Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives


{kind=link}
{kind=link}
{kind=link}
Abstract
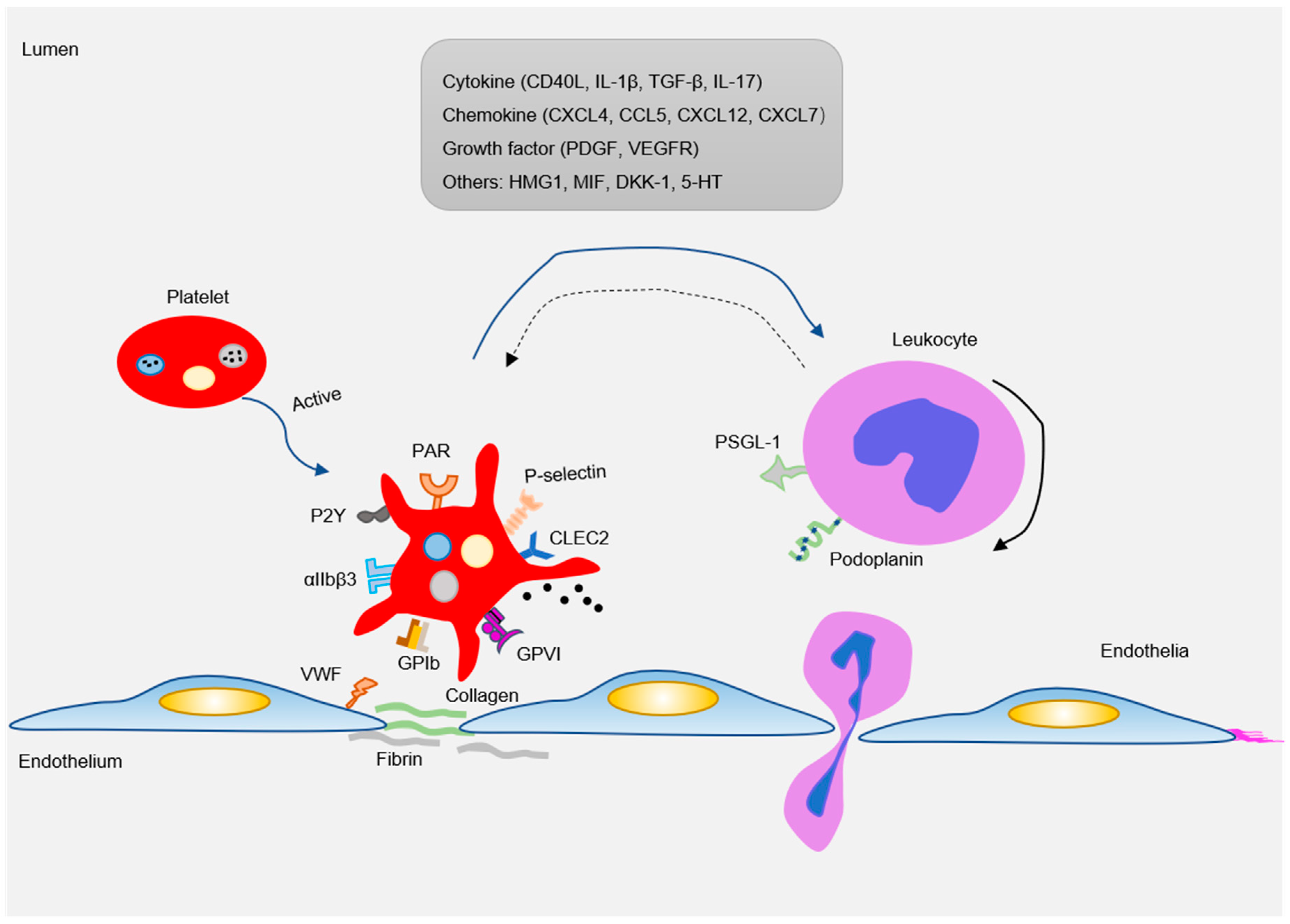
:1. Introduction
2. Platelets and Risk Factors of Atherosclerosis
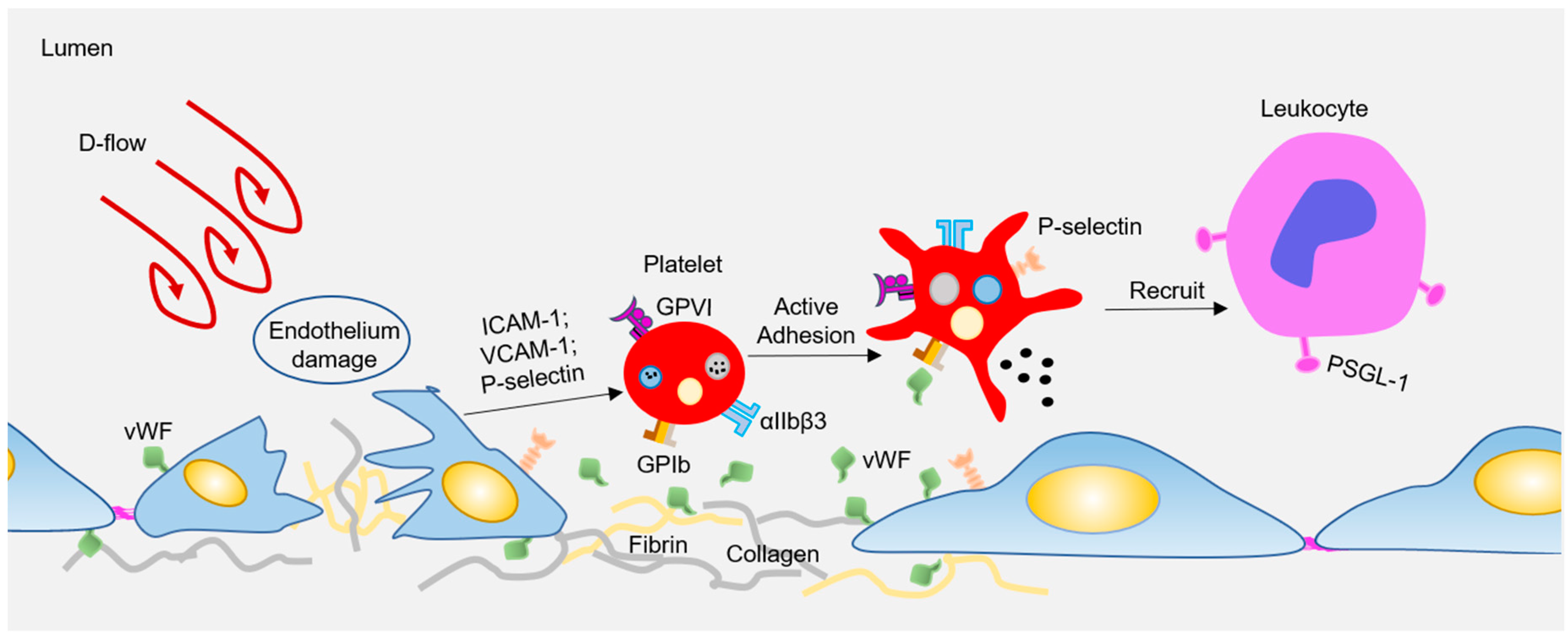
2.1. Disturbed Flow Modulated Platelets in AS
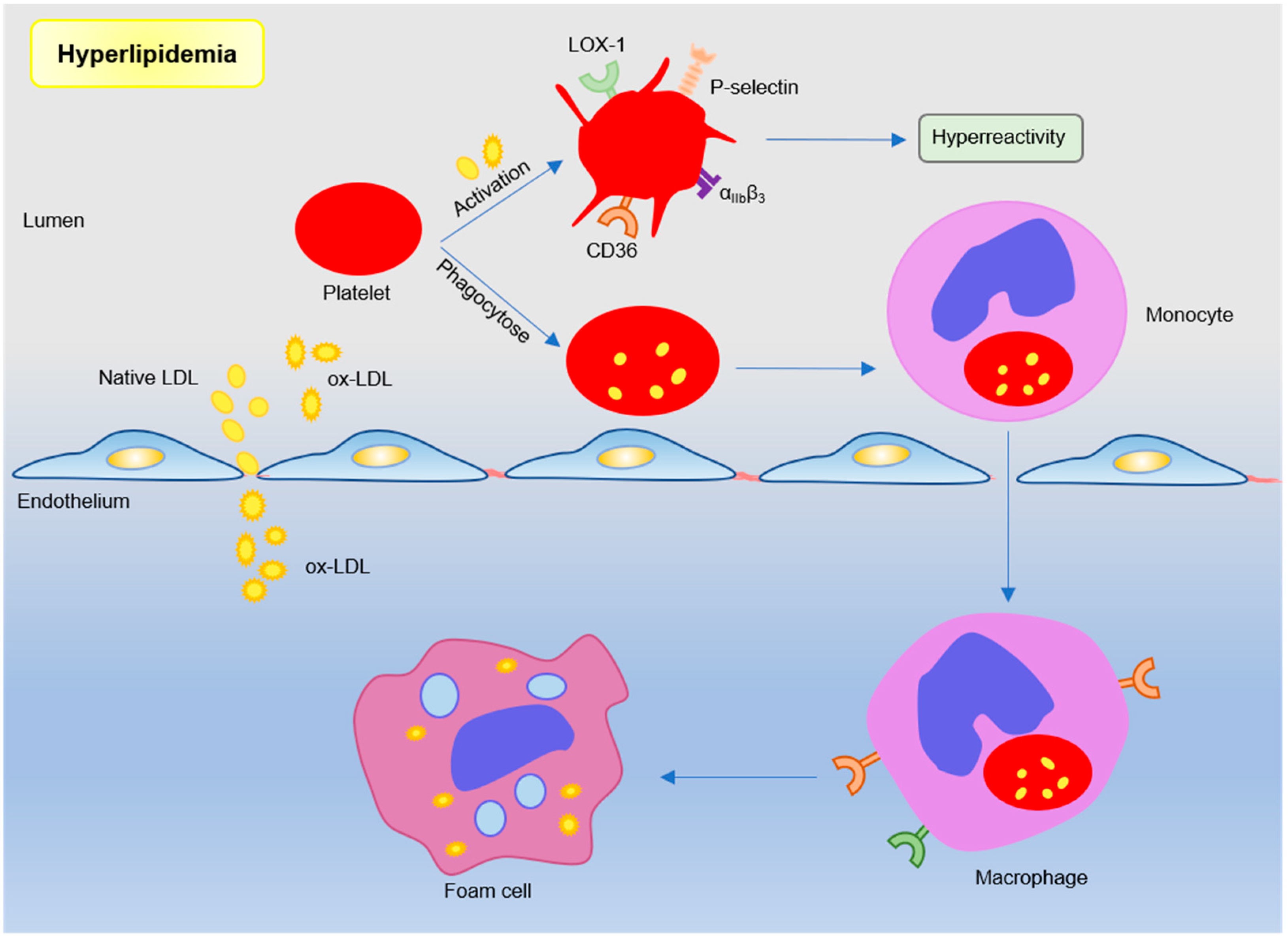
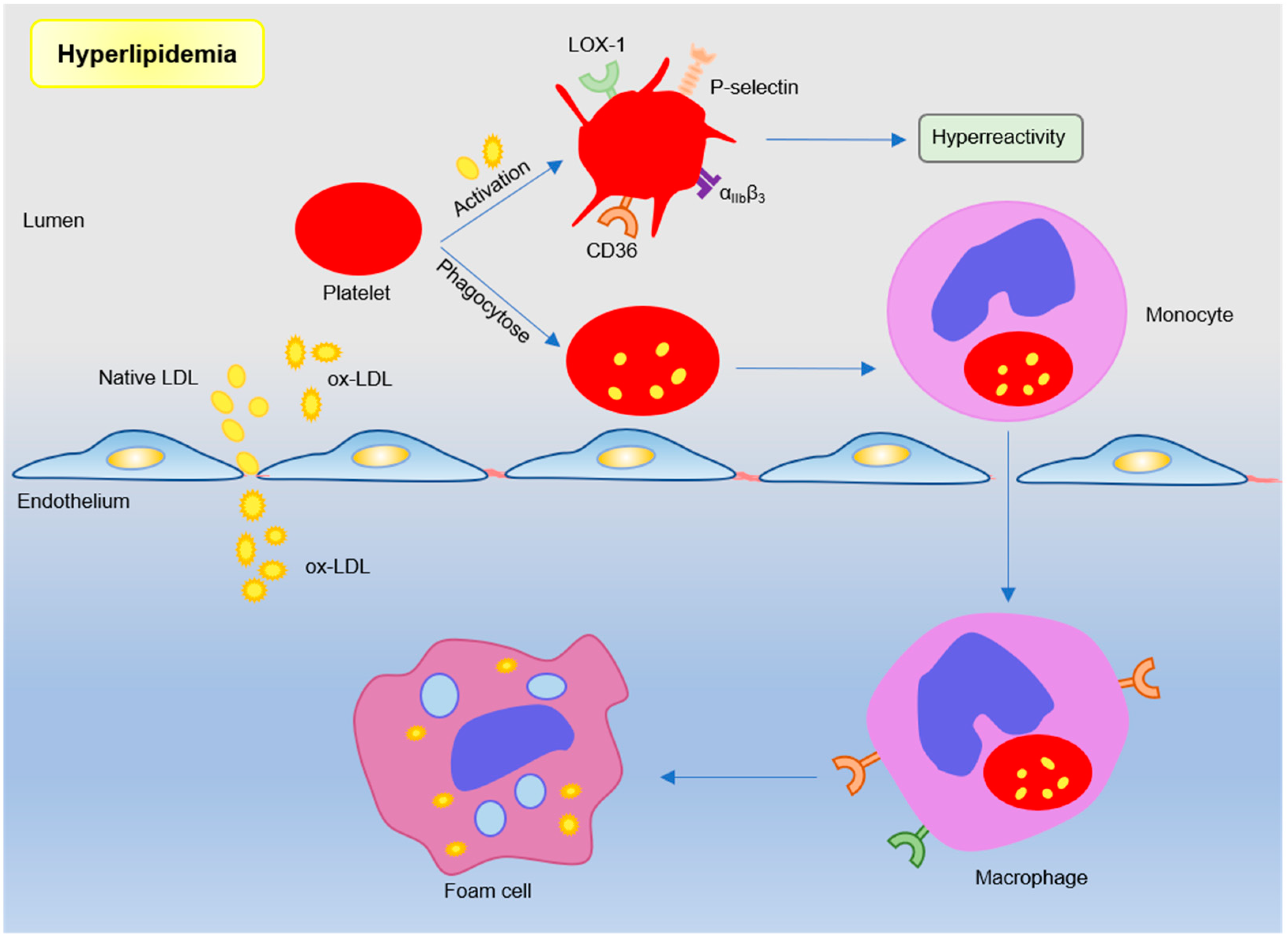
2.2. Hyperlipidemia Modulated Platelets in AS
2.3. Hyperglycemia-Modulated Platelets in AS
3. Platelet Receptors in Atherogenesis
3.1. Integrin αIIbβ3
3.2. GPIb-V-IX Complex
3.3. GPVI
3.4. P-Selectin
3.5. Thrombin Receptors
3.6. ADP Receptors
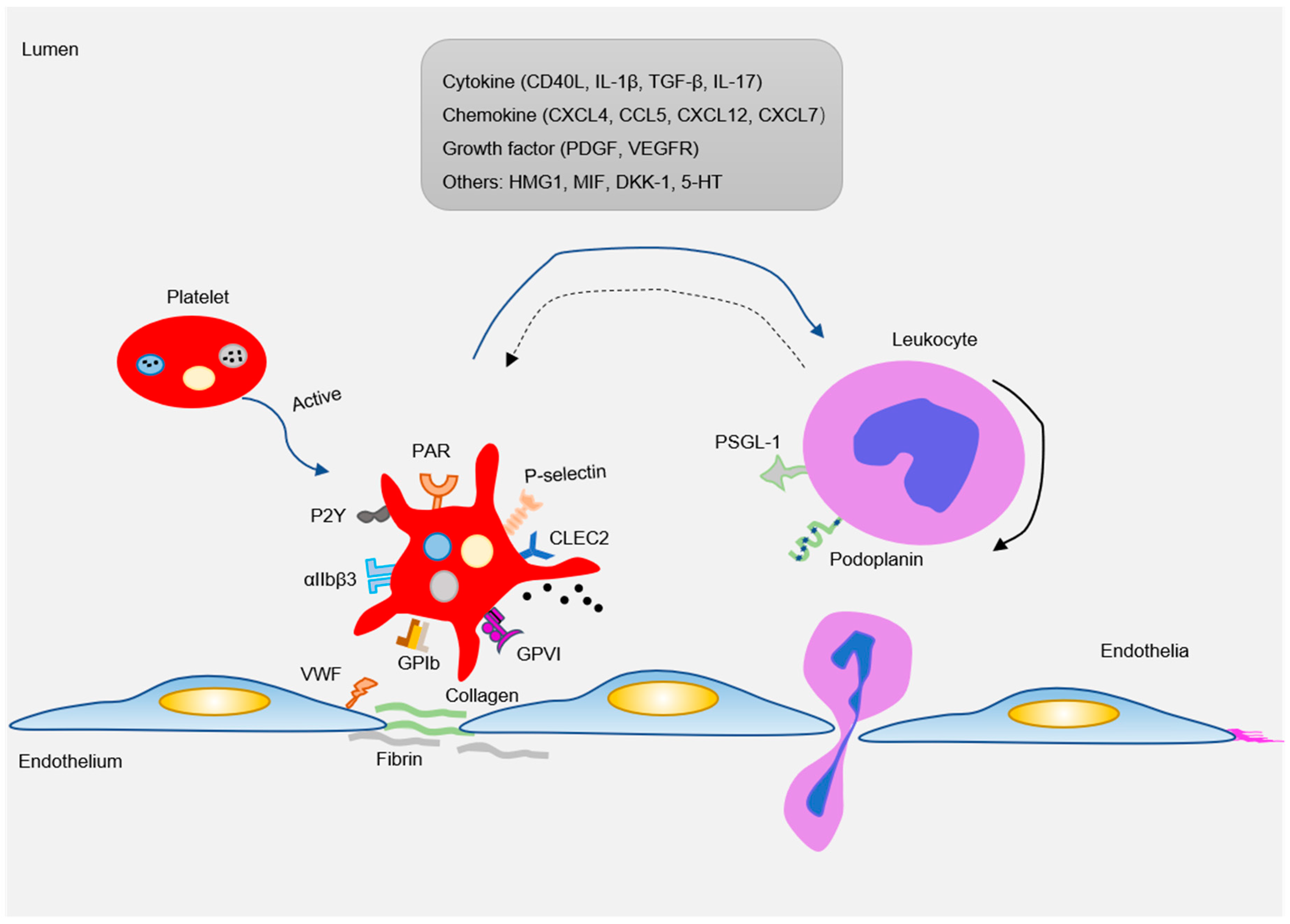
4. Platelet-Derived Proinflammatory Mediators in Atherogenesis
4.1. Cytokines
4.2. Chemokines
4.2.1. CXCL4
4.2.2. CCL5
4.2.3. CXCL7
4.2.4. CXCL12
4.3. Other Platelet-Derived Inflammatory Mediators
5. Platelet Migration in Atherogenesis
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS | Atherosclerosis |
| D-flow | Disturbed flow |
| ECs | Endothelial cells |
| ICAM-1 | Intercellular adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| vWF | Von Willebrand factor |
| GPIbα | Glycoprotein (GP) Ibα |
| GPVI | Glycoprotein VI |
| αIIbβ3 | Integrin αIIbβ3 |
| FLIPRs | Flow-induced protrusions |
| PMPs | Platelet microparticles |
| PECAM-1 | Platelet endothelial cell adhesion molecule-1 |
| MACE | Major cardiovascular adverse event |
| LDL | Low-density lipoprotein |
| HDL | High-density lipoprotein |
| LRP | LDL receptor-related protein |
| FAK | Focal adhesion kinase |
| oxLDL | Oxidized low-density lipoprotein |
| VSMCs | Vascular smooth muscle cells |
| SMCs | Smooth muscle cells |
| LOX-1 | Lectin-like oxidized LDL receptor 1 |
| DM | Diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| PGI2 | Prostacyclin |
| glycLDL | Glycosylated LDL |
| MPV | Mean platelet volume |
| sGPVI | Soluble dimeric GPVI |
| CAD | Coronary atherosclerotic heart disease |
| CEU | Contrast-enhanced ultrasound |
| PSGL-1 | P-selectin glycoprotein ligand 1 |
| DCs | Dendritic cells |
| PARs | Protease-activated receptors |
| IL-1β | Interleukin (IL)-1β |
| TRAF | TNF receptor-related factor |
| PF-4 | Platelet factor 4 |
| NETs | Neutrophil extracellular traps |
| PBP | Platelet basic protein |
| CTAP-III | Connective tissue-activating peptide III |
| β-TG | β thrombin |
| NAP-2 | Neutrophil-activating peptide 2 |
| SDF-1 | Stromal cell-derived factor-1 |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptors |
| CyPA | Cyclophilin A |
| HMG1 | Amphoterin |
References
- Huo, Y.; Ley, K.F. Role of platelets in the development of atherosclerosis. Trends Cardiovasc. Med. 2004, 14, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar]
- Aukrust, P.; Halvorsen, B.; Ueland, T.; Michelsen, A.E.; Skjelland, M.; Gullestad, L.; Yndestad, A.; Otterdal, K. Activated platelets and atherosclerosis. Expert Rev. Cardiovasc. Ther. 2010, 8, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Xing, R.; Cui, X.; Xiao, Y.; Xie, L.; You, P.; Wang, T.; Zeng, L.; Peng, W.; et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis 2018, 271, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Rayfield, E.J. How hyperglycemia promotes atherosclerosis: Molecular mechanisms. Cardiovasc. Diabetol. 2002, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hundelshausen, P.; Weber, C. Platelets as Immune Cells Bridging Inflammation and Cardiovascular Disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef]
- Siegel-Axel, D.; Daub, K.; Seizer, P.; Lindemann, S.; Gawaz, M. Platelet lipoprotein interplay: Trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008, 78, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, J.H. The Platelet: Form and Function. Semin. Hematol. 2006, 43, S94–S100. [Google Scholar] [CrossRef]
- Li, Z.; Yang, F.; Dunn, S.; Gross, A.K.; Smyth, S.S. Platelets as immune mediators: Their role in host defense responses and sepsis. Thromb. Res. 2011, 127, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Von Hundelshausen, P.; Duchene, J. Platelet-derived chemokines in atherosclerosis. Hämostaseologie 2017, 35, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nording, H.; Baron, L.; Langer, H.F. Platelets as therapeutic Targets to prevent Atherosclerosis. Atherosclerosis 2020, 307, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Ni, C.W.; Rezvan, A.; Suo, J.; Budzyn, K.; Llanos, A.; Harrison, D.; Giddens, D.; Jo, H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1535–H1543. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C. Atheroprotective signaling mechanisms activated by steady laminar flow in endothelial cells. Circulation 2008, 117, 1082–1089. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chen, C.N.; Lee, P.L.; Yang, C.T.; Chuang, H.S.; Chien, S.; Usami, S. Analysis of the effect of disturbed flow on monocytic adhesion to endothelial cells. J. Biomech. 2003, 36, 1883–1895. [Google Scholar] [CrossRef]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M.E.M. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Glise, L.; Larsson, P.; Jern, S.; Borén, J.; Levin, M.; Ny, T.; Fogelstrand, P.; Bergh, N. Disturbed Laminar Blood Flow Causes Impaired Fibrinolysis and Endothelial Fibrin Deposition In Vivo. Thromb. Haemost. 2019, 119, 223–233. [Google Scholar] [CrossRef]
