell viability, SI architecture and barrier function |
| B6(Cg)-Tyrc-2 J/J, | |||
| Bmi1CreERT/+;Rosa26R/+ HopXCreERT/+;Rosa26R/+ | |||
| Lgr5EGFP-IRES-CreERT2/+;Rosa26R/+, | |||
| Lgr5EGFP-IRES-CreERT2/+ mice | |||
| [137] | MCF7-xenograft-bearing 6–8-week-old | Cylic FMD plus TMX | Counteraction of TMX-related increase in uterus size and weight FMD led to:
|
| female NOD/SCIDγ mice Six-to-eight-week-old female BALB/c mice | Weekly 48-h fasting (n = 5) or FMD (n = 5) plus TMX |
| Ref. | Materials | Mode of Fasting Plus CT | Outcome of Fasting Plus CT |
|---|---|---|---|
| [23] | Murine and human cancer cells | 24 h Starvation before and 24 h Starvation during DXR or CP | More intense delay of progression of melanoma, glioma, and breast cancer cells compared to CT alone |
| Subcutaneous allografts of murine breast cancer (4T1), melanoma (B16), glioma (GL26), metastatic neuroblastoma models (NXS2, Neuro-2a), and xenografts of human neuroblastoma (ACN), breast cancer (MDA-MB-231), and ovarian cancer (OVCAR3) cell lines | 48 to 60 h fasting combined with DXR or CP | ||
| [130] | ZL55 mesothelioma cancer cells | CDDP with serum starvation | Sensitization of cancer cells, human mesothelioma xenografts, and human lung adenocarcinoma xenografts to CDDP via stimulation of ATM/Chk2/p53 signaling pathway |
| Human ZL55 mesothelioma xenografts | 48 h Fasting combined with CDDP | ||
| Human lung carcinoma A549 xenografts | |||
| [137] | MCF7-xenograft-bearing 6–8-wk-old female NOD/SCIDγ mice | Cylic FMD plus TMX | Potentiation of FULV, TMX, palbociclib (or revertion of acquired resistance to FULV plus palbociclib via:
|
| 6-to-8-wk-old female BALB/c mice | Weekly 48 h fasting (n = 5) or FMD (n = 5) plus TMX | ||
| [141] | Balb/c mice orthotopically injected with a syngeneic triple negative breast cancer cell line (4T1) | 30% CR combined with cisplatin/docetaxel |
|
| [142] | Primary mouse glia, murine GL26, rat C6 and human U251, LN229 and A172 glioma cells | STS combined with temozolomide |
|
| Mice with subcutaneous or intracranial models of GL26 glioma | 48 h Starvation prior to chemotherapy | ||
| [143] | CT26 colon carcinoma cell | 48 h STS combined with OXP | Amplification of the toxicity and the DNA damage-dependent proapoptotic effect of OXP |
| BALB/c mice models bearing subcutaneous CT26 colon cancer | |||
| [146] | Murine | FMD or STS combined with FMD with DXR or CP | Additive effect on tumor suppression via T cell-dependent killing of cancer cells |
| Br east cancer (4T1) | FMD combined with DXR | ||
| murine melanoma (B16) model | |||
| [148] | BxPC-3, MiaPaca-2, and Panc-1 cells | Culture in combination of gemcitabine and FMM | Reinforcement of the efficacy of gemcitabine via increasing levels of h ENT1, and reducing RRM1 levels |
| Pancreatic cancer xenograft mice | 24 h Starvation prior to gemcitabine | ||
| [149] | C57BL/6J mice bearing B16 melanoma tumors | 48 h Fasting combined with DXR or CP | Efficacy of two fasting cycles in terms of inhibition of tumor growth equal to that of DXR or CP. Reinforcement of the efficacy of chemotherapy via induction of the proapoptotic effect of p53 due to disruption of REV1-p53 interaction |
| [Ref] Study Type (Clinical Trials Gov. Identifier) | Patients/Methods | Outcome of Fasting Plus CT |
|---|---|---|
| [106] Phase II/III randomized trial (NCT02126449) | 131 patients with HER2-negative stage II/III breast cancer (no diabetes and BMI > 18 kg m−2) were randomized to receive either FMD or regular diet for 3 d prior to and during neoadjuvant CT |
|
| [107] Randomized-controlled pilot trial (NCT01304251) | HER2 negative breast cancer patients on 48 h STF (24 h before and after CT) were compared to patients on healthy nutrition |
|
| [108] Partially randomized clinical trial (NCT00936364) | Cancer patients a on platinum-based CT fasted for 24 h or 48 h prior CT or 72 h (48 h prior to and 24 h post CT) |
|
| [109] Case series | 10 cancer patients voluntarily fasted prior to (48–140 h) and/or following (5–56 h) CT |
|
| [110] Individually randomized cross-over trial (NCT01954836) | 34 women with breast cancer or ovarian cancer were randomized to a 60 h STF (36 h before and 24 h after CT) in the first half of CT followed by normocaloric diet (group A; n = 18) or vice versa (group B; n = 16) |