- Skilbeck, C.A.; Walker, P.G.; David, T.; Nash, G.B. Disturbed flow promotes deposition of leucocytes from flowing whole blood in a model of a damaged vessel wall. Br. J. Haematol. 2004, 126, 418–427. [Google Scholar] [CrossRef]
- Tersteeg, C.; Heijnen, H.F.G.; Eckly, A.; Pasterkamp, G.; Urbanus, R.T.; Maas, C.; Hoefer, I.E.; Nieuwland, R.; Farndale, R.W.; Gachet, C. FLow-Induced PRotrusions (FLIPRs): A Platelet-Derived Platform for the Retrieval of Microparticles by Monocytes and Neutrophils. Circ. Res. 2014, 114, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Z.; Hu, Y. Possible roles of platelet-derived microparticles in atherosclerosis. Atherosclerosis 2016, 248, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Meza, D.; Shanmugavelayudam, S.K.; Mendoza, A.; Sanchez, C.; Rubenstein, D.A.; Yin, W. Platelets modulate endothelial cell response to dynamic shear stress through PECAM-1. Thromb Res. 2017, 150, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Stevens, H.Y.; Melchior, B.; Bell, K.S.; Yun, S.J.; Yeh, J.C.; Frangos, J.A. PECAM-1 is a critical mediator of atherosclerosis. Dis. Model Mech. 2008, 1, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Halcox, J.P.; Banegas, J.R.; Roy, C.; Dallongeville, J.; De Backer, G.; Guallar, E.; Perk, J.; Hajage, D.; Henriksson, K.M.; Borghi, C. Prevalence and treatment of atherogenic dyslipidemia in the primary prevention of cardiovascular disease in Europe: EURIKA, a cross-sectional observational study. BMC Cardiovasc. Disord. 2017, 17, 160. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cai, G.; Wang, Y.; Liu, R.; Xi, Z.; Li, G.; Wen, W.; Wu, Y.; Wang, C.; Ji, Q.; et al. Comparison of long-term outcomes of young patients after a coronary event associated with familial hypercholesterolemia. Lipids Health Dis. 2019, 18, 131. [Google Scholar] [CrossRef] [Green Version]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. BioMed Res. Int. 2018, 2018, 6508709. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, P.K.; Hughes, S.M.T.; Plumb, R.D.; Devine, A.; Mcveigh, G.E. Statins have beneficial effects on platelet free radical activity and intracellular distribution of GTPases in hyperlipidaemia. Clin. Sci. 2010, 118, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Surya, I.I.; Akkerman, J.N. The influence of lipoproteins on blood platelets. Am. Heart J. 1993, 125, 272–275. [Google Scholar] [CrossRef]
- Sener, A.; Enc, E.; Ozsavci, D.; Vanizor-Kural, B.; Demir, M. Exogenous L-Arginine and HDL Can Alter LDL and ox-LDL-Mediated Platelet Activation: Using Platelet P-Selectin Receptor Numbers. Clin. Appl. Thromb./Hemost. 2010, 17, E79–E86. [Google Scholar] [CrossRef]
- Koller, E.; Koller, F.; Doleschel, W. Specific Binding Sites on Human Blood Platelets for Plasma Lipoproteins. Biol. Chem. 1982, 363, 395–406. [Google Scholar] [CrossRef]
- Riddell, D.R.; Vinogradov, D.V.; Stannard, A.K.; Chadwick, N.; Owen, J.S. Identification and characterization of LRP8 (apoER2) in human blood platelets. J. Lipid Res. 1999, 40, 1925–1930. [Google Scholar] [PubMed]
- Relou, I.A.M.; Hackeng, C.M.; Akkerman, J.N.; Malle, E. Low-density lipoprotein and its effect on human blood platelets. Cell. Mol. Life Sci. 2003, 60, 961–971. [Google Scholar] [CrossRef] [PubMed]
- van der Stoep, M.; Korporaal, S.J.; Van Eck, M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc. Res. 2014, 103, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Sauter, R.; Fahrleitner, M.; Grimm, J.; Stakos, D.; Emschermann, F.; Panagiota, V.; Gnerlich, S.; Perk, A.; Schonberger, T. Binding of Oxidized Low-Density Lipoprotein on Circulating Platelets Is increased in Patients With Acute Coronary Syndromes and Induces Platelet Adhesion to Vascular Wall In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2017–2020. [Google Scholar] [CrossRef] [Green Version]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.S.; Mcneil, C.; Wheatcroft, S.B.; Yuldasheva, N.; Febbriao, M. Oxidized LDL activates blood platelets through CD36/NOX2–mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- Zimman, A.; Titz, B.; Komisopoulou, E.; Biswas, S.; Graeber, T.G.; Podrez, E.A. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS ONE 2014, 9, e84488. [Google Scholar] [CrossRef]
- Daub, K.; Seizer, P.; Stellos, K.; Kramer, B.F.; Bigalke, B.; Schaller, M.; Fatehmoghadam, S.; Gawaz, M.; Lindemann, S. Oxidized LDL-Activated Platelets Induce Vascular Inflammation. Semin. Thromb. Hemost. 2010, 36, 146–156. [Google Scholar] [CrossRef]
- Yue, H.; Febbraio, M.; Klenotic, P.A.; Kennedy, D.J.; Wu, Y.; Chen, S.; Gohara, A.F.; Li, O.; Belcher, A.; Kuang, B. CD36 Enhances Vascular Smooth Muscle Cell Proliferation and Development of Neointimal Hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-Dependent Surface Expression of LOX-1 in Human Platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Ogura, S.; Little, P.J.; Xu, S.; Sawamura, T. Targeting LOX-1 in atherosclerosis and vasculopathy: Current knowledge and future perspectives. Ann. N. Y. Acad. Sci. 2019, 1443, 34–53. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capes, S.E.; Hunt, D.L.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Woo, V.; Bose, R. Platelet hyperactivity and abnormal Ca2+ homeostasis in diabetes mellitus. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H1480–H1489. [Google Scholar] [CrossRef] [PubMed]
- Keating, F.K.; Sobel, B.E.; Schneider, D.J. Effects of Increased Concentrations of Glucose on Platelet Reactivity in Healthy Subjects and in Patients With and Without Diabetes Mellitus. Am. J. Cardiol. 2003, 92, 1362–1365. [Google Scholar] [CrossRef]
- Ferroni, P.; Basili, S.; Falco, A.; Davi, G. Platelet activation in type 2 diabetes mellitus. J. Thromb. Haemost. 2004, 2, 1282–1291. [Google Scholar] [CrossRef]
- Winocour, P.D. Platelet Abnormalities in Diabetes Mellitus. Diabetes 1992, 41, 26–31. [Google Scholar] [CrossRef]
- Ferretti, G.; Rabini, R.A.; Bacchetti, T.; Vignini, A.; Salvolini, E.; Ravaglia, F.; Curatola, G.; Mazzanti, L. Glycated Low Density Lipoproteins Modify Platelet Properties: A Compositional and Functional Study. J. Clin. Endocrinol. Metab. 2002, 87, 2180–2184. [Google Scholar] [CrossRef]
- Ju, L.; McFadyen, J.D.; Al-Daher, S.; Alwis, I.; Chen, Y.; Tønnesen, L.L.; Maiocchi, S.; Coulter, B.; Calkin, A.C.; Felner, E.I.; et al. Compression force sensing regulates integrin αIIbβ3 adhesive function on diabetic platelets. Nat. Commun. 2018, 9, 1087. [Google Scholar] [CrossRef] [Green Version]
- Rusak, T.; Misztal, T.; Rusak, M.; Branskajanuszewska, J.; Tomasiak, M. Involvement of hyperglycemia in the development of platelet procoagulant response: The role of aldose reductase and platelet swelling. Blood Coagul. Fibrinolysis 2017, 28, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, N.; Yngen, M.; Ostenson, C.; Wallen, N.H.; Hjemdahl, P. Enhanced leukocyte–platelet cross-talk in Type 1 diabetes mellitus: Relationship to microangiopathy. J. Thromb. Haemost. 2004, 2, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patko, Z.; Csaszar, A.; Acsady, G.; Őry, I.; Takacs, E.; Fűresz, J. Elevation of monocyte-platelet aggregates is an early marker of type 2 diabetes. Interv. Med. Appl. Sci. 2012, 4, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Lee, M.K.S.; Alsharea, A.; Dragoljevic, D.; Barrett, T.J.; Montenont, E.; Basu, D.; Heywood, S.E.; Kammoun, H.L.; Flynn, M.C. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J. Clin. Investig. 2017, 127, 2133–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.H.; Bergmeier, W. Sugar makes neutrophils RAGE: Linking diabetes-associated hyperglycemia to thrombocytosis and platelet reactivity. J. Clin. Investig. 2017, 127, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.D.; Echagarruga, C.; Ogando, Y.; Montenont, E.; Chen, Y.; Fisher, E.A.; Berger, J.S. Hyperglycemia enhances arsenic-induced platelet and megakaryocyte activation. J. Transl. Med. 2017, 15, 55. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J. Interleukin 1 Receptor 1 and Interleukin 1β Regulate Megakaryocyte Maturation, Platelet Activation, and Transcript Profile During Inflammation in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, L.; Thomson, G.J.A.; Adams, R.C.M.; Nell, T.A.; Laubscher, W.A.; Pretorius, E. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, H.; Kleiman, N.S. Platelet pathophysiology, pharmacology, and function in coronary artery disease. Coron. Artery Dis. 2017, 28, 614–623. [Google Scholar] [CrossRef]
- Shattil, S.J. Signaling through platelet integrin αIIbβ3: Inside-out, outside-in, and sideways. Thromb. Haemost. 1999, 82, 318–325. [Google Scholar] [CrossRef]
- Shpilberg, O.; Rabi, I.; Schiller, K.; Walden, R.; Harats, D.; Tyrrell, K.S.; Coller, B.; Seligsohn, U. Patients With Glanzmann Thrombasthenia Lacking Platelet Glycoprotein αIIbβ3 (GPIIb/IIIa) and αvβ3 Receptors Are Not Protected From Atherosclerosis. Circulation 2002, 105, 1044–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, S.; Zemany, L.; Standley, K.N.; Novack, D.V.; La Regina, M.; Bernalmizrachi, C.; Coleman, T.; Semenkovich, C.F. β3 integrin deficiency promotes atherosclerosis and pulmonary inflammation in high-fat-fed, hyperlipidemic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 6730–6735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, A.; Feng, Z.; Chandran, R.R.; Kabir, I.; Rotllan, N.; Aryal, B.; Sheikh, A.Q.; Ding, L.; Qin, L.; Fernandezhernando, C. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat. Commun. 2018, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Enders, G.; Matos, F.C.; Tomic, L.I.; Leiderer, R.; Eisenmenger, K.; Krombach, F. Fibrinogen Deposition at the Postischemic Vessel Wall Promotes Platelet Adhesion During Ischemia-Reperfusion In Vivo. Blood 1999, 94, 3829–3838. [Google Scholar] [CrossRef]
- Massberg, S.; Schurzinger, K.; Lorenz, M.; Konrad, I.; Schulz, C.; Plesnila, N.; Kennerknecht, E.; Rudelius, M.; Sauer, S.; Braun, S. Platelet Adhesion Via Glycoprotein IIb Integrin Is Critical for Atheroprogression and Focal Cerebral Ischemia An In Vivo Study in Mice Lacking Glycoprotein IIb. Circulation 2005, 112, 1180–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maile, L.A.; Busby, W.H.; Xi, G.; Gollahan, K.P.; Flowers, W.L.; Gafbacik, N.; Gafbacik, S.; Stewart, K.; Merricks, E.P.; Nichols, T.C. An anti-αVβ3 antibody inhibits coronary artery atherosclerosis in diabetic pigs. Atherosclerosis 2017, 258, 40–50. [Google Scholar] [CrossRef]
- Lopez, J.A. The platelet glycoprotein Ib-IX complex. Blood Coagul. Fibrinolysis 2017, 5, 97–120. [Google Scholar] [CrossRef]