|
| [129] Phase I clinical trial (NCT00936364) | Patients with malignant solid tumors were treated with platinum-based doublet chemotherapy combined with 72 h fasting (48 h before and 24 h after CT) |
|
| [137] Interventional clinical trials NCT03595540 (24 patients) NCT03340935 (12 patients) | 35 patients with HR+ breast cancer treated with FULV or TMX, as adjuvant or as palliative strategy, and 1 patient treated with fulvestrant plus palbociclib for advanced disease followed 5 d FMD (Xentigen) Q4W (NCT03595540) (average 6.8 FMD cycles, max 14 cycles) or 5 d FMD regimen Q3-4W (average 5.5 cycles) |
|
| Cancer Type Clinical Trials Gov. Identifier | Arm I: Intervention | Arm II: Active Comparator | Primary Endpoint | Time Frame |
|---|---|---|---|---|
| Breast Cancer Prostate Cancer NCT01802346 | Low-calorie diet 3 d before and 24 h after CT during the 12 wks of CT | Normal diet |
| 12 wks |
| Non-small Cell Lung Cancer NCT03700437 | Chemolieve® in patients on carboplatin/pemetrexed and pembrolizumab 3 d before CT/immunotherapy and on the 1st day of CT/immunotherapy for the first 4 C | Control arm: regular diet |
| Screening baseline and on:
|
| Prostatic Neoplasms NCT02710721 | 60 h-FMD (36 h before and 24 h after CT) | MD | Change of FACT-P/-Taxane/-A sum score from baseline to day 8 after each CT |
|
| Breast cancer Ovarian cancer NCT03162289 | Intermittent fasting 60–72 h (36–48 h before and 24 h after CT) | 60–72 h Vegan d 36–48 h before and 24 h after CT during the first 4 C of CT and thereafter 2 d (24 h before and after CT) vegan and sugar-restricted diet | Change of FACT-G score |
|
| Cancer Breast Cancer Colorectal Cancer NCT03595540 | Monthly C of Prolon FMD (L-Nutra) in patients under active cancer treatment | NA |
| 6 mo |
| Breast cancer Melanoma malignant NCT03454282 | 5-day FMD followed for 1 C (Cohorts A and B) or for 4 consecutive every-4-week C, postop. | NA | Changes in PBMCs | 3 years |
| Glioblastoma NCT03451799 | 16-wk KD while on standard of care cancer treatment (Radiation + Temozolomide) | NA | Safety of KD | 4 mo |
| Advanced LKB1-inactive Lung Adenocarcinoma NCT03709147 | Every-three wks, 5-d-FMD up to 4 C in patients receiving: Metformin Hydrochloride Cisplatin Carboplatin Pemetrexed | Metformin Hydrochloride Cisplatin Carboplatin Pemetrexed | Progression-free survival | 60 mo |
| Malignant Neoplasm Cancer NCT03340935 | FMD | NA | Safety of FMD | 2 years |
| Cancer NCT03840213 | Behavioral: Filling a questionnaire and interview focused on diet | NA | N of patients who voluntarily changed eating habits or followed fasting or restrictive diet during CT | 1 year |
| Glioblastoma Multiforme NCT01865162 | KD as adjuvant for treatment-refractory glioblastoma multiforme | NA | Safety of KD | 1 year |
| Glioblastoma Multiforme NCT02302235 | KD adjunctive to standard radiation and temozolomide CT | Phase 2 |
| 6 mo |
| Glioblastoma Multiforme NCT01535911 | KD in adults with newly diagnosed glioblastoma while being on RT and CT | NA | Changes in brain tumor size assessed by MRI | 6 wks after RT completion |
| Childhood cancer survivors NCT03523377 | Overnight fasting (12h) after completion of therapy | NA | Measure of Glu metabolism | 6 mo |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deligiorgi, M.V.; Liapi, C.; Trafalis, D.T. How Far Are We from Prescribing Fasting as Anticancer Medicine? Int. J. Mol. Sci. 2020, 21, 9175. https://doi.org/10.3390/ijms21239175
Deligiorgi MV, Liapi C, Trafalis DT. How Far Are We from Prescribing Fasting as Anticancer Medicine? International Journal of Molecular Sciences. 2020; 21(23):9175. https://doi.org/10.3390/ijms21239175
Chicago/Turabian StyleDeligiorgi, Maria V., Charis Liapi, and Dimitrios T. Trafalis. 2020. "How Far Are We from Prescribing Fasting as Anticancer Medicine?" International Journal of Molecular Sciences 21, no. 23: 9175. https://doi.org/10.3390/ijms21239175
APA StyleDeligiorgi, M. V., Liapi, C., & Trafalis, D. T. (2020). How Far Are We from Prescribing Fasting as Anticancer Medicine? International Journal of Molecular Sciences, 21(23), 9175. https://doi.org/10.3390/ijms21239175