- Massberg, S.; Brand, K.; Gruner, S.; Page, S.; Muller, E.; Muller, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 2002, 196, 887–896. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Sundd, P.; Zarpellon, A.; Ouyang, H.; Mikulski, Z.; Zampolli, A.; Ruggeri, Z.M.; Ley, K. Genetic deletion of platelet glycoprotein Ib alpha but not its extracellular domain protects from atherosclerosis. Thromb. Haemost. 2014, 112, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Methia, N.; Andre, P.; Denis, C.V.; Economopoulos, M.; Wagner, D.D. Localized reduction of atherosclerosis in von Willebrand factor–deficient mice. Blood 2001, 98, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Manka, D.; Forlow, S.B.; Sanders, J.M.; Hurwitz, D.; Bennett, D.K.; Green, S.A.; Ley, K.; Sarembock, I.J. Critical Role of Platelet P-Selectin in the Response to Arterial Injury in Apolipoprotein-E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Strassel, C.; Hechler, B.; Bull, A.; Gachet, C.; Lanza, F. Studies of mice lacking the GPIb-V-IX complex question the role of this receptor in atherosclerosis. J. Thromb. Haemost. 2009, 7, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- Mangin, P.H.; Onselaer, M.; Receveur, N.; Lay, N.L.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Loyau, S.; Dupuis, A.; Babar, A.K. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshehri, O.; Hughes, C.E.; Montague, S.J.; Watson, S.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonberger, T.; Siegel-Axel, D.; Bussl, R.; Richter, S.; Judenhofer, M.S.; Haubner, R.; Reischl, G.; Klingel, K.; Munch, G.; Seizer, P.; et al. The immunoadhesin glycoprotein VI-Fc regulates arterial remodelling after mechanical injury in ApoE-/- mice. Cardiovasc. Res. 2008, 80, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawaz, M.; Vogel, S.; Pfannenberg, C.; Pichler, B.J.; Langer, H.F.; Bigalke, B. Implications of glycoprotein VI for theranostics. Thromb. Haemost. 2014, 112, 26–31. [Google Scholar]
- Villmann, J.M.; Burkhardt, R.; Teren, A.; Villmann, T.; Thiery, J.; Drogies, T. Atherosclerosis, myocardial infarction and primary hemostasis: Impact of platelets, von Willebrand factor and soluble glycoprotein VI. Thromb. Res. 2019, 180, 98–104. [Google Scholar] [CrossRef]
- Massberg, S.; Konrad, I.; Bultmann, A.; Schulz, C.; Munch, G.; Peluso, M.; Lorenz, M.; Schneider, S.; Besta, F.; Muller, I. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2003, 18, 397–399. [Google Scholar] [CrossRef]
- Bultmann, A.; Li, Z.; Wagner, S.; Peluso, M.; Schonberger, T.; Weis, C.; Konrad, I.; Stellos, K.; Massberg, S.; Nieswandt, B.; et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis—A possible role of fibronectin. J. Mol. Cell Cardiol. 2010, 49, 532–542. [Google Scholar] [CrossRef]
- Jamasbi, J.; Megens, R.T.A.; Bianchini, M.; Uhland, K.; Munch, G.; Ungerer, M.; Sherman, S.; Faussner, A.; Brandl, R.; John, C. Cross-Linking GPVI-Fc by Anti-Fc Antibodies Potentiates Its Inhibition of Atherosclerotic Plaque- and Collagen-Induced Platelet Activation. JACC Basic Transl. Sci. 2016, 1, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Metzger, K.; Vogel, S.; Chatterjee, M.; Borst, O.; Seizer, P.; Schonberger, T.; Geisler, T.; Lang, F.; Langer, H.F.; Rheinlaender, J. High-frequency ultrasound-guided disruption of glycoprotein VI-targeted microbubbles targets atheroprogressison in mice. Biomaterials 2015, 36, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, J.; Megens, R.T.A.; Bianchini, M.; Munch, G.; Ungerer, M.; Faussner, A.; Sherman, S.; Walker, A.; Goyal, P.; Jung, S.M. Differential Inhibition of Human Atherosclerotic Plaque-Induced Platelet Activation by Dimeric GPVI-Fc and Anti-GPVI Antibodies Functional and Imaging Studies. J. Am. Coll. Cardiol. 2015, 65, 2404–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestweber, D.; Blanks, J.E. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999, 79, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.L.; Huo, Y.; Jung, U.; Ghosh, S.; Manka, D.; Sarembock, I.J.; Ley, K. Direct Demonstration of P-Selectin– and VCAM-1–Dependent Mononuclear Cell Rolling in Early Atherosclerotic Lesions of Apolipoprotein E–Deficient Mice. Circ. Res. 1999, 84, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, P.C.; Wagner, D.D. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003, 101, 2661–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, M.; Saffaripour, S.; Watson, S.R.; Mayadas, T.N.; Hynes, R.O.; Wagner, D.D. Reduced recruitment of inflammatory cells in a contact hypersensitivity response in P-selectin-deficient mice. J. Exp. Med. 1995, 181, 2277–2282. [Google Scholar] [CrossRef]
- Johnson, R.C.; Schaefer, E.J.; Wagner, D.D. Absence of P-selectin delays fatty streak formation in mice. J. Clin. Investig. 1997, 99, 1037–1043. [Google Scholar] [CrossRef]
- Dong, Z.M.; Brown, A.A.; Wagner, D.D. Prominent Role of P-Selectin in the Development of Advanced Atherosclerosis in ApoE-Deficient Mice. Circulation 2000, 101, 2290–2295. [Google Scholar] [CrossRef]
- Ye, Z.; Zhong, L.; Zhu, S.; Wang, Y.; Zheng, J.; Wang, S.; Zhang, J.; Huang, R. The P-selectin and PSGL-1 axis accelerates atherosclerosis via activation of dendritic cells by the TLR4 signaling pathway. Cell Death Dis. 2019, 10, 507. [Google Scholar] [CrossRef]
- Sarma, J.; Laan, C.A.; Alam, S.; Jha, A.; Fox, K.A.A.; Dransfield, I. Increased Platelet Binding to Circulating Monocytes in Acute Coronary Syndromes. Circulation 2002, 105, 2166–2171. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, A.S.; Mcintyre, T.M.; Mcever, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Ay, C.; Riedl, J.; Kopp, C.W.; Eichelberger, B.; Koppensteiner, R.; Panzer, S. Platelet-specific markers are associated with monocyte-platelet aggregate formation and thrombin generation potential in advanced atherosclerosis. Thromb. Haemost. 2015, 115, 615–621. [Google Scholar] [PubMed] [Green Version]
- Zhang, N.; Liu, Z.; Yao, L.; Mehtadsouza, P.; Mcever, R.P. P-Selectin Expressed by a Human SELP Transgene Is Atherogenic in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorell, L.; Martinez-Gonzalez, J.; Rodriguez, C.; Gentile, M.; Calvayrac, O.; Badimon, L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb. Haemost. 2008, 99, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.; Huang, T.; Koukos, G.; Fletcher, E.K.; Turner, S.E.; Shearer, A.M.; Gurbel, P.A.; Rade, J.J.; Kimmelstiel, C.; Bliden, K.P. Noncanonical Matrix Metalloprotease 1–Protease-Activated Receptor 1 Signaling Drives Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1368–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, S.; Singh, N.K.; Mani, A.M.; Rao, G.N. Protease-activated receptor 1 inhibits cholesterol efflux and promotes atherogenesis via cullin 3–mediated degradation of the ABCA1 transporter. J. Biol. Chem. 2018, 293, 10574–10589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.M.; Mann, A.; Conrad, K.; Saum, K.; Hall, D.; Mckinney, L.M.; Robbins, N.; Thompson, J.C.; Peairs, A.; Camerer, E. PAR2 (Protease-Activated Receptor 2) Deficiency Attenuates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Phuong, P.T.; Fukuda, D.; Yamaguchi, K.; Murata, C.; Nishimoto, S.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T. Protease-Activated Receptor-2 Plays a Critical Role in Vascular Inflammation and Atherosclerosis in Apolipoprotein E–Deficient Mice. Circulation 2018, 138, 1706–1719. [Google Scholar] [CrossRef]
- Hikita, T.; Mirzapourshafiyi, F.; Barbacena, P.; Riddell, M.; Pasha, A.; Li, M.; Kawamura, T.; Brandes, R.P.; Hirose, T.; Ohno, S. PAR-3 controls endothelial planar polarity and vascular inflammation under laminar flow. EMBO Rep. 2018, 19, e45253. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Nakanishimatsui, M.; Zheng, Y.; Sulciner, D.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Covic, L.; Kuliopulos, A. Protease-Activated Receptors in Cardiovascular Diseases. Circulation 2006, 114, 1070–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelken, N.A.; Soifer, S.J.; Okeefe, J.; Vu, T.H.; Charo, I.F.; Coughlin, S.R. Thrombin receptor expression in normal and atherosclerotic human arteries. J. Clin. Investig. 1992, 90, 1614–1621. [Google Scholar] [CrossRef]
- Ruf, W. Proteases, Protease-Activated Receptors, and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1252–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, S.; Singh, N.K.; Gali, S.; Mani, A.M.; Rao, G.N. Protein Kinase Cθ Via Activating Transcription Factor 2-Mediated CD36 Expression and Foam Cell Formation of Ly6Chi Cells Contributes to Atherosclerosis. Circulation 2018, 138, 2395–2412. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Cornelissen, I.; Mountford, J.K.; Coughlin, S.R. Atherosclerosis proceeds independently of thrombin-induced platelet activation in ApoE-/- mice. Atherosclerosis 2009, 205, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Scridon, A.; Mărginean, A.; Huțanu, A.; Chinezu, L.; Gheban, D.; Perian, M.; Vântu, A.; Gherțescu, D.; Fișcă, P.C.; Șerban, R.C. Vascular protease-activated receptor 4 upregulation, increased platelet aggregation, and coronary lipid deposits induced by long-term dabigatran administration—results from a diabetes animal model. J. Thromb. Haemost. 2019, 17, 538–550. [Google Scholar] [CrossRef]
- Hechler, B.; Lenain, N.; Marchese, P.; Vial, C.; Heim, V.; Freund, M.; Cazenave, J.; Cattaneo, M.; Ruggeri, Z.M.; Evans, R.J. A role of the fast ATP-gated P2X1 cation channel in thrombosis of small arteries in vivo. J. Exp. Med. 2003, 198, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef] [Green Version]
- Hechler, B.; Leon, C.; Vial, C.; Vigne, P.; Frelin, C.; Cazenave, J.; Gachet, C. The P2Y1 receptor is necessary for adenosine 5’-diphosphate-induced platelet aggregation. Blood 1998, 92, 152–159. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hechler, B.; Nonne, C.; Roh, E.J.; Cattaneo, M.; Cazenave, J.; Lanza, F.; Jacobson, K.A.; Gachet, C. MRS2500 [2-Iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a Potent, Selective, and Stable Antagonist of the Platelet P2Y1 Receptor with Strong Antithrombotic Activity in Mice. J. Pharmacol. Exp. Ther. 2006, 316, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, C.; Ravanat, C.; Freund, M.; Cazenave, J.; Gachet, C. Differential Involvement of the P2Y1 and P2Y12 Receptors in Platelet Procoagulant Activity. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1941–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amison, R.T.; Momi, S.; Morris, A.; Manni, G.; Keir, S.; Gresele, P.; Page, C.P.; Pitchford, S.C. RhoA signaling through platelet P2Y1 receptor controls leukocyte recruitment in allergic mice. J. Allergy Clin. Immunol. 2015, 135, 528–538. [Google Scholar] [CrossRef]
- Zerr, M.; Hechler, B.; Freund, M.; Magnenat, S.; Lanois, I.; Cazenave, J.; Leon, C.; Gachet, C. Major Contribution of the P2Y1 Receptor in Purinergic Regulation of TNFα-Induced Vascular Inflammation. Circulation 2011, 123, 2404–2413. [Google Scholar] [CrossRef] [Green Version]
- Hechler, B.; Freund, M.; Ravanat, C.; Magnenat, S.; Cazenave, J.P.; Gachet, C. Reduced atherosclerotic lesions in P2Y1/apolipoprotein E double-knockout mice: The contribution of non-hematopoietic-derived P2Y1 receptors. Circulation 2008, 118, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Gachet, C. Regulation of platelet functions by p2 receptors. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 277–300. [Google Scholar] [CrossRef]
- Cattaneo, M. Platelet P2 receptors: Old and new targets for antithrombotic drugs. Expert Rev. Cardiovasc. Ther. 2007, 5, 45–55. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Zhang, L.; Luo, X.; Li, J.; Chen, X.; Niu, H.; Wang, K.; Sun, Y.; Wang, X. Roles of Purinergic Receptor P2Y, G Protein–Coupled 12 in the Development of Atherosclerosis in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e81–e89. [Google Scholar] [CrossRef] [Green Version]
- West, L.; Steiner, T.; Judge, H.M.; Francis, S.E.; Storey, R.F. Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc. Res. 2014, 102, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yu, C.; Pi, S.; Mao, L.; Hu, B. The role of P2Y 12 receptor in ischemic stroke of atherosclerotic origin. Cell. Mol. Life Sci. 2019, 76, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Boulaftali, Y.; Owens, A.P.; Beale, A.; Piatt, R.; Casari, C.; Lee, R.H.; Conley, P.B.; Paul, D.S.; Mackman, N.; Bergmeier, W. CalDAG-GEFI Deficiency Reduces Atherosclerotic Lesion Development in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; Mcintyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rechciński, T.; Grębowska, A.; Kurpesa, M.; Sztybrych, M.; Peruga, J.Z.; Trzos, E.; Rudnicka, W.; Krzemińskapakuła, M.; Chmiela, M. Interleukin-1b and interleukin-1 receptor inhibitor gene cluster polymorphisms in patients with coronary artery disease after percutaneous angioplasty or coronary artery bypass grafting. Kardiologia Polska 2009, 67, 601. [Google Scholar]
- Gorący, I.; Kaczmarczyk, M.; Ciechanowicz, A.; Lewandowska, K.; Jakubiszyn, P.; Bodnar, O.; Kopijek, B.; Brodkiewicz, A.; Cyrylowski, L. Polymorphism of Interleukin 1B May Modulate the Risk of Ischemic Stroke in Polish Patients. Med.-Buenos Aires 2019, 55, 558. [Google Scholar] [CrossRef] [Green Version]
- Garlichs, C.D.; Kozina, S.; Fatehmoghadam, S.; Tomandl, B.; Stumpf, C.; Eskafi, S.; Raaz, D.; Schmeiser, A.; Yilmaz, A.; Ludwig, J. Upregulation of CD40-CD40 Ligand (CD154) in Patients with Acute Cerebral Ischemia. Stroke 2003, 34, 1412–1418. [Google Scholar] [CrossRef]
- Varo, N.; De Lemos, J.A.; Libby, P.; Morrow, D.A.; Murphy, S.A.; Nuzzo, R.; Gibson, C.M.; Cannon, C.P.; Braunwald, E.; Schonbeck, U. Soluble CD40L: Risk prediction after acute coronary syndromes. Circulation 2003, 108, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Sanguigni, V.; Pignatelli, P.; Lenti, L.; Ferro, D.; Bellia, A.; Carnevale, R.; Tesauro, M.; Sorge, R.; Lauro, R.; Violi, F. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation 2005, 111, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Mullerberghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- Mach, F.; Schonbeck, U.; Sukhova, G.K.; Atkinson, E.; Libby, P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 1998, 394, 200–203. [Google Scholar] [CrossRef]
- Lutgens, E.; Gorelik, L.; Daemen, M.J.A.P.; Ed, D.M.; Grewal, I.S.; Koteliansky, V.; Flavell, R.A. Requirement for CD154 in the progression of atherosclerosis. Nat. Med. 1999, 5, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, M.; Dworacki, G.; Kufelgrabowska, J.; Watala, C.; Kozubski, W. Upregulation of CD40 ligand and enhanced monocyte-platelet aggregate formation are associated with worse clinical outcome after ischaemic stroke. Thromb. Haemost. 2012, 107, 346–355. [Google Scholar] [PubMed]
- Bavendiek, U.; Zirlik, A.; Laclair, S.; Macfarlane, L.A.; Libby, P.; Schonbeck, U. Atherogenesis in Mice Does Not Require CD40 Ligand From Bone Marrow–Derived Cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Prasad, K.; Denis, C.; Papalia, J.; Wagner, D. CD40L stabilizes arterial thrombi by a 3 integrin-dependent mechanism. Nat. Med. 2001, 8, 247–252. [Google Scholar] [CrossRef]
- Henn, V.; Steinbach, S.; Buchner, K.; Presek, P.; Kroczek, R.A. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood 2001, 98, 1047–1054. [Google Scholar] [CrossRef]
- Zirlik, A.; Bavendiek, U.; Libby, P.; Macfarlane, L.A.; Gerdes, N.; Jagielska, J.; Ernst, S.; Aikawa, M.; Nakano, H.; Tsitsikov, E. TRAF-1, -2, -3, -5, and -6 Are Induced in Atherosclerotic Plaques and Differentially Mediate Proinflammatory Functions of CD40L in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1101–1107. [Google Scholar] [CrossRef]
- Lutgens, E.; Lievens, D.; Beckers, L.; Wijnands, E.; Soehnlein, O.; Zernecke, A.; Seijkens, T.; Engel, D.; Cleutjens, J.P.M.; Keller, A.M. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J. Exp. Med. 2010, 207, 391–404. [Google Scholar] [CrossRef]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Nassar, T.; Sachais, B.S.; Akkawi, S.; Kowalska, M.A.; Bdeir, K.; Leitersdorf, E.; Hiss, E.; Ziporen, L.; Aviram, M.; Cines, D.B. Platelet Factor 4 Enhances the Binding of Oxidized Low-density Lipoprotein to Vascular Wall Cells. J. Biol. Chem. 2003, 278, 6187–6193. [Google Scholar] [CrossRef] [Green Version]
- Pitsilos, S.; Hunt, J.L.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.M.; Mitchell, M.E. Platelet factor 4 localization in carotid atherosclerotic plaques: Correlation with clinical parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef]
- O’brien, J.R.; Etherington, M.D.; Pashley, M.A. Intra-Platelet Platelet Factor 4 (IP.PF4) and the Heparin-Mobilisable Pool of PF4 in Health and Atherosclerosis. Thromb. Haemost. 1984, 51, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Scheuerer, B.; Ernst, M.; Durrbaumlandmann, I.; Fleischer, J.; Gragegriebenow, E.; Brandt, E.; Flad, H.D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Tyka, M.; Helmes, C.M.; Akhavanpoor, M.; Rupp, G.; Domschke, G.; Linden, F.; Wolf, A.; Doesch, A.O.; Lasitschka, F. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+in vitro and in vivo. Innate Immun. 2015, 21, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachais, B.S.; Turrentine, T.; Mckenna, J.M.D.; Rux, A.H.; Rader, D.J.; Kowalska, M.A. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb. Haemost. 2007, 98, 1108–1113. [Google Scholar] [PubMed]
- Gleissner, C.A.; Shaked, I.; Erbel, C.; Bockler, D.; Katus, H.A.; Ley, K. CXCL4 Downregulates the Atheroprotective Hemoglobin Receptor CD163 in Human Macrophages. Circ. Res. 2010, 106, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hundelshausen, P.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; Weber, C. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Schober, A.; Manka, D.; Von Hundelshausen, P.; Huo, Y.; Hanrath, P.; Sarembock, I.J.; Ley, K.; Weber, C. Deposition of Platelet RANTES Triggering Monocyte Recruitment Requires P-Selectin and Is Involved in Neointima Formation After Arterial Injury. Circulation 2002, 106, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Veillard, N.R.; Kwak, B.R.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.I.; Mach, F. Antagonism of RANTES Receptors Reduces Atherosclerotic Plaque Formation in Mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Steffens, S.; Arnaud, C.; Pelli, G.; Burger, F.; Proudfoot, A.E.I.; Mach, F. A Novel RANTES Antagonist Prevents Progression of Established Atherosclerotic Lesions in Mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Koperlenkiewicz, O.M.; Kaminska, J.; Lisowska, A.; Milewska, A.J.; Hirnle, T.; Dymickapiekarska, V. Factors Associated with RANTES Concentration in Cardiovascular Disease Patients. BioMed Res. Int. 2019, 2019, 3026453. [Google Scholar]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Doring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.; Baxter, S.A.; Dreau, D.; Nesmelova, I.V. The heterodimerization of platelet-derived chemokines. Biochim. Biophys. Acta 2013, 1834, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R.; Von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sonmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Mollmann, J. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 2018, 8, 10647. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Uchida, O.; Kajiwara, N.; Kim, E.Y.; Patel, A.C.; Osullivan, M.P.; Walter, M.J.; Schwendener, R.A.; Cook, D.N.; Danoff, T.M. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 2005, 11, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; Von Hundelshausen, P.; Ley, K. Platelet Chemokines in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [Green Version]
- Ghasemzadeh, M.; Kaplan, Z.S.; Alwis, I.; Schoenwaelder, S.M.; Ashworth, K.J.; Westein, E.; Hosseini, E.; Salem, H.H.; Slattery, R.; McColl, S.R.; et al. The CXCR1/2 ligand NAP-2 promotes directed intravascular leukocyte migration through platelet thrombi. Blood 2013, 121, 4555–4566. [Google Scholar] [CrossRef] [Green Version]
- Orn, S.; Breland, U.M.; Mollnes, T.E.; Manhenke, C.; Dickstein, K.; Aukrust, P.; Ueland, T. The Chemokine Network in Relation to Infarct Size and Left Ventricular Remodeling Following Acute Myocardial Infarction. Am. J. Cardiol. 2009, 104, 1179–1183. [Google Scholar] [CrossRef]
- Abiyounes, S.; Sauty, A.; Mach, F.; Sukhova, G.K.; Libby, P.; Luster, A.D. The Stromal Cell–Derived Factor-1 Chemokine Is a Potent Platelet Agonist Highly Expressed in Atherosclerotic Plaques. Circ. Res. 2000, 86, 131–138. [Google Scholar] [CrossRef]
- Merckelbach, S.; Der Vorst, E.P.C.V.; Kallmayer, M.; Rischpler, C.; Burgkart, R.; Doring, Y.; De Borst, G.; Schwaiger, M.; Eckstein, H.H.; Weber, C. Expression and Cellular Localization of CXCR4 and CXCL12 in Human Carotid Atherosclerotic Plaques. Thromb. Haemost. 2018, 118, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Von Ungernsternberg, S.N.I.; Seizer, P.; Schlegel, F.; Buttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.F.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4–CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.; Gremse, F.; Kiessling, F.; Weber, C.; Schober, A. CXCL12 Promotes the Stabilization of Atherosclerotic Lesions Mediated by Smooth Muscle Progenitor Cells in Apoe -Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raines, E.W. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004, 15, 237–254. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folestad, E.; Kunath, A.; Wågsäter, D. PDGF-C and PDGF-D signaling in vascular diseases and animal models. Mol. Asp. Med. 2018, 62, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Ferri, N. Naturally occurring PDGF receptor inhibitors with potential anti-atherosclerotic properties. Vasc. Pharmacol. 2015, 70, 1–7. [Google Scholar] [CrossRef]
- Hu, W.; Huang, Y. Targeting the platelet-derived growth factor signalling in cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1221–1224. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Sudo, T.; Yokode, M.; Murayama, T.; Kataoka, H.; Takakura, N.; Nishikawa, S.; Nishikawa, S.; Kita, T. Functional Blockade of Platelet-Derived Growth Factor Receptor-β but Not of Receptor-α Prevents Vascular Smooth Muscle Cell Accumulation in Fibrous Cap Lesions in Apolipoprotein E–Deficient Mice. Circulation 2001, 103, 2955–2960. [Google Scholar] [CrossRef] [Green Version]
- Kozaki, K.; Kaminski, W.E.; Tang, J.; Hollenbach, S.; Lindahl, P.; Sullivan, C.M.; Yu, J.C.; Abe, K.; Martin, P.J.; Ross, R. Blockade of Platelet-Derived Growth Factor or Its Receptors Transiently Delays but Does Not Prevent Fibrous Cap Formation in ApoE Null Mice. Am. J. Pathol. 2002, 161, 1395–1407. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Medley, S.C.; Hu, T.; Hinsdale, M.E.; Lupu, F.; Virmani, R.; Olson, L.E. PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 2015, 6, 7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhiainen, A.; Imai, S.; Rauvala, H.; Parkkinen, J. Occurrence of Amphoterin (HMG1) as an Endogenous Protein of Human Platelets that Is Exported to the Cell Surface upon Platelet Activation. Thromb. Haemost. 2000, 84, 1087–1094. [Google Scholar] [PubMed]
- Ahrens, I.; Chen, Y.; Topcic, D.; Bode, M.; Haenel, D.; Hagemeyer, C.E.; Seeba, H.; Duerschmied, D.; Bassler, N.; Jandeleitdahm, K. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb. Haemost. 2015, 114, 994–1003. [Google Scholar] [PubMed] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.; Borst, O. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; Dangelo, A.; Bianchi, M. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; Mcredmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, P.; Satoh, K.; Odell, M.R.; Soe, N.N.; Cui, Z.; Mohan, A.; Abe, J.I.; Alexis, J.D.; Sparks, J.D.; Berk, B.C. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E–deficient mice. J. Exp. Med. 2011, 208, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seizer, P.; Von Ungernsternberg, S.N.I.; Schonberger, T.; Borst, O.; Munzer, P.; Schmidt, E.; Mack, A.F.; Heinzmann, D.; Chatterjee, M.; Langer, H.F. Extracellular Cyclophilin A Activates Platelets Via EMMPRIN (CD147) and PI3K/Akt Signaling, Which Promotes Platelet Adhesion and Thrombus Formation In Vitro and In Vivo. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.J.; Schlegel, M.; Zhou, F.; Gorenchtein, M.; Bolstorff, J.; Moore, K.J.; Fisher, E.A.; Berger, J.S. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci. Transl. Med. 2019, 11, eaax0481. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, A.; Rhyn, P.; Schoedon, G.; Schaer, D.J. Regulated expression of platelet factor 4 in human monocytes—Role of PARs as a quantitatively important monocyte activation pathway Abstract: Human mononuclear phagocytes have recently been shown to express constitutively and even more so, upon stimul. J. Leukoc. Biol. 2005, 78, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382.e23. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Momi, S.; Baglioni, S.; Casali, L.; Giannini, S.; Rossi, R.; Page, C.P.; Gresele, P. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am. J. Respir Crit. Care Med. 2008, 177, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Borst, O.; Gehring, E.M.; Schoenberger, T.; Urban, B.; Ninci, E.; Seizer, P.; Schmidt, C.; Bigalke, B.; Koch, M. PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J. Mol. Med.-JMM 2010, 88, 1277–1288. [Google Scholar] [CrossRef]
- Witte, A.; Rohlfing, A.-K.; Dannenmann, B.; Dicenta, V.; Nasri, M.; Kolb, K.; Sudmann, J.; Castor, T.; Rath, D.; Borst, O. The chemokine CXCL14 mediates platelet function and migration via direct interaction with CXCR4. Cardiovasc. Res. 2020, 80, cvaa080. [Google Scholar] [CrossRef]
- Witte, A.; Chatterjee, M.; Lang, F.; Gawaz, M. Platelets as a Novel Source of Pro-Inflammatory Chemokine CXCL14. Cell Physiol. Biochem. 2017, 41, 1684–1696. [Google Scholar] [CrossRef]
- Bradfield, P.F.; Scheiermann, C.; Nourshargh, S.; Ody, C.; Luscinskas, F.W.; Rainger, G.E.; Nash, G.B.; Miljkovic-Licina, M.; Aurrand-Lions, M.; Imhof, B.A. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood 2007, 110, 2545–2555. [Google Scholar] [CrossRef]
- Gils, J.M.V.; Martins, P.A.D.C.; Mol, A.; Hordijk, P.L.; Zwaginga, J.J. Transendothelial migration drives dissociation of plateletmonocyte complexes. Thromb. Haemost. 2008, 99, 271–279. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Tang, C. Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 9760. https://doi.org/10.3390/ijms21249760
Wang L, Tang C. Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. International Journal of Molecular Sciences. 2020; 21(24):9760. https://doi.org/10.3390/ijms21249760
Chicago/Turabian StyleWang, Lei, and Chaojun Tang. 2020. "Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives" International Journal of Molecular Sciences 21, no. 24: 9760. https://doi.org/10.3390/ijms21249760
APA StyleWang, L., & Tang, C. (2020). Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. International Journal of Molecular Sciences, 21(24), 9760. https://doi.org/10.3390/ijms21249760